


8
Movement Disorders
Note: Significant diseases are indicated in bold and syndromes in italics.
I. Parkinson’s Disease (Box 8.1)
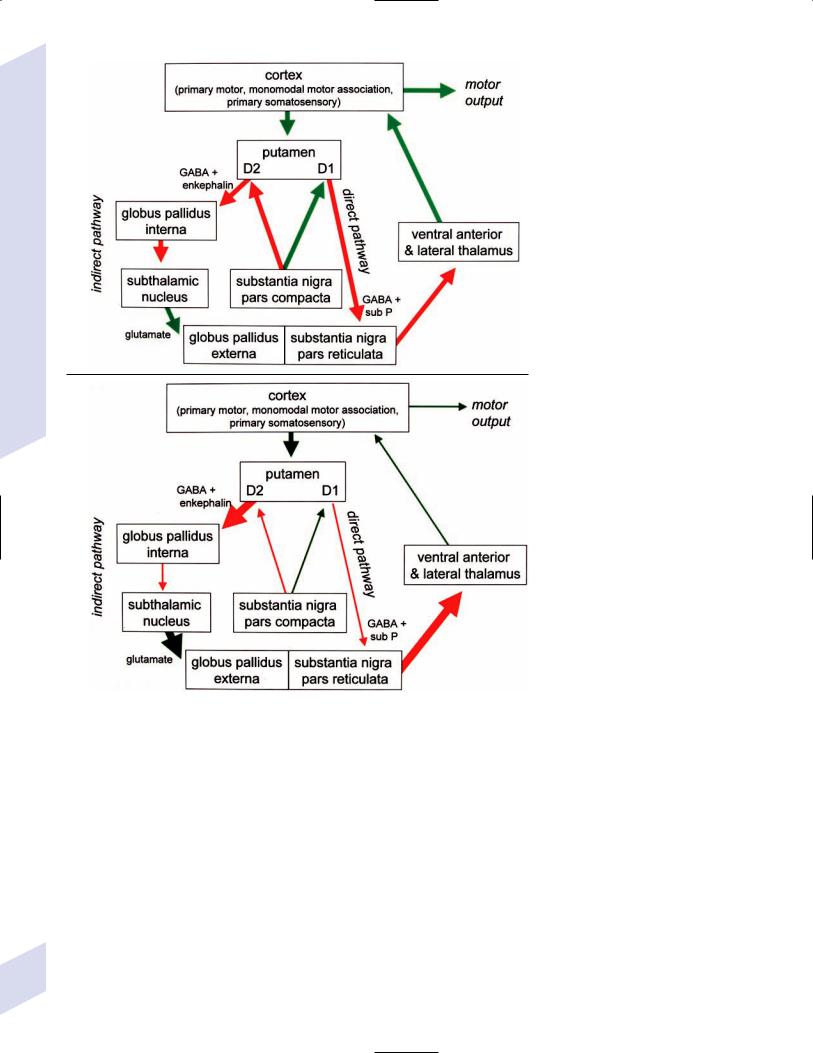
1.Pathophysiology: increased inhibition of the ventral anterior and lateral thalamic output to the motor cortex caused by loss of dopaminergic input to the striatum (Fig. 8–1)
a.D2 receptors in the striatum are expressed on aspiny and spiny neurons (see p. 11); activation of the D2 receptor causes acetylcholine release onto muscarinic receptors that inhibits glutamatergic input to the striatum from the cortex and the GABAergic neurotransmission within the basal ganglia (i.e., D2 receptor activation inhibits most of the basal ganglia circuit)
b.histology: degeneration of the dopaminergic substantia nigra pars compacta neurons and, to a lesser extent, the basal forebrain (e.g., nucleus basalis of Meynert), the hypothalamus, and brainstem pigmented neurons (locus coeruleus, dorsal vagal motor nucleus) probably due to dysfunction in the ubiquitin–proteosome system
i.neuron loss is pronounced in lateroventral part of pars compacta, and is usually asymmetric; symptoms appear typically when 80% of substantia nigra pars compacta neurons are lost
ii.surviving neurons often exhibit eosinophilic inclusions surrounded by a halo that contain -synuclein and ubiquitin {Lewy body (see p. 162)} (Box 8.1)
(1)-synuclein is a presynaptic protein thought to be involved in dopamine release and/or synaptic plasticity; in Parkinson’s disease it becomes insoluble and aggregates into fibrils that attract ubiquitin and neurofilaments, thereby forming Lewy bodies
c.genetics: the majority of cases are sporadic, but several inherited forms exist (Table 8–1)
i.parkin: acts to covalently link proteins targeted for destruction to ubiquitin, which then can be recognized by proteosomes; mutations cause accumulation of intracellular proteins that form Alzheimer’s-like neurofibrillary tangles instead of Lewy bodies
(1)accounts for the majority of early-onset Parkinson’s disease
ii.ubiquitin C-terminal hydrolase L1 acts to release ubiquitin from its conjugated protein after delivery of the conjugated protein to the proteosome
iii.PINK1: a mitochondrial protein kinase that regulates the electron transport chain
2.Epidemiology: exhibits increased incidence according to geographic (industrialized countries nonindustrialized countries) and racial factors
a.risk factors for sporadic Parkinson’s disease include drinking well water, rural residence, farming (possibly related to pesticide exposure in industrialized countries), and employment in wood pulp mills or steel industries; risk may be reduced by cigarette smoking and caffeine use
Box 8.1
Synucleinopathies
Parkinson’s disease, multisystem atrophy, Lewy body dementia
Parkinson’s Disease
183
8 Movement Disorders
184
A
B
Figure 8–1 (A) normal basal ganglia function. (B) Basal ganglia function in Parkinson’s disease.
Table 8–1 Familial Parkinson’s Disease
Major subtypes |
Chromosome |
Mutated gene/protein |
Notes |
|
|
|
|
Autosomal dominant |
|
|
|
PARK 1,4 |
4 |
-synuclein |
Usually a de novo |
|
|
|
mutation |
PARK 3 |
2 |
? |
|
PARK 5 |
4 |
Ubiquitin C-terminal |
|
|
|
hydrolase L1 |
|
Autosomal recessive |
|
|
|
PARK 2 |
6 |
Parkin (E3 ubiquitin |
Onset 20–40 years |
|
|
ligase) |
of age |
PARK 6 |
1 |
PINK-1 |
|
PARK 7 |
1 |
DJ-1 |
|
|
|
|
|

3.Symptoms: onset is typically 50–60 years of age in patients with sporadic Parkinson’s disease; a younger age of onset suggests an inherited form
a.initially presents as a unilateral resting tremor, bradykinesia, or limb stiffness; progressive rigidity and bradykinesia eventually will become bilateral and will involve freezing episodes and dysphagia
i.progression involves hypophonia, dysarthria, dysphagia, and an expressionless face
ii.symptoms are slowly progressive, presenting asymmetrically or unilaterally
iii.symptoms most commonly begin in the upper extremities, but occasionally may initiate in the lower extremities or jaw (particularly with tremor)
b.depression (50%)
c.dementia (30%): half of demented Parkinson’s disease patients have no other pathological cause, but 35% exhibit Alzheimer’s-like histological changes
d.restless legs syndrome; akathisia; REM sleep behavioral disorder (see pp. 169–170)
e.autonomic symptoms (urinary urgency, constipation {intestinal pseudoobstruction}, sexual dysfunction) and oculomotor abnormalities are mild and develop only late in the disease course
i.20% of patients develop significant orthostatic hypotension often due to medication side effects, but this can represent the misdiagnosis of a multisystem atrophy disorder
f.dementia develops in 30% of patients with advanced disease
4.Treatment (Table 8–2)
i.management of autonomic dysfunction
(1)orthostatic hypotension
(a)nonpharmacological treatments: graded position changes; avoid Valsalva maneuvers; squatting or flexing of the lower extremity musculature prior to standing; increase fluid and salt intake; lower extremity compressive garments
(b)pharmacological treatments
(i)fludrocortisone
(ii)midodrine, ephedrine, pseudoephedrine, methylphenidate
(iii)desmopressin acetate (ddAVP); erythropoietin; caffeine
(2)bladder hyperactivity: reduce drinking in the evening; anticholinergic medications (oxybutynin, propantheline)
(3)constipation: dietary modification and stool softeners; discontinuing anticholinergic medications
ii.management of side effects of medical treatment
(1)“off”/freezing phenomenon
(a)combine a dopamine agonist with Sinemet
(b)increase the frequency of combined medications
(c)substitute or add a sustained-release preparations of Sinemet
(2)dyskinesia/dystonia
(a)occurring with the peak in drug levels (“on” dyskinesia/ dystonia)
(i)lower the dose of dopamine agonists or Sinemet
(ii)substitute sustained-release preparations of Sinemet for shorter-acting medications
(iii)add a catechol-O-methyltransferase (COMT) antagonist
(iv)consider neurosurgical intervention
(b)occurring a minimal drug levels (“off” dyskinesia/dystonia)
(i)evening “off” dyskinesia: add sustained-release Sinemet in the evening
Parkinson’s Disease
185

8 Movement Disorders
186
Table 8–2 Medications for Bradykinesia, Tremor, and Rigidity in Parkinson’s Disease
Drug |
Mechanism of action |
Side effects |
Notes |
|
|
|
|
L-dopa carbidopa |
Dopamine precursor (L-dopa) |
Nausea, dystonia, orthostatic |
Regular Sinemet effect lasts 6 hr |
(Sinemet) |
peripheral MAO inhibitor (carbidopa) |
hypotension, hallucinations |
(less in late disease) |
|
Add domperidone if carbidopa is |
(late in disease) |
Keep carbidopa 150 mg/d to |
|
|
||
|
ineffective at a 1 : 4 ratio |
|
avoid CNS penetration |
|
|
|
Low-protein diet improves |
|
|
|
absorption from GI system |
|
|
|
|
Ergot DA agonists |
Agonists of 5-HT and |
As per Sinemet; anorexia, |
Side effects with pergolide are more |
|
adrenoceptors, as well as |
confusion |
mild than those with bromocriptine |
Bromocriptine |
D2 agonist, partial D1 agonist |
Painful swollen feet |
|
Pergolide (Permax) |
D1 and D2 agonist |
|
|
|
|
|
|
Non-ergot DA |
|
|
|
agonists |
|
|
|
Apomorphine |
|
Nausea |
Can be given as a nasal spray, |
|
|
|
sublingually, SQ, or PR |
Pramipexole |
D2 receptor agonist; minimal on |
As per Sinemet; drowsiness |
Cause less motor fluctuations than |
(Mirapex) |
D1 receptors |
and sleep attacks |
Sinemet, but more cognitive and |
Ropinirole (Requip) |
|
|
hypotensive side effects |
|
|
|
|
|
|
|
|
Selegiline (Deprenyl) |
Inhibits MAO-B breakdown of |
As per Sinemet |
May be neuroprotective, therefore |
|
L-DOPA in the brain |
|
is initial treatment for young |
|
|
|
patients |
|
|
|
Many drug interactions (particularly |
|
|
|
antidepressants) |
|
|
|
|
COMT inhibitors |
Inhibit COMT breakdown of L-DOPA |
As per Sinemet; liver failure |
Increase “on” time duration of |
Entacapone |
in the GI system and liver |
with tolcapone |
Sinemet |
|
|
|
|
Tolcapone |
|
|
|
|
|
|
|
Benztropine |
Muscarinic acetylcholine receptor |
Dry mouth, blurred vision, |
Generally are more effective |
(Cogentin) |
antagonists |
urinary retention, confusion |
against tremor and rigidity than |
Trihexyphenidyl |
|
|
bradykinesia |
|
|
|
|
(Artane) |
|
|
|
|
|
|
|
Amantadine |
? NMDA receptor antagonist, |
Restlessness, insomnia, |
Modest effect, useful in early/mild |
|
anticholinergic, MAO-B inhibition, |
depression, hallucinations |
disease |
|
↑ DA release |
|
May reduce freezing in late disease |
|
|
|
|
|
|
|
|
Abbreviations: CNS, central nervous system; COMT, catechol-O-methyltransferase; GI, gastrointestinal; MAO, monoamine oxidase; NMDA, N-methyl-D-aspartate; SQ, subcutaneous.
(ii)between dose “off” dyskinesia
1.add dopamine agonist or amantadine
2.increase the frequency of medication administration
3.consider neurosurgical intervention
(3)treatment-induced hallucinations
(a)identify and discontinue the offending medication in order of likely causation: anticholinergics amantadine selegiline dopamine agonists Sinemet
(b)lower the dose of dopamine agonists or Sinemet
(c)add a low-dose atypical antipsychotic medications (clozapine, quetiapine) (Box 8.2)
b.surgical treatment: best response to medication postoperatively is equal to the patient’s best response to medication preoperatively, and the chief improvement is in terms of reduced fluctuations and dyskinesias
i.lesion procedures: usually done with stereotactic and intraoperative electrophysiological confirmation of location, but can be done by means of radiosurgery (e.g., gamma knife)
Box 8.2
Lewy Body dementia’s Parkinsonism is very sensitive to antipsychotics.
(1)thalamotomy: targets the ventral intermediate nucleus
(a)improves only tremor, therefore not often used in Parkinson’s disease
(b)not effective against bradykinesia, and may exacerbate hypophonia and gait abnormalities; avoid bilateral procedures due to cognitive impairment
(2)pallidotomy: targets the posterolateral region of the internal globus pallidus to avoid impairment of memory and cognitive function
(a)effective against all major Parkinson’s disease symptoms and dyskinesias
(b)limited to a unilateral procedure due to high incidence of cognitive impairment with bilateral procedures
ii.deep brain stimulator procedures: over stimulation may produce dyskinesias
(1)thalamic stimulation: effectiveness as per thalamotomy, but fewer side effects
(2)globus pallidus stimulation: effectiveness as per pallidotomy, but fewer side effects; bilateral stimulation has pronounced benefits for gait abnormalities
(3)subthalamic stimulation: most effective surgical procedure for bradykinesia, and is effective for all other major Parkinson’s disease symptoms; bilateral stimulation has pronounced benefits for gait abnormalities
II.Progressive Supranuclear Palsy (PSP) (Box 8.3)
1.Pathophysiology: Gliosis and neuron loss predominantly in the frontal cortex, globus pallidus, substantia nigra, and mesencephalic nuclei
a.histology: affected areas exhibit Alzheimer’s disease-like neurofibrillary tangles (with hyperphosphorylated tau protein) and neuropil threads
i.D2 receptors in the basal ganglia are reduced, unlike Parkinson’s disease where they are increased; this likely reflects the loss of cholinergic striatal interneurons in PSP (see p. 11)
b.genetics
i.90% of sporadic cases are associated with homozygous H1 alleles of the tau gene, although 60% of normal people also are homozygous for the H1 allele
ii.families with autosomal dominant inheritance patterns have been described
2.Symptoms: develop between 60–70 years of age
a.axial rigidity limb rigidity, leading to an abnormal gait and falls early in the disease (particularly backward falls, unlike Parkinson’s disease patients who fall forward)
b.cortical-type dysarthria; hypophonia
c.reduced voluntary saccades, particularly a vertical gaze palsy involving downward upward gaze; voluntary smooth pursuit movements become impaired thereafter
i.involuntary (vestibulo-ocular) eye movements are preserved until late in the disease
3.Diagnostic testing: loss of saccadic fast-phase on optokinetic testing may distinguish early PSP from Parkinson’s disease
4.Treatment: none specific
5.Prognosis: usually fatal within 6 years
Progressive Supranuclear Palsy (PSP)
Box 8.3
The tau-opathies—Alzheimer’s disease, frontotemporal lobar degeneration/Pick’s disease; cortical-basal ganglionic degeneration; progressive supranuclear palsy
187

8 Movement Disorders
III. Cortical-Basal Ganglionic Degeneration
1.Pathophysiology: Gliosis with neuronal loss and vacuolation in cortical layers III–V (like frontotemporal lobar degeneration/Pick’s disease), the basal ganglia, and the substantia nigra; depigmentation of the substantia nigra also occurs
a.surviving neurons exhibit accumulations of hyperphosphorylated tau
b.genetics: associated with the H1 allele of the tau gene, as per PSP
2.Symptoms: develop 60 years of age; symptoms occur in a random order and may relapse and remit, but they all are concentrated initially in one limb
a.subcortical symptoms: Parkinsonism; postural and/or action tremor; limb dystonia; reflex myoclonus
b.cortical symptoms: dementia similar to frontotemporal lobar degeneration (see p. 160); apraxia (e.g., constructional dyspraxia); cortical sensory loss; weakness
i.Gerstmann’s syndrome and alien-hand syndrome is a rare complication of corticobasal ganglionic degeneration
3.Diagnostic testing: MRI may reveal regional degeneration in frontal and parietal cortex
4.Treatment: none specific
5.Prognosis: symptoms remain focally in one limb for several years, but eventually progress to involve other limbs; severe dysphagia eventually develops
IV. Multiple System Atrophy (MSA)
1.General histology: exhibits multiple types of inclusion bodies to varying degrees; the most typical for MSAs are
a.an argentophilic cytoplasmic inclusion in neurons and glia that resemble neuropil threads but that contain only ubiquitin and no tau protein
b.a flame-shaped glial cytoplasmic inclusion similar to the neurofibrillary tangles of Alzheimer’s disease but that do not contain tau protein
2.The old classification scheme
a.olivopontocerebellar degeneration (OPCD)—idiopathic degeneration of the inferior olivary nuclei, ventral pontine nuclei, and cerebellar cortex with relative preservation of the substantia nigra and striatum
b.striatonigral degeneration—idiopathic degeneration of the putamen and substantia nigra with relative preservation of the globus pallidus, caudate, and subthalamic nucleus
c.Shy-Drager syndrome—idiopathic degeneration of the intermediolateral cell column in thoracic and lumbar spinal cord, the dorsal motor nucleus of the vagus, and in the sympathetic chain ganglia
3.The new classification scheme: The Shy-Drager syndrome has been eliminated because autonomic dysfunction is present in all types of multiple-system atrophy
a.multiple system atrophy with predominant parkinsonian features (MSA-P)/striatonigral degeneration form of MSA
i.histology: the pattern of degeneration is as for striatonigral degeneration Shy-Drager syndrome
(1)despite pronounced Parkinsonian features, does not have Lewy bodies, neurofibrillary tangles, or even many of the MSA inclusion bodies in the surviving neurons of substantia nigra
ii.epidemiology: sporadic occurrence only
iii.symptoms: bradykinesia, rigidity, autonomic dysfunction; less commonly involves myoclonus, neck anteroflexion, or dysarthria
188
(1)autonomic dysfunction usually predates parkinsonian features
(2)hyperpigmentation of the extremities
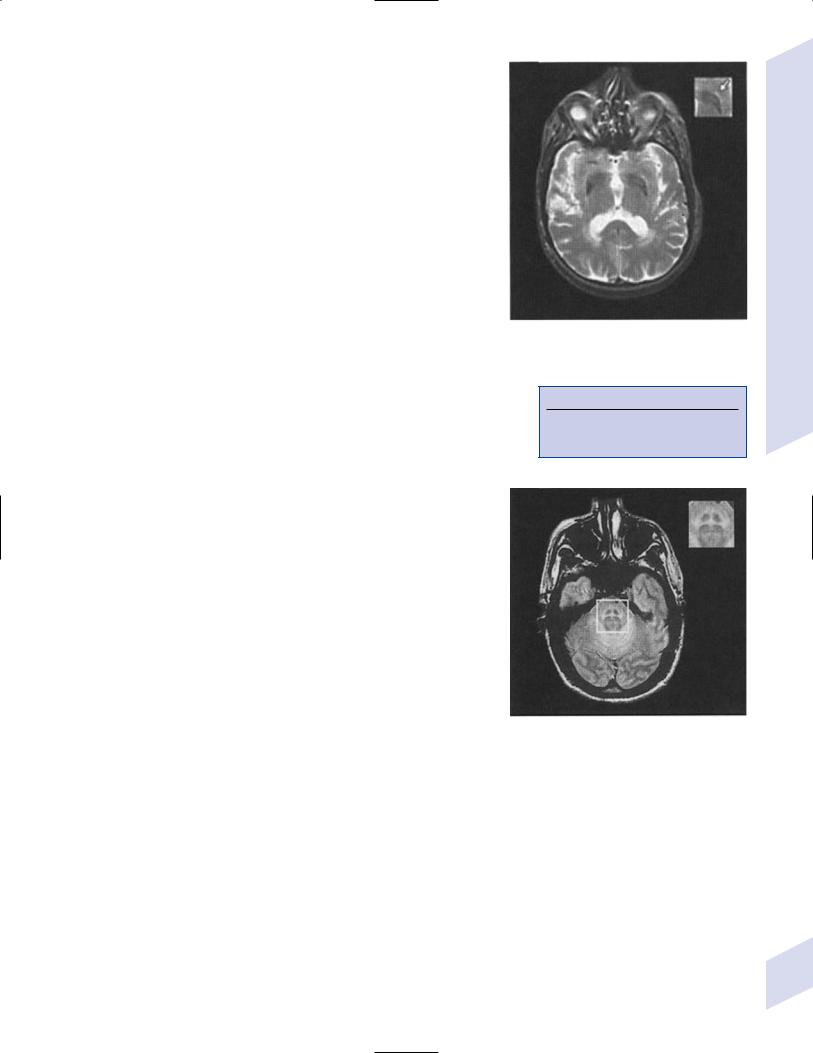
iv.diagnostic testing: late in the disease, MRI demonstrates
(1)T2 hypointensity in the globus pallidus with a lateral linear hyperintensity, and putamen atrophy (Fig. 8–2)
(2)loss of pontine fibers {hot-cross bun sign} (Fig. 8–3)
v.treatment: dopaminergic agents usually have little effect against the parkinsonism because D2 receptors are lost in the striatum; management of autonomic dysfunction as per Parkinson’s disease
b.multiple system atrophy with predominant cerebellar features (MSA-C)/OPCD form of MSA
i.histology: pattern of degeneration is as for olivopontocerebellar degeneration Shy-Drager syndrome
ii.epidemiology: peak incidence at 50–60 years of age; usually sporadic, but may have autosomal dominant or recessive patterns of inheritance (usually such cases are classified under spinocerebellar ataxias)
iii.symptoms: ataxia in the lower extremities upper extremities; cerebellar-type dysarthria; autonomic dysfunction (Box 8.4)
(1)subtypes of MSA-C include
(a)parkinsonian symptoms (50%)
(b)retinal degeneration
(c)spastic paraplegia and areflexia
(d)dementia, ophthalmoplegia, and extrapyramidal signs
iv.diagnostic testing: late in the disease, neuroimaging demonstrates atrophy of the brainstem and cerebellum
v.treatment: none specific; management of autonomic dysfunction as per Parkinson’s disease
Figure 8–2 T2 MRI of MSA-P. (From Kaufmann H, Biaggioni I. Autonomic failure in neurodegenerative disorders. Semin Neurol 2003, 23:357, Fig. 3A. Reprinted by permission.)
Box 8.4
MSA-C usually occur in a hereditary pattern, and thus are considered spinocerebellar ataxias (see p. 198).
V. Dystonias
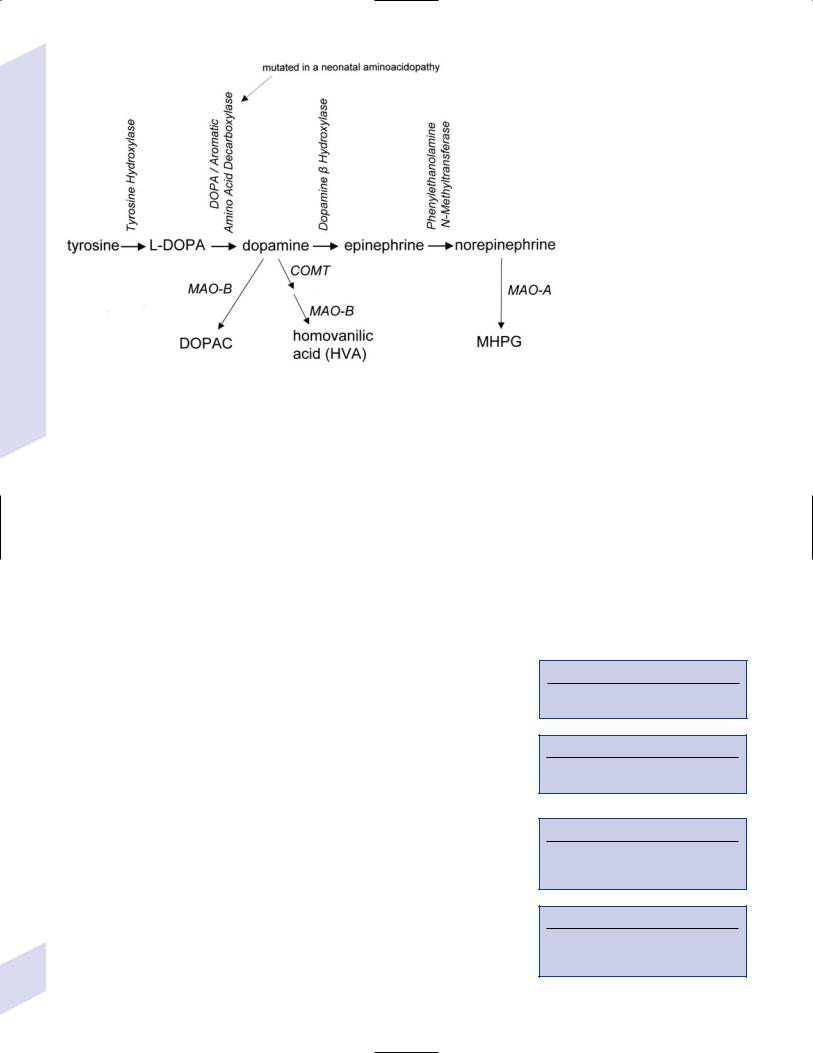
1.Pathophysiology: primary dystonias result from dysfunction the dopaminergic systems (Fig. 8–4) in the of basal ganglia without apparent neuron loss or neurochemical deficit; abnormal function of the motor and sensory cerebral cortex and in lower motor neuron reflexes are likely reactions to the basal ganglia dysfunction
a.primary and secondary motor and sensory cerebral cortex exhibit abnormal activation patterns even when dystonic movements are not occurring
b.lower motor neuron reflexes are apparently abnormal because the dystonic movements involve unnecessary activation of co-agonist muscles and also antagonist muscles
Figure 8–3 Hot-cross bun sign of MSA-P. (From Kaufmann H, Biaggioni I. Autonomic failure in neurodegenerative disorders. Semin Neurol 2003, 23:357, Fig. 3C. Reprinted by permission.)
i.even when dystonic movements are not occurring, dystonia patients exhibit reduced skeletal muscle inhibition (e.g., reduced reciprocal inhibition of antagonist muscles during voluntary movement in focal dystonia patients, reduced suppression of the blink reflex in blepharospasm patients)
c.focal lesions can cause dystonia when they are located in the contralateral putamen globus pallidus caudate, thalamus, brainstem
i.rarely dystonia presents after peripheral nerve injury, likely by altering basal ganglia function in genetically susceptible individuals
2.Causes of dystonia
Dystonias
189
8 Movement Disorders
190
a.primary dystonias (Table 8–3)
b.secondary dystonias
i.neurodegenerative disorder-related dystonias: Parkinson’s disease, multiple-system atrophies, progressive supranuclear palsy, corticobasal ganglionic degeneration, spinocerebellar atrophies (particularly
Table 8–3 Primary Dystonias
Type |
Genetics |
Notes |
|
|
|
Early-onset dystonia |
Mutation in torsin A |
Childhood onset |
(DYT1) |
gene, an ATP-binding |
Progresses to other limbs |
|
protein |
|
|
Accounts for most primary |
|
|
(AD, 60% penetrance) |
|
|
dystonia |
|
|
|
|
|
|
|
Dystonia-Parkinsonism/ |
Unknown gene (X-linked) |
Segmental or diffuse dystonia |
Lubag syndrome |
|
Common only in Philippines |
|
|
|
|
|
|
DOPA-responsive dystonia- |
Mutation in GTP |
Treat with levodopa–carbidopa |
Parkinsonism |
cyclohydrolase I gene |
(Sinemet) |
|
(AD) (Box 8.5) |
|
Segawa syndrome |
Mutation in tyrosine |
Treat with levodopa–carbidopa |
|
hydroxylase gene (AR) |
(Sinemet) |
|
(Box 8.6) |
Dystonia is worse in evening, |
|
|
|
|
|
better or absent in morning |
|
|
|
Rapid-onset dystonia- |
(AD) |
Adult onset |
parkinsonism |
|
Develops over several hours to |
|
|
|
|
|
weeks |
|
|
|
Paroxysmal nonkinesigenic |
(AD) |
Dystonia is not triggered by |
choreoathetosis |
|
activity |
|
|
|
Paroxysmal choreoathetosis |
(AD) |
Dystonia occurs with diplopia, |
with spasticity and ataxia |
|
paresthesias, and ataxia |
|
|
Spastic paraplegia develops |
|
|
between episodes of dystonic |
|
|
symptoms |
|
|
|
Myoclonic dystonia |
Mutation of -sarcoglycan |
Symptoms reduced by alcohol use |
(Box 8.7) |
gene (AD) (Box 8.8) |
|
|
|
|
Note: Dark gray primary dystonias with parkinsonian features; light gray primary dystonias with choreoathetosis.
Abbreviations: AD, autosomal dominant; GTP, guanosine 5 -triphosphate.
Figure 8–4 Biosynthesis of dopamine, norepinephrine, and epinephrine.
Box 8.5
GTP cyclohydrolase I synthesizes tetrahydrobiopterin cofactor for DA synthesis.
Box 8.6
Tyrosine hydroxylase is directly involved in DA synthesis.
Box 8.7
Localize to the same region of chromosome 16 as benign familial infantile convulsions
Box 8.8
Other sarcoglycans are mutated in autosomal recessive limb-girdle muscular dystrophy

Machado-Joseph’s disease/SCA-3), ataxic telangiectasia, Huntington’s disease, dentatorubropallidoluysian atrophy (DRPLA)
ii.autoimmune: related to antibasal ganglia antibodies, which occur in 65% of patients with atypical dystonia; probably these antibodies are a reaction to a Streptococcus infection similar to Sydenham’s chorea and pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS syndrome; see p. 193)
iii.disorders of metal overload: Wilson’s disease, Hallervorden-Spatz disease
iv.leukodystrophies, particularly metachromatic leukodystrophy
v.metabolic diseases of the nervous system: ceroid lipofuscinosis, methylmalonic academia, aromatic amino acid decarboxylase deficiency, homocystinuria, Lesch-Nyhan’s disease
vi.acquired conditions: biopterin deficiency (due to failure of dopamine synthesis), basal ganglia injury
vii.neuroacanthocytosis (see p. 193)
viii.complex regional pain syndrome I/reflex sympathetic dystrophy
ix.iatrogenic: dopamine antagonists (cocaine, amphetamines, antipsychotics); dopamine agonists
3.Subtypes of dystonia by age groups
a.childhoodand adolescent-onset dystonias typically develop in lower extremities and progress to involve other body parts
b.adult-onset dystonias typically develop in the upper extremities or head, and do not progress
4.Symptoms
a.slow, sustained, and repetitive twisting and flexion movements and/or posturing, with normal muscle tone between episodes; may involve a degree of tremulousness, but unlike true tremor dystonic movements have a significant directional component
i.often dystonia movements become painful, particularly if they are prolonged
ii.dystonic movements can be triggered by specific activities involving the affected muscles (e.g., writer’s cramp triggered by the act of writing) or by emotional states
iii.dystonic movements may be aborted or broken by sensory stimulation (usually touch) to specific parts of the body (usually the dystonic region)
b.subtypes of dystonia by symptoms
i.focal dystonias
(1)cervical dystonias: rotation {torticollis}, extension {retrocollis}, flexion {anterocollis}, or abduction {laterocollis} of the head
(2)blepharospasm—blinking and squeezing of the orbicularis oculi muscles, may be preceded by sensation of eye irritation: often triggered by bright light
(a)typically bilateral but with a significant asymmetry
(3)Meige’s syndrome—orolingual dystonias with blepharospasm
(4)Brueghel syndrome—jaw opening with lip retraction, tongue protrusion, and platysma contraction
(5)facial/oromandibular dystonias—can involve facial, masticatory, lingual, and/or oropharynx muscles; commonly occurs with blepharospasm or cervical dystonias
(6)spastic dysphonia—dystonia of the laryngeal muscles causing the voice to become hypophonic and strained with frequent tone breaks
(7)limb dystonias
ii.multifocal dystonia, segmental dystonia, hemidystonia (rare)
iii.generalized dystonia (Box 8.9)
Box 8.9
Status dystonicus—A severe, prolonged period of dystonic posturing often preceded by increasingly frequent and prolonged episodes
Can develop out of any type of dystonia, often precipitated by changes in medications or intercurrent illness
May develop rhabdomyolysis due to prolonged muscle contraction
Requires sedation or even paralysis and ventilation if fatigue or severe pain develop
Dystonias
191
8 Movement Disorders
192
5.Diagnostic testing
a.levodopa–carbidopa or anticholinergic medication trial to distinguish DOPA-responsive dystonias
b.genetic analysis for torsin-A gene (DYT1) mutations
c.routine evaluations to evaluate for the possibility of secondary causes of dystonia
i.ceruloplasmin, slip-lamp evaluation for Kayser-Fleischer rings, and 24-hour urine copper to evaluate for the possibility of Wilson’s disease
ii.neuroimaging to evaluate for basal ganglia lesions
6.Treatment
a.medical treatment
i.botulinum toxin: effective in 75% of focal dystonias
(1)clinical resistance may develop due to blocking antibodies, especially with frequent injections; this can be overcome by changing botulinum toxin serotypes
(2)the only frequent side effect is excessive local weakness or weakness in neighboring muscles
ii.carbidopa–levodopa: effective in 15% of dystonias
iii.anticholinergic agents (benztropine, trihexyphenidyl): effective in 40% of dystonias but limited use because of side effects
iv.muscle relaxants: baclofen, tizanidine, cyclobenzaprine
(1)intrathecal baclofen is unlikely to be of benefit if baclofen given orally was not effective
v.clonazepam, gabapentin
b.surgical treatment: for patients who have failed medical therapy
i.lesion procedures
(1)rhizotomy, myomectomy: for focal dystonia
(2)thalamotomy: has a high rate of complications, particularly dysarthria and cognitive impairment
(a)bilateral thalamotomy for generalized dystonia
(b)contralateral thalamotomy for segmental dystonia
(3)pallidotomy: fewer side effects than thalamotomy
ii.deep brain stimulation in the globus pallidus: particularly effective for early-onset dystonia (DYT1), cervical dystonias, and blepharospasm
VI. Tic Disorders
1.Pathophysiology: likely related to abnormal dopaminergic activity in the basal ganglia, leading to inappropriate cortical arousal; mild abnormalities in the size and symmetry of the basal ganglia and cortical regions are apparent, as are abnormal accumulations of dopamine and D2 receptors in the basal ganglia
2.Subtypes
a. idiopathic tic disorders
i.simple or multiple motor tics: e.g., blinking, snarling, tongue protrusion, neck stretching, finger or toe curling
ii.complex motor tics: e.g., spitting, foot stomping, teeth grinding, obscene gesturing {copropraxia}
iii.simple phonic tics: e.g., grunting, throat clearing, coughing
iv.complex phonic tics: e.g., unintelligible wordor stuttering-like utterances
v.Tourette’s syndrome: combination of multiple motor tics and at least one phonic tic that begins at 20 years of age
(1)most cases are sporadic, but can exhibit polygenic inheritance with variable penetrance

b.secondary tic disorders
i.posttraumatic
ii.postinfectious: occurring after group A -hemolytic Streptococcus infections, typically pharyngitis
(1)PANDAS infection syndrome: develops acutely shortly after an infection; no relation to rheumatic fever
(a)neuropsychiatric symptoms is typically an obsessive– compulsive disorder
(2)Sydenham’s chorea—develops several months after an infection, spontaneously resolves after several weeks; involves both chorea and tic behaviors
(a)typically occurs in conjunction with symptoms of rheumatic fever (carditis, arthritis, subcutaneous nodules, erythema marginatum), and chorea may recur within a few years
(b)particularly common in patients with B cell alloantigen D8/17
iii.neuroacanthocytosis—a disorder characterized by spiny erythrocytes {acanthocytes} caused by an abnormality in membrane lipids; neurological abnormalities include orofacial dystonias involving mutilation of the facial soft tissues, choreiform movements, tic behaviors, behavioral changes, and a motor neuropathy
(1)autosomal recessive inheritance with linkage to chromosome 9
iv.drug-induced: amphetamines, dopaminergic agonists, antipsychotics, antiepileptics
3.Symptoms: idiopathic and postinfectious tic disorders generally have onset in childhood, whereas noninfectious secondary tic disorders have an adult onset
a.involuntary movements or vocalizations that can be transiently suppressed although they are associated with a subjective urge to perform them that builds up over time as they are suppressed; movements are jerky in nature (i.e., resembling myoclonus or clonus), but are characteristically stereotypical and coordinated
i.the type of tic behavior and its severity may change over time
ii.tics are exacerbated by stress (as are all dyskinesias), but also by relaxation
iii.vocalization of understandable words indicates Tourette’s syndrome, which also must involve multiple motor tics
b.tic disorder is associated with choreiform movements, dystonias, learning disorders, attention-deficit hyperactivity disorder, and obsessive–compulsive disorder (50%)
4.Treatment: antipsychotics (haloperidol, pimozide, risperidone), clonidine, clonazepam
a.selective serotonin reuptake inhibitors (SSRIs) reduce obsessive–compulsive behaviors but they do not reduce tic severity
5.Prognosis: idiopathic forms usually peak in severity during adolescence then spontaneously remit in early adulthood
VII. Huntington’s Disease
1.Pathophysiology: expansion of a trinucleotide repeat in the huntingtin gene on chromosome 4 that appears to cause cell death by NMDA excitotoxicity and/or abnormal activity of mitochondrial oxidative phosphorylation
a.requires 40 repeats for symptoms; 30–45 repeats may not produce symptoms, but are thought likely to increase in size in future generations
i.a larger number of repeats causes symptomatic presentation at a younger age {anticipation} and also a more severe course
Huntington’s Disease
193
8 Movement Disorders
b.an autosomal dominant inherited disease; inheritance of the affected huntingtin gene on the paternal chromosome produces more severe disease
c.histology: loss of spiny neurons (see p. 11) and glia in the striatum and of deep cortical neurons (particularly layer V, like Alzheimer’s disease) causes generalized atrophy that is characteristically pronounced in the caudate nuclei; surviving neurons exhibit intranuclear inclusions containing huntingtin protein and ubiquitin
2.Symptoms
a.chorea, which is pronounced during ambulation
b.psychiatric disturbance: personality change, hypersexuality, psychosis
c.progressive dementia
d.oculomotor abnormalities
i.hypometric saccades progressing to a complete inability to initiate voluntary saccades, thereby requiring head thrusting to change visual fixation
ii.inability to suppress involuntary saccades causes gaze impersistence
e.orolingual apraxia
f.parkinsonism: develops late in disease and replaces the hyperkinetic features of chorea
g.dysarthria, dysphagia, and respiratory insufficiency are preterminal symptoms
3.Diagnostic testing
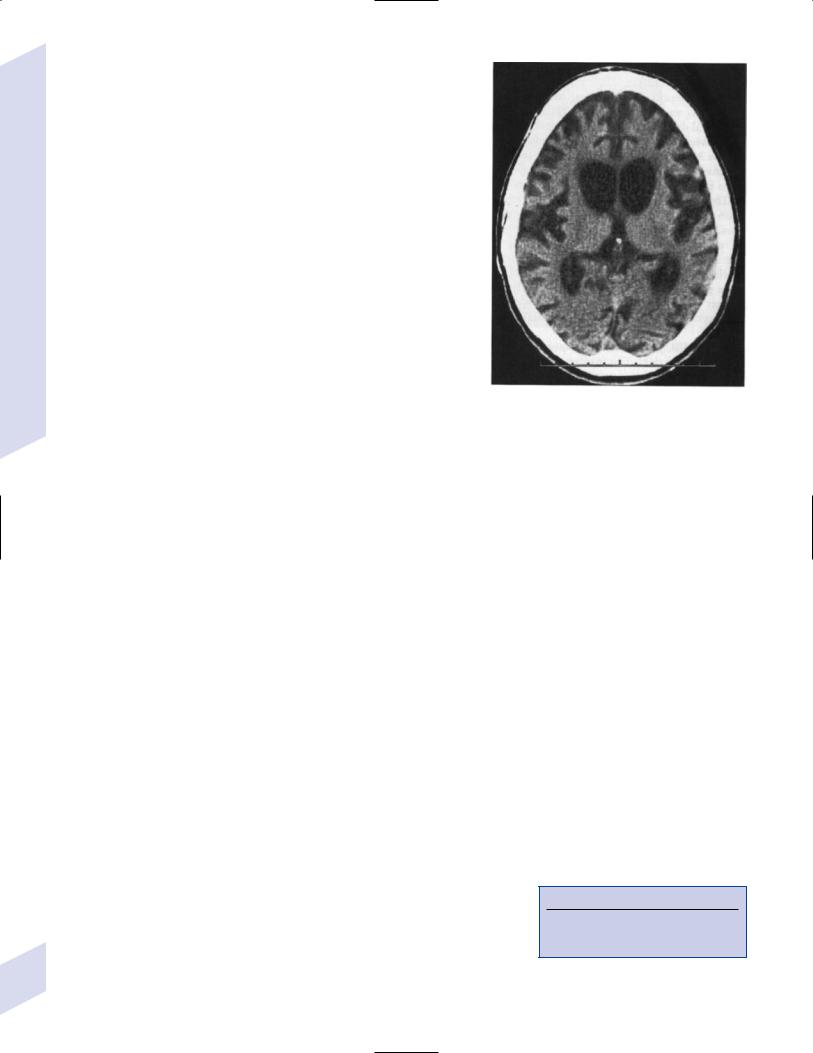
Figure 8–5 Huntington’s disease, with diffuse cortical atrophy and caudate atrophy identified by rounded lateral surfaces of the frontal horns. (From McKhann GM et al. Q&A Color Review of Clinical Neurology and Neurosurgery. Stuttgart, Germany: Georg Thieme; 2003:23, Fig. 13. Reprinted by permission.)
a.genetic testing for the trinucleotide repeat
b.neuroimaging demonstrates caudate atrophy (Fig. 8–5), similar to some frontotemporal lobar degeneration disorders
c.positron emission tomography (PET) scan demonstrates hypometabolism in the striatum
d.blood smear shows no acanthocytes, to rule out acanthocytosis
4.Treatment
a.antipsychotic medications for severe chorea
b.nondopaminergic anti-Parkinsonian medications for Parkinsonian features
c.psychiatric disturbances are managed as per routine
5.Prognosis: Death within 15 years from onset
VIII. Wilson’s Disease/Hepatolenticular Degeneration
1.Pathophysiology: Autosomal recessive mutations in a membrane-bound cop- per-transporting ATPase that secretes excess copper into the bile; accumulation of copper causes degeneration of the putamen globus pallidus, caudate, cortex mesencephalic nuclei, dentate nucleus
a.histology: hyperpigmented areas from copper deposition are associated with neuronal degeneration and local demyelination that eventually will form cavitary lesions; reactive gliosis in these areas involves
i.Alzheimer glia type 1: a multinucleated astrocyte; specific for Wilson’s disease
ii.Alzheimer glia type 2: greatly enlarged astrocytes with oval nuclei (Box 8.10)
iii.Opalski cells: large cells with coarsely granular cytoplasm that are possibly derived from histiocytes (Fig. 8–6)
2.Epidemiology: Bimodal distribution
194 |
i. early-onset (7–15 years of age): rapidly progressive symptoms |
Box 8.10
Alzheimer glia type 2 are also seen in Canavan’s disease, hepatic encephalopathy, and urea cycle disorders.
ii.late-onset (21–40 years of age): slowly progressive symptoms
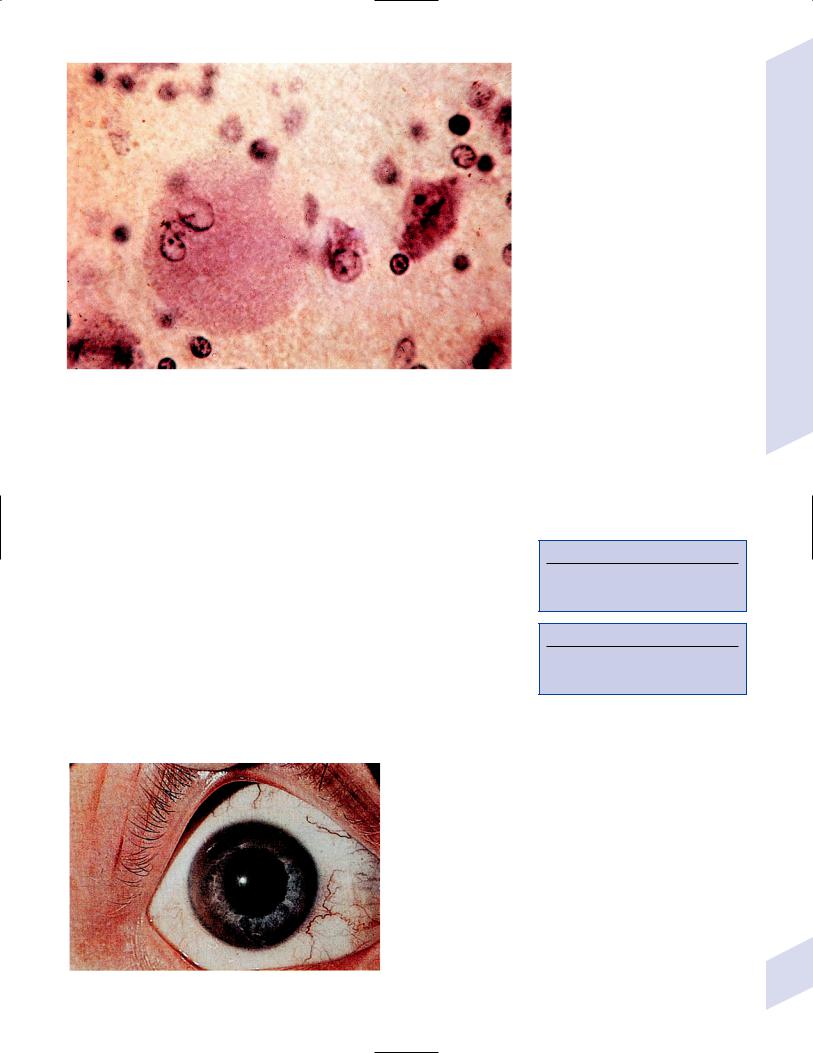
Figure 8–6 Opalski cell (left) characterized by its large size and lobulated nucleus. (From Hirano A. Color Atlas of Pathology of the Nervous System, 2nd Ed. Tokyo/New York: Igaku-Shoin Press; 1988:117, Fig. 283. Reprinted by permission.)
3.Symptoms: majority of patients begin with liver disease; however, neurological and/or psychiatric symptoms can be the initial presentation
a.neurological symptoms: dysarthria progressing to anarthria; mood lability, psychosis; rest and wing-beating tremor, ataxia, dystonia; loss of postural reflexes; seizures
b.systemic symptoms
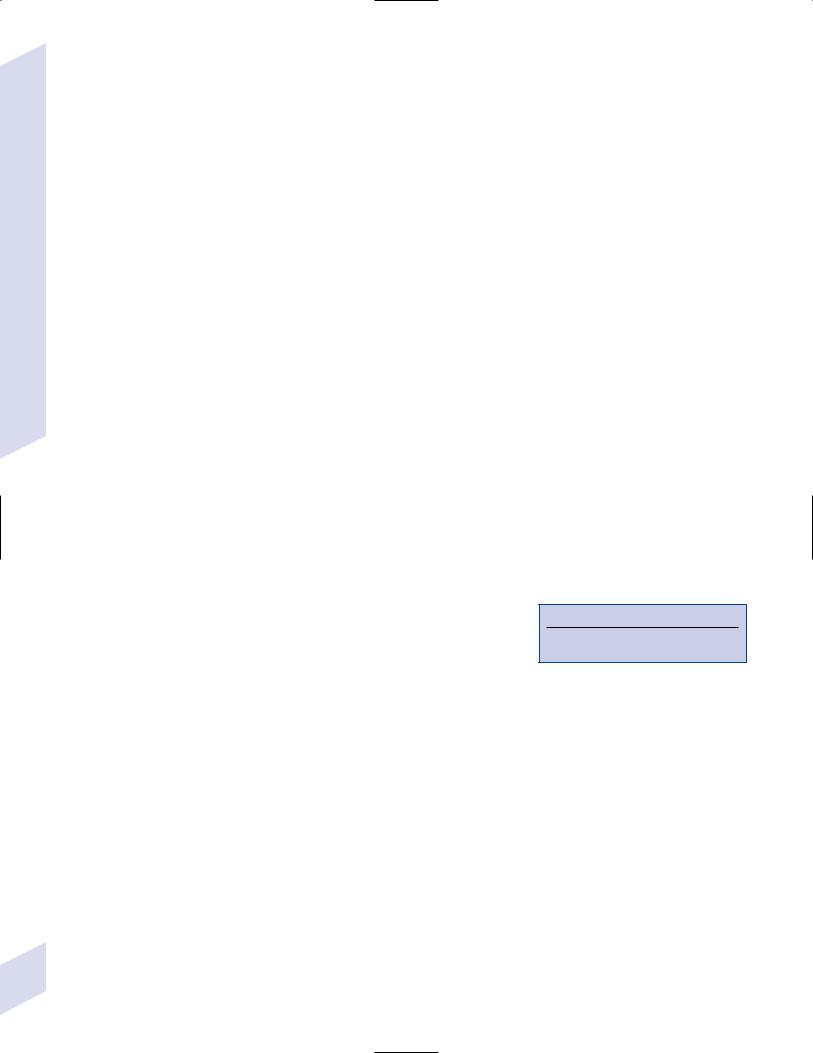
i.eye: copper deposits in Descemet’s membrane of cornea {KayserFleischer rings} are asymptomatic (Fig. 8–7) (Box 8.11)
ii.hepatitis leading to liver cirrhosis
iii.splenomegaly with hemolytic anemia
iv.bone fragility due to osteoporosis
4.Diagnostic testing: decreased serum ceruloplasmin levels (Box 8.12); elevated urine copper excretion
5.Treatment: tetrathiomolybdate; chronic penicillamine vitamin B6; zinc supplements; low copper diet
6.Prognosis: uniformly fatal without treatment; with treatment, neuropsychiatric disease improves in 50% and remains stable in 15%, whereas liver disease is reversible in only 25%
Figure 8–7 Kayser-Fleischer ring. (From Lee DA, Higginbotham. Clinical Guide to Comprehensive Ophthalmology. Stuttgart, Germany: Georg Thieme; 1999:9, Fig. 1–10. Reprinted by permission.)
Wilson’s Disease/Hepatolenticular Degeneration
Box 8.11
Kayser-Fleischer rings are present in 100% of patients with neuropsychiatric Wilson’s disease, 85% of patients with liver disease
Box 8.12
Aceruloplasminemia is similar to Wilson’s disease, but central nervous system damage is due to iron accumulation.
195
IX. Amyotrophic Lateral Sclerosis (ALS)
|
1. |
Pathophysiology: unknown, but may relate to |
|
|
|
a. |
glutamate excitotoxicity: patients exhibit increased cerebrospinal fluid |
|
|
|
(CSF) glutamate levels, reduced levels of the astrocyte excitatory amino acid |
|
|
|
transporter (EAAT-2), and abnormal messenger ribonucleic acid (mRNA) |
|
|
|
processing of the EAAT-2; excessive glutamate stimulation of S-alpha- |
|
|
|
amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors |
|
|
|
may increase intracellular calcium and allow the generation of free radicals |
|
|
|
that then damage cellular proteins by tyrosine nitration, causing apoptosis |
|
|
b. |
autoimmune injury: patients exhibit an increased incidence of parapro- |
|
|
|
teinemia; some patients exhibit specific antibodies against the L-type |
|
|
|
voltage-gated calcium channels, and leukocyte invasion of the affected |
Disorders |
|
b. |
spinal cord |
2. |
familial: 20% are caused by mutations in superoxide dismutase (SOD)-1 |
||
|
Subtypes |
||
Movement |
|
a. |
sporadic: includes all those disorders listed under ALS variants |
|
|
gene, which can be inherited in an autosomal dominant or recessive |
|
|
|
|
|
|
|
|
pattern |
|
3. |
Histology: loss of motoneurons in anterior horn, motor cranial nerve nuclei, |
|
|
|
and primary motor cortex of spinal cord with astrocytosis and macrophage |
|
8 |
|
invasion |
|
|
|
|
|
|
|
a. |
surviving motoneurons are swollen and exhibit high levels of phosphorylated |
|
|
|
intermediate filaments |
|
|
b. |
surviving motoneurons (particularly in the spinal cord) exhibit three types |
|
|
|
of inclusion bodies, all of which include ubiquitin |
|
|
|
i. Lewy body-like inclusions |
|
|
|
ii. Bunina bodies: eosinophilic inclusions arranged as a chain; stain for |
|
|
|
cystatin-C |
|
|
|
iii. basophilic inclusions |
|
4. |
Symptoms |
|
|
|
a. |
slow development of focal weakness, often in the distal extremity (e.g., grip |
|
|
|
weakness, foot drop); can develop initially in the bulbar muscles causing |
|
|
|
dysarthria and dysphagia, in the respiratory muscles, or in the axial |
|
|
|
extensor muscles (causing head drop, like a myopathy) (Box 8.13) |
|
|
|
i. spread of weakness occurs along the organization of the motor |
|
|
|
homunculus, although new weakness often jumps contralaterally into |
|
|
|
the opposite limb |
|
|
|
ii. progresses to involve pseudobulbar dysfunction (i.e., spontaneous |
|
|
|
laughing or crying without the perception of emotion), severe |
|
|
|
dysphagia that prohibits swallowing, and ultimately respiratory |
|
|
|
impairment |
|
|
|
iii. weakness involves fasciculations and cramping, often in unusual |
|
|
|
places (e.g., the neck muscles) |
|
|
|
iv. preserves ocular motor and bowel/bladder function |
|
|
b. |
dementia (5%), of a frontotemporal lobar degeneration/Pick’s disease type |
|
|
c. |
absence of pain, sensory impairment, bowel/bladder dysfunction, or |
|
|
|
impairment of ocular motility |
|
5. |
Diagnostic testing |
|
|
|
a. |
EMG: demonstrates fasciculation potentials and a reduced number of |
|
|
|
active motor units that fire disproportionately fast given the muscle |
|
|
|
contraction (evidence of denervation), as well as abnormally shaped motor |
|
|
|
unit potentials (evidence of reinnervation) |
|
|
b. |
neuroimaging, mostly to exclude cervical spine stenosis |
|
|
c. |
serology: may demonstrate anti-GM1 antibodies, paraproteinemia |
196 |
|
d. |
genetic testing: for SOD-1 mutations, in cases of familial inheritance |
Box 8.13
Common causes of head drop—Myasthenia, polymyositis, ALS

Table 8–4 Amyotrophic Lateral Sclerosis Variants
ALS variants: Children (the spinal muscular atrophies [SMA])
Disease |
Pathophysiology |
Distinguishing features |
|
|
|
Infantile-onset SMA/Werdnig- |
Mutations in the SMN protein, which assembles |
Death 2 years of age from respiratory |
Hoffman disease |
snRNPs |
failure and aspiration |
|
mutations in NAIP |
Diaphragm strength is specifically preserved |
|
|
Tongue atrophy, with normal face and eye |
|
|
function |
|
|
|
Arrested infantile-onset SMA |
|
Can survive to adulthood |
|
|
Delayed motor milestones; rarely can walk |
|
|
Hand tremor |
|
|
|
Juvenile-onset SMA/Kugelberg- |
|
Onset 5–15 years of age; survives to |
Welander disease |
|
adulthood |
|
|
Progressive limb-girdle weakness |
|
|
Preservation of calf muscles |
ALS variants: Adults |
|
|
|
|
|
Disease |
Pathophysiology |
Distinguishing features |
|
|
|
Spinal muscular atrophy type IV |
Rarely has mutations in SMN or NAIP; autosomal |
Onset 20 years of age |
|
dominant & recessive forms |
Slow and benign course, mostly in shoulders |
|
|
|
|
|
Spares bulbar muscles |
|
|
|
Primary lateral sclerosis |
Sporadic occurrence, ? cause |
Pure UMN weakness |
|
|
Involves bulbar muscles last |
|
|
Can involve emotional lability (bulbar affect) |
|
|
|
Progressive muscular atrophy |
As per ALS, including gene mutations |
Pure LMN weakness |
|
|
Often occurs in members of families with |
|
|
familial ALS |
|
|
Younger age of onset, slower course than ALS |
|
|
|
Progressive bulbar palsy |
Autosomal recessive inheritance |
Motoneuron loss from lower cranial nerve |
|
|
nuclei and/or cervical spine (e.g., respiratory |
|
|
weakness) |
|
|
|
Bulbospinal neuronopathy/ |
Trinucleotide repeat in the androgen receptor |
Gynecomastia, testicular atrophy |
Kennedy’s disease |
(X-linked) |
LMN weakness in face, tongue, oropharynx, |
|
|
|
|
|
proximal shoulder & hip muscles |
|
|
|
Abbreviations: LMN; NAIP, neuronal apoptosis inhibitory protein; SMN, survival of motoneuron; UMN, upper motor neuron.
6.Treatment: supportive; riluzole (Rilutek) prolongs survival 6 months
7.Prognosis: rate of spread of weakness is steady but variable from patient to patient
a.average survival from diagnosis 3 years
b.ALS variants all have a better prognosis (Table 8–4)
X. Hereditary Spastic Paraplegias (Box 8.14)
1.Pathophysiology: Degeneration of the corticospinal tracts and dorsal columns at their distal ends (e.g., thoracolumbar spinal cord for the corticospinal tract, medulla for dorsal columns); degeneration is primary axonal with minimal neuron loss in motor cortex, anterior horn, or dorsal root ganglion
2.Symptoms: highly variable severity even among members in an affected family
a.begins as exertionand cold-triggered calf stiffness and spasms, but progresses to bilateral, symmetric lower extremity weakness with minimal atrophy
Box 8.14
Other diseases with spastic paraplegia—
Focal spinal cord disease, subacute combined degeneration, tethered cord syndrome, DOPA-responsive dystonia, leukodystrophies, spinocerebellar ataxia type 3, Friedreich’s ataxia
Hereditary Spastic Paraplegias
197

8 Movement Disorders
Table 8–5 Hereditary Spastic Paraplegias
Locus |
Chromosome |
Gene name & protein function |
Notes |
|
|
|
|
SPG-4 |
2 |
Spastin (Box 8.15), a nuclear |
|
|
|
protein with an unknown function |
|
SPG-3 |
14 |
Atlastin, a GTPase with an |
|
|
|
unknown function |
|
SPG-17 |
11 |
Seipin, an endoplasmic reticulum |
Silver syndrome: SPG-17 with |
|
|
protein |
weakness and atrophy of |
|
|
|
hand muscles |
SPG-11 |
15 |
? |
Hypoplastic corpus callosum, |
|
|
|
mental retardation |
SPG-7 |
16 |
Paraplegin, a nuclear-encoded |
Ragged-red muscle fibers, |
|
|
mitochondrial chaperone protein |
strokes, mental retardation, |
|
|
|
cerebellar atrophy |
SPG-1 |
X |
L1CAM (Box 8.16), a neuron- |
Mental retardation, |
|
|
specific cell adhesion molecule |
hydrocephalus |
SPG-2 |
X |
Proteolipoprotein (PLP) (Box 8.17), |
Motor aphasia, mental |
|
|
an intrinsic myelin protein |
retardation, vision loss |
|
|
|
|
Note: White autosomal dominant; light gray autosomal recessive; dark gray sex-linked.
i.weakness is predominantly in tibialis anterior, biceps femoris, and iliopsoas (i.e., muscles of the triple flexion response)
ii.spasticity, particularly of the biceps femoris that causes a characteristic bent-kneed gait
b.mild lower extremity large fiber sensory loss, with preservation of small fiber sensation
c.urinary urgency due to spastic bladder
d.pes cavus, hyperlordosis
3.Subtypes (Table 8–5)
4.Diagnostic testing
a.MRI may show atrophy of thoracolumbar spinal cord
b.somatosensory evoked potentials demonstrate delayed conduction and cortical potential from the lower extremities but normal responses from the upper extremities
c.genetic testing: available only for spastic paraplegia genes SPG-4 and SPG-3; SPG-2 does not involve gene duplication of the proteolipid PLP gene as does Pelizaeus-Merzbacher disease, therefore it cannot be tested with the available PLP genetic tests
5.Treatment: none specific
6.Prognosis: progressive gait disturbance may ultimately require wheelchair; normal lifespan
a.childhood-onset subtypes generally have mild progression despite their early onset
b.severity of disease cannot be predicted by course in other family members
Box 8.15
Spastin is also mutated in distal spinal muscular atrophy.
Box 8.16
LICAM is also mutated in mental retardation, aphasia, shuffling gait, adducted thumbs (MASA) disorder and X-linked hydrocephalus disorders
Box 8.17
PLP is also mutated in Pelizaeus-Merzbacher disease
XI. Ataxia Disorders
1.Autosomal dominant ataxia disorders a. spinocerebellar ataxia
i.pathophysiology: atrophy primarily of the cerebellar afferents (inferior olive, pons, spinocerebellar tract, Clarke’s nucleus) and deep cerebellar nuclei, and to a lesser extent the cerebellar cortex and other brain regions (e.g., anterior horn, dorsal columns, substantia nigra, red nucleus, globus pallidus); associated with an axonal neuropathy producing sensory or sensory-motor symptoms
198 |
(1) does not have inclusions bodies, unlike MSA-C |

Table 8–6 Spinocerebellar Ataxias
Subtype |
Genetics |
Specific symptoms |
Notes |
|
|
|
|
SCA-1 |
Trinucleotide repeat in |
Hypermetric saccades, hyperreflexia despite the |
SCA-1 & SCA-2 account for 50% of all |
|
ataxin-1 gene |
neuropathy |
spinocerebellar ataxias |
SCA-2 |
Trinucleotide repeat in |
Hypometric saccades, parkinsonism, myoclonus, |
|
|
the ataxin-2 gene |
neuropathy |
|
SCA-3 |
Trinucleotide repeat in |
Gaze-evoked nystagmus, spasticity, neuropathy; |
Type I: German heritage |
(Machado-Joseph |
the ataxin-3 gene, a |
dementia (type I), exophthalmos (type II) |
Type II: Portuguese heritage |
disease) |
ubiquitin-degrading |
|
|
|
|
||
|
protease |
|
|
SCA-6 |
Trinucleotide repeat in |
Downbeat and gaze-evoked nystagmus, |
Onset 50 years of age, usually |
|
P/Q-type Ca channel |
dysphagia |
without a family history |
|
(CACNA1A)* |
|
|
SCA-7 |
Trinucleotide repeat in |
Retinal degeneration, hearing loss |
|
|
ataxin-7 gene |
|
|
SCA-10 |
Pentanucleotide repeat |
Seizures |
Common in Mexicans |
SCA-14 |
Protein kinase C |
Myoclonus |
|
|
mutation |
|
|
|
|
|
|
*CACNA1A gene mutations also cause episodic ataxia type 2 and familial hemiplegic migraine.
ii.symptoms: onset typically in childhood or young adult; most exhibit anticipation with successive generations
(1)general symptoms: gait and limb ataxia, dysarthria
(2)subtypes: all have noncerebellar motor dysfunction and some sort of ocular dysfunction (Table 8–6)
b.dentatorubropallidoluysial atrophy (DRPLA)—could be considered an autosomal dominant spinocerebellar ataxia
i.pathophysiology: caused by a trinucleotide repeat in the DRPLA gene of chromosome 12, which has no known function but is located inside the nucleus
(1)histology: atrophy and gliosis in the cerebellum (particularly the dentate nucleus) and globus pallidus mesencephalon with sparing of the red nucleus, caudate, putamen, substantia nigra, and inferior olivary nucleus
(a)cerebellar atrophy may be based upon a preexisting hypoplasia
(b)subcortical demyelination is also observed
(2)more severe disease occurs with paternal inheritance
ii.epidemiology: common in Japan
iii.symptoms: onset between 10–60 years of age; involves ataxia, dementia, choreoathetosis (usually in adult-onset disease), seizures (65%), myoclonus (in 55%, usually in juvenile-onset disease), and myelopathy (only in homozygotes with long repeats)
iv.diagnostic testing: genetic testing for the DRPLA gene repeat
v.treatment: none specific
2.Autosomal recessive ataxia disorders
a.Friedreich’s ataxia—could be considered an autosomal recessive spinocerebellar ataxia
i.pathophysiology: caused by mutations in the frataxin gene, the protein of which may function in iron transport into the mitochondria
(1)mutations include
(a)trinucleotide repeats (95%): age of onset and severity proportionate to number of repeats
(b)point mutations (5%)
(2)histology: degeneration of projections from dentate nucleus, corticospinal tract, spinocerebellar tract, dorsal columns
(a)minimal involvement of cerebral or cerebellar cortices
Ataxia Disorders
199

8 Movement Disorders
200
ii.symptoms (Box 8.18)
(1)neurological symptoms: ataxia (trunk limbs), nystagmus, dysarthria; spasticity; large-fiber sensory loss with hyporeflexia (despite a Babinski reflex); weakness
(2)systemic symptoms: hypertrophic cardiomyopathy, subaortic stenosis; scoliosis, pes cavus; diabetes mellitus
iii.diagnostic testing: genetic testing of the frataxin gene
iv.treatment: none specific for the neurological disorder; idebenone for hypertrophic cardiomyopathy
v.prognosis: death by 40 years of age; time to wheelchair dependence is shorter in females
b.ataxia-telangiectasia (AT)
i.pathophysiology: caused by mutations of AT mutant (ATM) gene, the protein of which likely acts in the control of mitosis as a tumor suppressor
(1)histology: degeneration of cerebellar cortex and dentate nucleus, inferior olive, substantia nigra, and posterior columns
ii.symptoms: onset at 1–2 years of age
(1)neurological symptoms: ataxia (trunk limbs), dysarthria; oculomotor apraxia; dystonia, choreoathetosis; large-fiber sensory neuropathy with hyporeflexia that can occur in the presence of Babinski signs
(2)systemic symptoms
(a)ocular and cutaneous telangiectasias
(b)cancer (40%), typically leukemia or lymphoma may be presenting feature of the disease
(c)diabetes mellitus; gonadal hypoplasia
(d)chronic anemia and lymphopenia; recurrent infections
(e)hirsutism
iii.diagnostic testing
(1)serology demonstrates reduced immunoglobulins (Ig) (except IgM), an oligoclonal or monoclonal gammopathy (10%), and increased-fetoprotein
(2)chromosomal analysis for 7 → 14 translocation
(3)neuroimaging demonstrates an atrophic cerebellum
iv.treatment: none specific; frequent evaluation for cancer; avoid radiation exposure
v.prognosis: 20–50 year lifespan
c.Cockayne’s syndrome (see p. 286)
3.Episodic ataxias
a.type I inherited episodic ataxia
i.pathophysiology: caused by mutations of voltage-gated potassium channel (Box 8.19)
ii.symptoms: onset between 20–30 years of age
(1)episodic symptoms: last 1–2 minutes; triggered by changes in position or emotion
(a)ataxia without nystagmus or vertigo
(b)blurred vision
(2)chronic symptoms: myokymia
iii.treatment: antiepileptics; avoid acetazolamide
b.type II inherited episodic ataxia
i.pathophysiology: caused by mutations of P/Q-type voltage-gated calcium channel (CACNA1A) gene (Box 8.20)
Box 8.18
Friedrich’s ataxia is similar to vitamin E deficiency and to abetalipoproteinemia (which also has vitamin E deficiency)
Box 8.19
This voltage-gated potassium channel is bound by paraneoplastic antibodies in Isaac’s syndrome/neuromyotonia and limbic encephalitis.
Box 8.20
The P/Q-type voltage-gated calcium channel is bound by paraneoplastic antibodies in Isaac’s syndrome/neuromyotonia and limbic encephalitis.
ii.symptoms: onset between 20–30 years of age
(1)episodic symptoms: last 10 minutes to hours; triggered by exercise or stress
(a)ataxia, nystagmus, vertigo
(b)dysarthria
(c)generalized weakness
(d)headache
(2)chronic symptoms: nystagmus, ataxia
iii.treatment: acetazolamide
XII. Stiff Man’s Syndrome
1.Pathophysiology: unknown, but is associated with
a.anti-glutamate decarboxylase (-GAD) antibodies (40%)
b.autoimmune diseases, particularly diabetes (40%)
i.80% of type I diabetics exhibit antibodies against GAD, as well as other autoantibodies
2.Symptoms: onset in adulthood
a.painful muscle spasms with rigidity in axial and facial muscles extremities that initially is intermittent but eventually becomes continuous
i.spasms are exacerbated by voluntary movements and startle, and may be associated with myoclonus
3.Diagnostic testing: serology for anti-GAD antibodies; EMG demonstrates continuous motor unit potentials in agonist and antagonist muscles at rest
4.Treatment: benzodiazepines, clonidine, baclofen, prednisone, plasma exchange, IV Ig
XIII. Essential Tremor
1.Pathophysiology: none established, but may be a neurodegenerative disease a. risk factors: increasing age, White, family history
2.Symptoms: Kinetic tremor (Box 8.21) that typically develops in the arms and spreads to the head, neck, and oropharynx over several years; severe disease also involves postural tremor and/or an irregular, jerky resting tremor (20%)
3.Diagnostic testing: Evaluate for hyperthyroidism and Wilson’s disease
4.Treatment: Propranolol, primidone
Box 8.21
In comparison to physiological tremor, essential tremor’s amplitude is higher and its frequency is lower.
Iatrogenic Movement Disorders
XIV. Iatrogenic Movement Disorders (Table 8–7)
1.Tardive dyskinesia
a.pathophysiology: caused by chronic use of typical antipsychotic drugs and other phenothiazines (e.g., metoclopramide, perphenazine, prochlorperazine); rarely caused by atypical antipsychotics
i.20% overall prevalence with typical antipsychotic use, up to 50% in older patients
ii.5% annual incidence in patients on phenothiazine medications
iii.the previous hypothesis that tardive dyskinesia was due to an upregulation and supersensitivity of dopamine receptors in the basal ganglia is not well substantiated, and instead it may involve dysregulation of the sympathetic nervous system or a direct effect of the musculature
b.symptoms
i. stereotyped movements of the |
201 |
8 Movement Disorders
Table 8–7 Neuroleptic Malignant Syndrome and Other Hyperthermic Disorders
Neuroleptic |
|
|
|
malignant syndrome |
Malignant hyperthermia |
Serotonin syndrome |
Acute lethal catatonia |
Site of disorder |
Central nervous system |
Muscle |
Common |
Antipsychotic drugs, |
Use of halogenated anesthetics |
cause |
particularly high- |
or depolarizing anesthetics in |
|
potency or depot |
patients with mutations in |
|
preparations |
ryanodine calcium channel |
Other |
Rigidity |
Jaw clenching at the time of |
symptoms |
Encephalopathy |
medication administration |
|
Respiratory acidosis, cyanosis, |
|
|
Autonomic instability |
|
|
myoglobinuria |
|
|
Dystonia, tremor, or |
|
|
|
|
|
chorea |
|
Treatment |
Withdraw offending |
Withdraw offending drug |
|
drug |
Dantrolene |
|
Dantrolene |
|
|
Bicarbonate |
|
|
Dopaminergic agonists |
|
|
|
Central nervous system
Overdose on SSRIs, or combination treatment with MAO inhibitor, TCA, or meperidine
Shivering
Ataxia, hyperreflexia
Nausea, diarrhea
Hypertension
Seizures, coma
Supportive
Central nervous system
An exacerbated psychotic disorder
Preceding psychosis and dyskinesias
Waxy flexibility, not true rigidity
Electroconvulsive therapy
Abbreviations: MAO, monoamine oxidase; SSRIs, selective serotonin reuptake inhibitors; TCA, tricyclic antidepressant.
(1)face, tongue, and masticatory muscles causing lip smacking or pursing, tongue protrusion, and chewing
(2)extremities, producing choreiform movements or (less frequently) dystonias
(3)trunk and abdomen, producing pelvic thrusting and grunting
ii.tardive movements are suppressed by voluntary movements of the affected area but are exacerbated by voluntary movements of nonaffected areas or by distraction
iii.movements are rarely bothersome to the patient
c.treatment
i.discontinuation of the offending drug occasionally resolves the tardive movements years after discontinuation but usually they are permanent; tardive movements may dramatically worsen after removal of the drug but this is a transient effect
ii.benzodiazepines, reserpine, vitamin E
2.Drug-induced parkinsonism
a.causative medications
i.antipsychotics: the most common offending agents; parkinsonism
develops in 15% of patients taking antipsychotics, usually within 3 months of beginning the medication
ii.GI prokinetic agents, particularly metoclopramide
iii.others: lithium, amiodarone, meperidine, amphotericin, diltiazem
b.management: parkinsonian side effects generally resolve with continued use of medication
i.may lower the dose of typical antipsychotics
ii.may change to an atypical antipsychotic (clozapine, olanzapine, quetiapine)
iii.may use anticholinergic anti-parkinsonian medications (trihexyphenidyl, benztropine) or amantadine to counteract the effect of the causative medication
(1)withdraw anticholinergic medication after 3-month period to determine need of continuing this supplementation
202