

- •Contents
- •Preface
- •Contributors
- •01 Neuroanatomy
- •03 Seizures and Epilepsy
- •04 Disorders of Myelination
- •05 Tumors of the Nervous System
- •06 Headache and Pain Disorders
- •I. Episodic Headache
- •A. Episodic Headaches Lasting More than Four Hours
- •07 Behavioral Neurology
- •08 Movement Disorders
- •09 Diseases of the Nerves
- •10 Diseases of the Muscles
- •I. Bacteria
- •I. Diabetes Mellitus (DM)
- •Index

11
Infections of the
Nervous System
Note: Significant diseases are indicated in bold and syndromes in italics.
I. Bacteria
1.Bacterial meningitis
a.General symptoms: Only subtle behavioral changes may be apparent early in the disease course, but progression of the disease typically involves
i.fever, headache, meningismus, photophobia, somnolence
ii.focal neurological dysfunction (15%), particularly hearing loss
iii.seizures (20%)
b.general diagnostic testing
i.blood cultures identify causative pathogens in 50% of cases
ii.cerebrospinal fluid analysis
(1)increased intracranial pressure, protein, and lactate
(2)decreased glucose
(3)pleocytosis is typically 100 cells/ L, and 60% neutrophils; may be lower early in disease course
(4)Gram stain is positive in 60% (Box 11.1)
(5)cultures identify causative pathogen in 75%
iii.neuroimaging: usually is normal, but it may demonstrate hydrocephalus, parenchymal edema, and/or meningeal enhancement; in complicated cases, venous thrombosis or collections of pus between the dura and arachnoid layers of the meninges {empyema} may develop
c.pathophysiology (Table 11–1)
d.general treatment
i.antibiotics, as in Table 11–1; adjust as the causative bacteria are identified
(1)always use bactericidal agents, not bacteriostatic agents
(2)meningeal inflammation makes the brain permeable to essentially all antibiotics
ii.dexamethasone: reduces morbidity and mortality by 40% when used in children or adults with meningitis; give first dose prior to initiation of antibiotic therapy
iii.surgical drainage of any empyema, otitis media, or sinusitis
iv.prophylaxis with the Haemophilus influenza vaccine routinely in children, and with the Neisseria meningitidis vaccine in persons at high-risk for exposure
e.prognosis: 20% mortality; 40% of survivors will develop seizures
2.Brain abscess
a.pathophysiology: causes include
i.contiguous spread: the most common route for brain abscess formation; always results in a single abscess (Box 11.2)
Bacteria
Box 11.1
Yield of CSF Gram stain in bacterial meningitis is reduced only with 2 hour of antibiotic administration.
Box 11.2
Metastic tumors more commonly spread to the brain by a hematogenous route
249
11 Infections of the Nervous System
250
Table 11–1 Common Causes of Bacterial Meningitis
Patient group |
Causative bacteria (in order) |
Empiric treatment |
|
|
|
Neonates |
Group B Streptococcus |
Ampicillin third-generation |
|
Escherichia coli |
cephalosporin |
|
|
|
|
Listeria monocytogenes |
|
|
|
|
Children, adolescents, |
Neissera meningitidis |
Third-generation cephalosporin |
young adults |
Streptoccus pneumoniae |
vancomycin/rifampin |
|
|
|
|
(H. influenza is rare due to |
|
|
vaccination) |
|
|
|
|
Older adults |
Neissera meningitidis |
Ampicillin third-generation |
( 50 years of age) |
Listeria monocytogenes |
cephalosporin vancomycin/ |
|
rifampin |
|
|
|
|
|
|
|
Immunosuppressed |
Listeria monocytogenes |
Ampicillin third-generation |
|
Gram-negative bacilli |
cephalosporin vancomycin/ |
|
rifampin |
|
|
Klebsiella |
|
|
|
|
|
|
|
Shunt infection |
Staphylococcus epidermidis |
Vancomycin third-generation |
|
Staphylococcus aureus |
cephalosporin |
|
|
|
|
(methacillin resistant) |
|
|
|
|
Head trauma |
Staphylococcus epidermidis |
Vancomycin third-generation |
|
Staphylococcus aureus |
cephalosporin metronidazole |
|
|
|
|
Gram-negative bacilli |
|
|
anaerobic bacteria |
|
|
|
|
Note: Local flora and sensitivity patterns require consultation with an infectious diseases expert.
(1)the initial infection is located in a parameningeal site, such as
(a)purulent sinusitis, which produces frontal lobe abscesses
(b)otitis media or mastoiditis, which produce temporal lobe or cerebellar abscesses
ii.hematogenous spread: abscesses preferentially target areas of old brain injury (e.g., infarction), and are multiple in 15% of cases (Box 11.3)
(1)in adults, hematogenous spread of bacteria is usually from a pulmonary infection or (less commonly) endocarditis
(2)in children, hematogenous spread of bacteria is usually from some type of cyanotic congenital heart disease that provides a hypoxic environment and a right–left shunt thereby bypassing normal lung filtration
iii.head injury: closed head trauma is a rare cause of abscesses except in cases that involve unrepaired cerebrospinal fluid leaks
iv.neurosurgery, usually in cases where an air sinus has been opened
v.idiopathic (20%)
b.common pathogens: abscesses include multiple organisms in 90%, usually
Streptococcus species (50%), Bacteroides, and Enterobacteriaceae and other anaerobic bacteria
i.abscesses from trauma also include multiple Staphylococcal species
ii.abscesses in infants often include Gram-negative bacteria
c.symptoms: headache, nausea, and somnolence caused by increased intracranial pressure focal neurological injury
i.increased intracranial pressure may enlarge the head circumference in infants
ii.fever occurs in only 50% because the infection is isolated
d.diagnostic testing: ultimately requires tissue therapy
Box 11.3
Osler-Weber-Rendu syndrome/hereditary hemorrhagic telangiectasia—pulmonary arteriovenous fistulas bypass normal lung filtration; 5% of cases develop cerebral abscesses

i.neuroimaging staging
(1)early cerebritis (stage I): exhibits poorly demarcated ring enhancement that does not wash out and parenchymal edema
(2)late cerebritis (stage II): exhibits a developing necrotic center
(3)early encapsulation (stage III): exhibits a vascularized, necrotic center
(4)late encapsulation (stage IV): exhibits a thin collagen capsule that enhances but then washes out; gliosis develops around capsule
ii.diagnosis requires tissue biopsy
iii.blood cultures are rarely helpful; avoid lumbar puncture due to herniation risk
e.treatment
i.medical treatment: most effective in neuroimaging stages I–II, before the development of an abscess capsule
(1)empiric therapy: vancomycin third-generation cephalosporin metronidazole or chloramphenicol
(2)glucocorticoids prevent fibrous encapsulation but also reduce penetration of antibiotics, therefore use only in patients who are clinically deteriorating
ii.surgical drainage in cases with mass effect, elevated intracranial pressure, poor neurological condition, or proximity of the abscess to a ventricle (i.e., to prevent ventriculitis)
3.Bacterial encephalitis: an uncommon form of bacterial infection in the brain
a.Bartonella henselae/cat scratch disease
i.pathophysiology: a Gram-negative bacillus transmitted by scratches from a colonized cat and possibly by cat fleas
ii.symptoms: fever; lymphadenopathy proximal to the scratch; encephalitis with seizures develops in only 1% of infected patients
(1)may also cause myelitis and/or radiculitis in conjunction with angiomatous skin lesions that are similar to Kaposi’s sarcoma in the immunosuppressed
iii.diagnostic testing: B. henselae enzyme linked immunosorbent assay (ELISA) or polymerase chain reaction (PCR); cerebrospinal fluid is normal in 70% and otherwise shows only a mild lymphocytosis; cultures are unrevealing
iv.treatment: gentamicin or trimethoprim-sulfamethoxazole (TMP-SMX)
v.prognosis: complete recovery in the immunocompetent
4.Bacterial vasculitis (see p. 77)
II. Mycobacteria
1.Mycobacterium tuberculosis/tuberculosis
a.pathophysiology: transmitted from person to person by infected respiratory droplets; infection of the central nervous system may develop during the initial infection (usually pulmonary) or after reactivation of a dormant infection (as in the newly immunosuppressed)
i.Mycobacterium usually spread from a peripheral site of infection to the brain by a hematogenous route or rarely by rupture of a mass of infected granulation tissue {tubercles} into the cerebrospinal fluid; may form abscesses {tuberculomas} directly in brain parenchyma as well
ii.target organs
(1)pulmonary, lymphatic, genitourinary
(2)bones and joints, including the vertebral bodies and intervertebral disks causing spondylosis {Pott’s disease}
(3)meninges brain parenchyma
Mycobacteria
251
b.epidemiology: neurological complications are most common in children5 years of age and in the immunosuppressed HIV infection, glucocorticoid use, diabetics
c.symptoms (neurological): neurological symptom from
|
|
i. |
meningitis, which has a relatively high rate of cranial neuropathy |
|
|
|
|
(25%, particularly CN VI III, IV, VII) and the syndrome of inappro- |
|
|
|
|
priate antidiuretic hormone secretion (SIADH) due to its preference |
|
System |
|
|
for the basal meninges |
|
|
|
(1) |
seizures, focal deficits (15%), and hydrocephalus may also occur |
|
|
|
|
||
|
|
|
(2) |
can infect the spinal meninges, producing radicular and local back |
Nervous |
|
iii. |
|
pain and an ascending paralysis {Foix-Alajouanine syndrome} |
|
vasculitis (see p. 77) |
|||
|
|
|
|
(Box 11.4) |
|
|
ii. |
tuberculoma development (rare), which causes focal neurological injury |
|
the |
d. |
diagnostic testing: the diagnosis of tuberculosis infection in the brain is |
||
|
||||
of |
|
supported by identifying tuberculosis elsewhere in the body |
||
|
i. |
for meningitis: cerebrospinal fluid demonstrates increased protein, |
||
Infections |
|
|||
|
|
(1) |
elevated total protein levels ( 100 mg/dL, often 1000) may |
|
|
|
|
low glucose, and a lymphocytic pleocytosis; immunosuppressed |
|
|
|
|
patients may not exhibit the pleocytosis |
|
11 |
|
|
|
form a web-like coagulant that is very characteristic of tubercu- |
|
|
|
losis meningitis {Froin’s syndrome}; removal of the coagulant |
|
|
|
|
|
|
|
|
|
|
leaves a supernatant with a very low protein level |
|
|
|
(2) |
cerebrospinal fluid smear is positive in only 30%; acid-fast bacilli |
|
|
|
|
cultures are positive in 70%, but take 8 weeks to complete |
|
|
|
(3) |
M. tuberculosis PCR is positive in 75% |
|
|
|
(4) neuroimaging may demonstrate skull base meningeal enhancement |
|
|
|
ii. |
for tuberculoma |
|
|
|
|
(1) |
neuroimaging demonstrates one or more ring-enhancing lesion(s), |
|
|
|
|
either high or low density |
|
|
|
(2) |
tissue biopsy is required if the diagnosis of systemic tuberculosis |
|
|
|
|
cannot be made |
|
e. |
treatment |
||
|
|
i. |
medical treatment: isoniazid rifampin pyrazinamide ethambutol |
|
|
|
|
or streptomycin, typically continued for 6–9 months; optimal drug |
|
|
|
|
regimen depends upon local resistance patterns |
|
|
|
|
(1) |
may consolidate into a two-drug regimen if the patient is clinically |
|
|
|
|
improved after 2 months on the four-drug regimen; continue the |
|
|
|
|
two-drug regimen for 10 more months |
|
|
|
(2) |
vitamin B6 supplementation is necessary to prevent isoniazid |
|
|
|
|
neuropathy |
|
|
|
(3) |
glucocorticoids can be used to control tissue edema or Froin’s |
|
|
|
|
syndrome that can obstruct the arachnoid granulations and cause |
|
|
|
|
communicating hydrocephalus |
|
|
ii. |
surgical treatment |
|
|
|
|
(1) |
cerebrospinal fluid shunting for any signs of hydrocephalus |
|
|
|
(2) |
excision for tuberculomas that are causing symptomatic mass effect |
|
|
|
|
despite glucocorticoid therapy |
|
f. prognosis: uniformly fatal within 6 weeks if untreated; 20% mortality in |
|||
|
|
the immunocompetent, 30% mortality in immunosuppressed |
||
|
2. Mycobacterium leprae/leprosy |
|||
|
a. |
pathophysiology: M. leprae is spread between persons by infected droplets, |
||
|
|
and it grows best at temperatures a few degrees below human body tem- |
||
252 |
|
perature; preferentially infects in the epineurium and perineurium of |
||
|
nerves of the extremities |
|||
Box 11.4
The Foix-Alajouanine syndrome is more commonly seen with spinal arteriovenous malformation (AVM) growth.

b.symptoms: exhibits tuberculoid, intermediate, and lepromatous forms, that can occur in the same patient at different times depending upon the level of cellular immunity
i.tuberculous leprosy (in patients with strong cell-mediated immunity): a few skin nodules develop that are associated regions of sensory loss; palpable nerve hypertrophy
ii.indeterminate leprosy: a single hypopigmented patch that has sensory loss
(1)progresses to either tuberculous or lepromatous forms depending upon the effectiveness of the patient’s cell-mediated immunity
iii.lepromatous leprosy (in patients with poor cell-mediated immunity): multiple skin nodules with cartilage erosion in the nose and ears {leonine face deformity}; distal small-fiber sensory loss in the extremities, nose, and ears with palpable nerve hypertrophy
(1)skin nodules are not initially associated with sensory loss, as they are in the tuberculous form of leprosy
(2)weakness and arthropathy develop late in the disease course
c.diagnostic testing
i.identifying bacterial particles in macrophages in skin biopsies, nasal mucosa, or nerve biopsies
ii.lepromin skin test is useful for distinguishing between lepromatous and tuberculous forms of the disease, but is not useful in diagnosis because it is commonly negative in the lepromatous form
d.treatment: dapsone clofazimine rifampin; treat for 1 year in patients with indeterminate or tuberculous forms, or for 2 years in patients with lepromatous form
i.treatment often induces an erythema nodosum-like reaction upon initiation
III. Spirochetes
1.Lyme disease
a.pathophysiology: caused by Borrelia burgdorferi in North American; ticks of Ixodes (Box 11.5) genus serve as the vectors and deer serve as the reservoir
i.the host reaction is largely directed against Borrelia outer-surface proteins (Osp), which are membrane lipoproteins encoded by 21 plasmids that have high antigenic variability, which may account for the organism’s prolonged infectious course
ii.Borrelia preferentially invades meninges, nerve roots, and peripheral nerves, but only rarely brain parenchyma
b.epidemiology: common in New England, Minnesota–Wisconsin, and the Pacific Northwest
c.symptoms: 50% of infections are asymptomatic
i.stage 1 (acute infection): localized erythema migrans
ii.stage 2 (within weeks): nonspecific flu-like symptoms or meningitis; heart block and myocarditis; arthritis
iii.stage 3 (within months): mild sensory polyneuropathy; subtle cognitive disturbances occur in 5% of cases {Lyme encephalitis}
(1)radiculoneuritis is prominent only with B. afzelii or B. garinii infection, which cause Lyme disease in Europe
iv.“chronic” Lyme disease: similar to chronic fatigue syndrome and fi- bromyalgia, however, there is no clear evidence for its existence as a distinct disease entity
d.diagnostic testing: cerebrospinal fluid B. burgdorferi ELISA and Western blotting, or else positive blood serology in the presence of a consistent clinical syndrome
i.serum or cerebrospinal fluid Borrelia PCR is not sufficiently reliable
Spirochetes
Box 11.5
Some Ixodes ticks also produce holocyclotoxin, causing tick paralysis.
253
11 Infections of the Nervous System
e.treatment
i.prior to development of neurological symptoms: doxycycline for 14–21 days
ii.with neurological symptoms: third-generation cephalosporin IV for 2–4 weeks
iii.prophylaxis with the Lyme disease vaccine, an OspA adjuvant
(1)single-dose doxycycline may prevent infection it administered within 72 hours of a tick bite
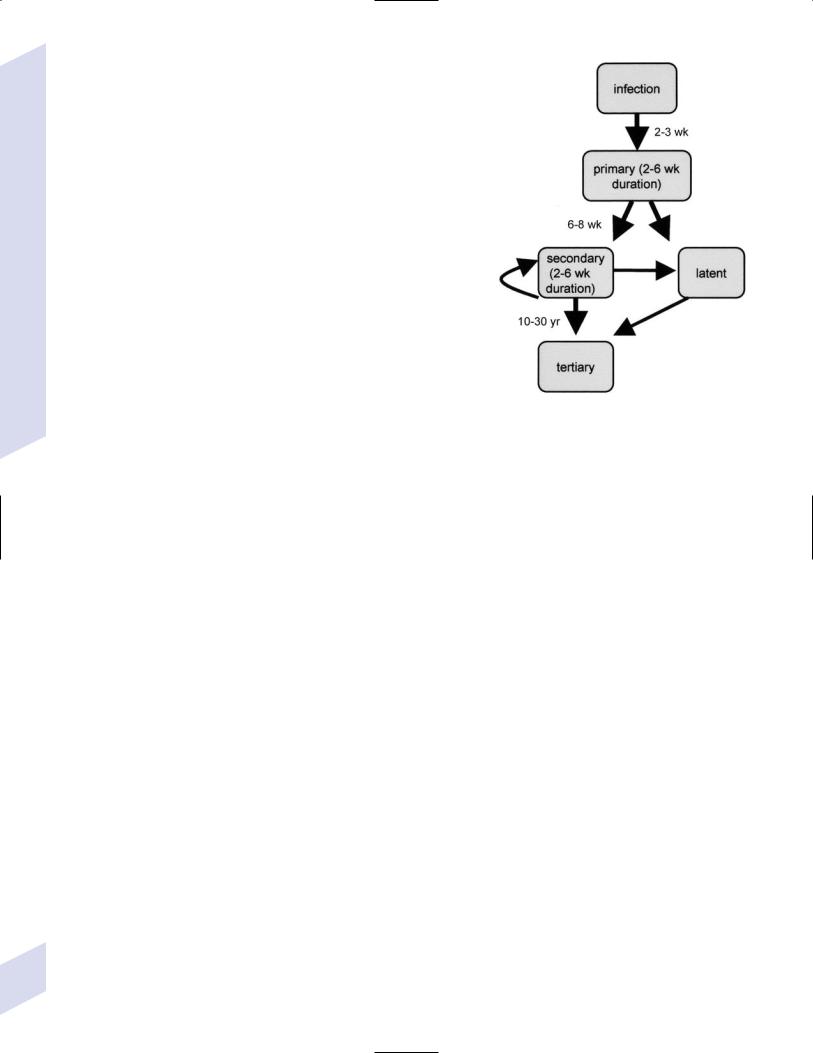
2.Syphilis: Caused by Treponema pallidum
a.primary syphilis: the only symptom is a painless chancre that is acquired by contact with an actively infected site on another person (i.e., another chancre, mucous membrane or skin rash, condyloma lata)
i.may exhibit asymptomatic central nervous system infection as documented by pleocytosis of the presence of spirochetes on dark field microscopy
b.secondary syphilis (Fig. 11–1)
i.symptoms
(1)flu-like syndrome
(2)rash with hyperkeratosis involving the palms and soles
(3) condyloma lata, particularly during relapses |
Figure 11–1 Stages of syphilis. |
(4)meningitis in only 2% of cases, although 30% of secondary syphilis patients have an asymptomatic central nervous system infection
ii.diagnostic testing: cerebrospinal fluid evaluation by a Venereal Disease Research Laboratory (VDRL) test is specific but not sensitive, and cerebrospinal fluid fluorescent treponemal antibody (FTA) testing is sensitive but not specific; therefore, evaluate with a VDRL, but treat even if these tests are negative so long as there is pleocytosis or the presence of oligoclonal bands in a patient with a compatible clinical syndrome
(1)baseline cerebrospinal fluid cell count, protein level, and VDRL titer should be obtained to compare with follow-up tests to assess the adequacy of treatment
iii.treatment: penicillin G 4 106 U IV q.4.h. for 14 days
(1)to determine the adequacy of treatment, reexamine cerebrospinal fluid every 6 months for 3 years whereas successful treatment is defined as
(a)reduction in the lymphocyte pleocytosis and protein level, which are the most sensitive and rapidly changing indicators of treatment efficacy
(b)fourfold reduction in VDRL titer, which may take 1 year to achieve
(2)repeated treatment is indicated if the cerebrospinal fluid studies fail to normalize or if symptoms persist
(3)in penicillin-allergic patients, attempt penicillin desensitization or else use doxycycline for 4 weeks
c.latent syphilis: an asymptomatic central nervous system infection that can be demonstrated in 40% of cases
i.diagnostic testing: as above; obtain baseline cerebrospinal fluid studies to assess adequacy of treatment, as per secondary syphilis
ii.treatment: penicillin G as described above
(1)reexamine the cerebrospinal fluid every 6 months for 3 years to determine adequacy of treatment, as per secondary syphilis
d.tertiary syphilis: aortitis, iritis, and painless granulomatous nodules {gummas}
254that may develop in numerous organ systems; neurological symptoms include i. meningitis
ii.meningovascular neurosyphilis, involving arteries of all sizes and causing strokes typically in the MCA distribution (see p. 77)
(1)diagnosis and treatment as per secondary syphilis
iii.parenchymatous neurosyphilis: develop years to decades after primary syphilis
(1)tabes dorsalis: inflammatory destruction of the lumbosacral dorsal root ganglia with subsequent loss of spinal sensory pathways
(a)symptoms: begin after a period of meningitis
(i)episodic pain in the lower extremities, or in the abdomen and thorax {visceral crises}
(ii)vision loss from optic atrophy
(iii)ataxia, areflexia, Charcot joints, and trophic ulcers from large-fiber sensory loss
(iv)insensitivity to pain, overflow incontinence, constipation, and sexual dysfunction from small-fiber sensory loss
(v)small, irregular pupils that accommodate but do not react to light {Argyll-Robertson pupil}; involves an unknown injury in the light reflex circuit
(b)diagnosis and treatment as per secondary syphilis
(2)general paresis: an encephalitic infection
(a)symptoms
(i)personality changes
(ii)affect abnormalities
(iii)reflex abnormalities (generally hyperreflexic)
(iv)eye: Argyll-Robertson pupils
(v)sensorium: illusions, delusions, hallucinations
(vi)intellectual dysfunction progressive dementia
(vii)speech dysarthria
(b)diagnosis and treatment as per secondary syphilis
IV. Rickettsia
1.Rocky Mountain spotted fever
a.pathophysiology: caused by Rickettsia rickettsii; Dermacentor (Box 11.6) ticks serve as the vector, and rodents and dogs serve as the reservoir
i.epidemiology: predominant in the north and east United States, Canada, and Mexico
b.symptoms: rash beginning on the ankles (Box 11.7); meningitis with focal neurological signs (15%) and seizures (5%)
c.diagnostic testing: immunofluorescence or immunoperoxidase staining of skin biopsy from the rash site; R. rickettsii serology
i.cerebrospinal fluid analysis is nondiagnostic
d.treatment: tetracycline or doxycycline continued until 2 days after the fever ends
e.prognosis: 20% mortality if treatment is delayed
2.Typhus
a.pathophysiology: caused by Rickettsia prowazekii, which is transmitted from human to human by lice; epidemic typhus occurs in overcrowded and unsanitary conditions during cold weather
b.symptoms: rash beginning in the axilla; meningitis focal neurological injury
c.diagnostic testing: R. prowazekii ELISA, PCR, or agglutination tests
d.treatment: tetracycline or doxycycline; vaccination for persons at exposure risk
Rickettsia
Box 11.6
Dermacentor also carries Colorado tick fever virus.
Box 11.7
Other types of meningitis with petechial rash: N. meningitidis, S. pneumoniae, S. aureus
255
11 Infections of the Nervous System
256
V. Viruses
1. Herpes viruses
a.herpes simplex (HSV) type I and II
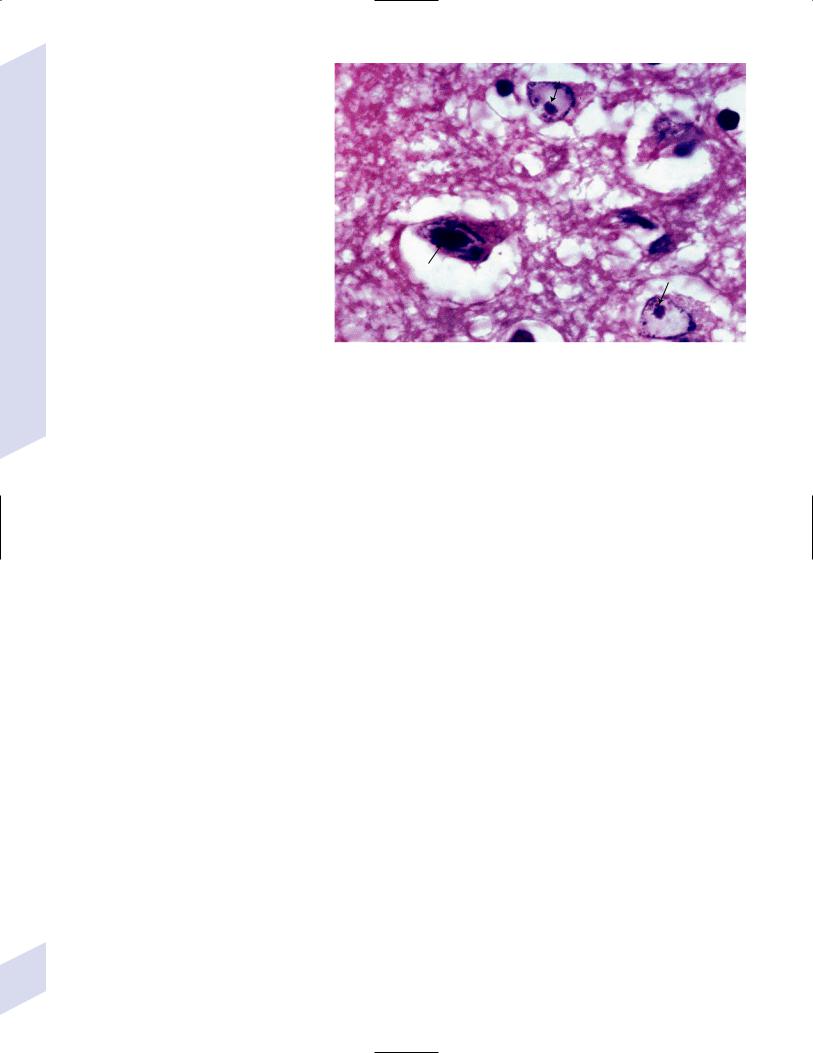
i.pathophysiology: causes hemorrhagic necrosis of the temporal lobes that is histologically characterized by large, solitary nuclear inclusions surrounded by a halo in neurons, astrocytes, and oligodendroglia {Cowdry type A
inclusions} (Fig. 11–2) and by Alzheimer’s disease-like neurofibrillary tangles
(1) |
acquired by person-to- |
|
|
|
|
|
person transmission of con- |
|
|
|
|
|
taminated body fluids, but |
|
|
|
|
|
most herpes encephalitis is |
|
|
|
|
|
likely caused by a reactiva- |
Figure 11–2 Cowdry type A inclusions (arrows). (From Hirano A. Color Atlas of Pathology |
|||
|
tion of latent infection of |
||||
|
of the Nervous System, 2nd Ed. Tokyo/New York: Igaku-Shoin Press; 1988:219, Fig. 550. |
||||
|
the trigeminal nerve (not by |
||||
|
Reprinted by permission.) |
|
|
|
|
|
primary infection) |
|
|
|
|
(2) |
viral subtypes |
|
|
|
|
|
(a) HSV-1: causes oral herpes and encephalitis |
|
|
|
|
|
(b) HSV-2: causes genital herpes and neonatal encephalitis occur- |
|
|
|
|
|
ring within a week of birth |
|
|
|
|
ii. symptoms: encephalitis that is often, but not always, acute in |
|
|
|
||
onset; slowly progressive cases may present only with personality |
|
|
|
||
changes |
|
|
|
|
|
(1) |
seizure and focal neurological injury occur in 50% of cases |
|
|
|
|
iii. diagnostic testing: cerebrospinal fluid may be frankly hemorrhagic, |
|
|
|
||
and the inflammatory leukocytosis is often not proportionate to the |
|
|
|
||
clinical severity |
|
|
|
|
|
(1) |
diagnosis requires HSV PCR, which can be serially evaluated to |
|
|
|
|
|
determine the effectiveness of treatment |
|
|
|
|
iv. treatment: acyclovir 10 mg/kg IV q.8.hr. for 14 days; side effects |
|
|
|
||
include reversible renal failure |
|
|
|
|
|
v. prognosis: 70% mortality untreated; 20% mortality treated |
|
|
|
||
b. varicella zoster virus: types of infections include |
|
|
|
||
i. chickenpox: 1% develop a self-limited encephalitis at the time of the |
|
|
|
||
rash; a transient meningitis-like syndrome with ataxia may develop |
|
|
|
||
3 weeks after the rash |
|
|
|
|
|
ii. herpes zoster/shingles: local reactivation of varicella infection in an |
|
|
|
||
infected dorsal root ganglia causes lymphocytic infiltration and local |
|
|
|
||
hemorrhage in the dorsal root ganglia and spinal roots; reactivation |
|
|
|
||
has been related to spinal trauma and immunosuppression, but often |
|
|
|
||
|
Box 11.8 |
||||
it just occurs spontaneously |
|
|
|||
(1) |
symptoms: pain and paresthesias in spinal or cranial der- |
|
Post-Herpetic Neuralgia |
|
|
|
matomes followed by a pruritic vesicular rash that develops |
|
Common in shingles patients 50 years |
||
|
within 3–4 days and crusts by 10 days; usually resolves within |
|
of age who had sensory symptoms prior |
||
|
2 weeks (Box 11.8) |
|
|
to rash |
|
|
(a) location: usually limited to 1–2 adjacent dermatomes in the |
|
Symptoms include various pains, dyses- |
||
|
|
thesia, and allodynia in the previously |
|||
|
thoracic cervical lumbosacral region |
|
|||
|
|
affected region |
|||
|
(i) 20% of cases have a cranial nerve distribution |
|
Develops 1–6 months after resolution |
||
|
(1) herpes zoster ophthalmicus: viral reactivation in the |
|
of the rash and lasting several months |
||
|
|
Treat with gabapentin, topical lidocaine |
|||
|
ophthalmic division of CN V causes conjunctivitis, |
|
|||
|
|
or capsaicin, or amitriptyline |
|||
|
keratitis, and uveitis, but rarely causes vision loss |
|
|||
|
|
|
|
||
|
|
|
|
||

(2)Ramsay-Hunt syndrome/otic zoster: viral reactivation in the geniculate ganglion of CN VII causes facial paresis and rash on the tympanic membrane and external auditory canal
(a)associated with a large-vessel vasculitis
(b)flaccid weakness develops in 5%, likely because the infection has involved the ventral root and spinal cord
(c)zoster sine herpete: radicular pain without rash; requires demonstration of varicella in the cerebrospinal fluid by PCR analysis or an increased ratio of cerebrospinal fluid-to-serum varicellazoster virus (VZV) titres
(2)treatment: acyclovir, famciclovir, valacyclovir for 7 days, started within 3 days of symptom onset; glucocorticoids for 3 weeks
iii.varicella encephalitis: dissemination to the brain occurs in 2% of immunocompetent patients and 50% of immunosuppressed patients who develop shingles; treat with acyclovir (Box 11.9)
c.cytomegalovirus (CMV)
i.histology: Cowdry type A inclusions are found in ependymal cells neurons, glia
ii.types of infection
(1)in utero infection: symptoms include retinitis and encephalitis, leading to vision loss, deafness, and mental retardation; cerebral calcifications, microcephaly, and migrational defects (lissencephaly/ pachygyria, polymicrogyria, cerebellar agenesis) can occur even without signs of systemic infection
(2)infection in the immunosuppressed (e.g., HIV, following bone marrow transplantation): usually spreads to the brain by a hematogenous route (causing encephalitis) or by direct spread through the cerebrospinal fluid (causing ventriculoencephalitis); often occurs with CMV retinitis, esophagitis, or colitis, and CMV viremia is invariably present
(a)symptoms: confusional state developing over weeks (encephalitis) or days (ventriculoencephalitis); cranial nerve palsies, nystagmus, hydrocephalus
(b)prognosis: usually fatal within a few months
iii.diagnostic testing: demonstration of CMV by PCR in the cerebrospinal fluid
iv.treatment: ganciclovir, foscarnet, cidofovir
2.Rabies virus
a.pathophysiology: viral inoculation occurs after a bite from an infected animal (dog, cat, fox, raccoon, bat); retrograde and transsynaptic transport carries the virus up the peripheral nerves and into the central nervous system (i.e., to the brain producing an encephalitic disease, or to the spinal cord producing a paralytic disease)
i.histology: characterized by microglial nodules {Babes nodes}, perivascular inflammation, and viral inclusions in the cytoplasm of Purkinje cells and hippocampal pyramidal cells {negri bodies} that appear as bullet-shaped particles on electron microscopy (Fig. 11–3)
b.symptoms: begins several days to weeks after the inoculating bite as a flulike syndrome that progresses to either
i.an encephalitic form, involving behavioral changes (agitation, hyperactivity), severe dysphagia producing frothing, and eventually seizures and psychosis before death
(1)hydrophobia is likely due to the inability to swallow
ii.a paralytic form, which develops over a period of days leading to death
Box 11.9
Reye’s Syndrome
A pediatric encephalopathy from cerebral edema and elevated ammonia levels caused by rapid liver failure with fatty infiltration of the liver
Induced by aspirin use during several types of viral infections, including varicella and influenza
Viruses
257

11 Infections of the Nervous System
A 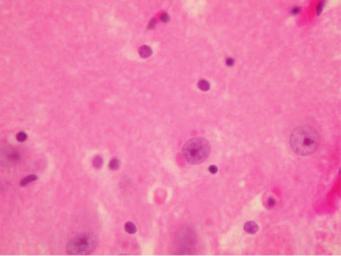 B
B
Figure 11–3 Pathological findings of rabies. (A) Negri body (arrows). (B) Babes nodules (arrows). Courtesy of Dr. C. Yamada and the Centers for Disease Control.
c.diagnostic testing: histological evaluation of the brains of the infected animal for viral particles; observe apparently healthy animals for 10 days after the bite to ensure they are not infected
d.treatment
i.wound cleansing specifically with benzyl ammonium chloride
ii.postexposure treatment and prophylaxis with the human diploid cell vaccine and local injection of rabies immunoglobulin
3.Arboviruses (Table 11–2)
a.general pathophysiology: carried by arthropods (ticks, mosquitoes); all exhibit an epidemic pattern in the summer
b.general symptoms: a flu-like syndrome followed in some cases by meningitis or encephalitis
c.treatment: none proven; may treat West Nile virus (WNV) with ribavirin, interferon, or WNV immunoglobulin, although these are of unproven value
Table 11–2 Arboviruses
Arbovirus |
Region |
Vector/reservoir |
Notes |
|
|
|
|
West Nile |
Entire US and |
Mosquito/birds |
10% mortality; 20% incidence |
|
Canada |
|
of focal neurological injury |
|
|
|
(e.g., poliomyelitis) |
St. Louis |
North and |
Mosquito/birds |
Slow development, |
encephalitis |
South America |
|
occasionally fatal; rarely causes |
|
|
|
focal neurological injury |
California-LaCross |
Midwest US |
Mosquito/many |
Frequently causes seizures that |
encephalitis |
|
mammals |
develop into epilepsy (10% of |
|
|
|
cases) but rarely causes focal |
|
|
|
neurological injury |
Eastern equine |
Eastern US |
Mosquito/birds |
60% mortality; neuroimaging |
encephalitis |
|
|
may show focal lesions in the |
|
|
|
basal ganglia and thalamus; |
|
|
|
exhibits simultaneous |
|
|
|
epidemics in horses |
Western equine |
Northern US |
Mosquito/birds |
10% mortality |
encephalitis |
and Canada |
|
|
Colorado tick |
Western US |
TIck (Dermacentor)/ |
Infected ticks live only at high |
fever |
|
rodents (Box 11.10) |
altitudes |
|
|
|
|
Box 11.10
Dermacentor also carries Rickettsia rickettsii, which causes Rocky Mountain spotted fever.
258

4.Polio viruses and polio-related syndromes
a.polio: commonly caused by one major (i.e., poliovirus) and several minor enteroviruses that directly infect the anterior horn lower motoneurons, causing their death; polio has been largely eradicated by vaccination
i.symptoms: developed 2–5 days after viral gastroenteritis with asymmetric and focal myalgias, fasciculations, and weakness in the lower extremity upper extremity bulbar muscles
ii.epidemiology: risk increased with age into young adulthood
iii.diagnostic testing: stool cultures for poliovirus
iv.treatment: prophylaxis with inactivated polio vaccine, which is routine in children
v.prognosis: significant improvement occurred in 80% after the acute illness
b.polio-related syndromes
i.postvaccination polio: caused by oral polio vaccine use, which was a live-attenuated virus that is no longer in use in the US; occurred in 0.04 per 100,000 vaccinations
ii.postpolio syndrome: progressive weakness beginning at least 10 years after polio that occurs in the previously affected musculature; a dubious disease entity that may reflect normal age-related loss of lower motoneurons
5.JC virus/progressive multifocal leukoencephalopathy (PML)
a.pathophysiology: a polyoma virus infection of oligodendroglia and astrocytes that causes cell death and demyelination; usually occurs in immunosuppressed patients after reactivation of a latent infection
b.histology: “ground glass” intranuclear inclusions in oligodendroglia; spheres and elongated rods (“spaghetti and meatballs”) on electron microscopy
c.symptoms: focal neurological injury with dementia progressing over months
d.diagnostic testing: identification of JC virus in cerebrospinal fluid by PCR
i.neuroimaging demonstrates multiple nonenhancing areas of decreased T1 and increased T2 signal without mass effect or edema; begin as small lesions that eventually form large confluent lesions
e.treatment: none specific against the JC virus, but reversing an immunosuppressed state (e.g., highly active antiretroviral therapy (HAART) therapy in HIV patients) may improve the symptoms
f.prognosis: 4-month survival, but 10% exhibit spontaneous remission
6.Measles virus
a.general pathophysiology: histologically characterized by Cowdry type A inclusion bodies in neurons and glia, and multinucleated giant cells
b.types of infection
i.acute self-limited encephalitis, associated with the rash
ii.measles inclusion body encephalitis: occurs in immunosuppressed patients several months after measles infection
(1)symptoms: rapidly-progressive dementia, seizures, and myoclonus progressing to coma
(2)treatment: reverse immunosuppressed state, if possible
iii.subacute sclerosing panencephalitis: a post-infection complication caused by abnormal assembly of the measles virus particles in the infected cells, leading to viral particle accumulation and transmission only at sites of cell–cell contact; develops in children several years after the viral rash
Viruses
259

11 Infections of the Nervous System
260
(1)symptoms: behavioral changes progressing slowly to dementia and ultimately to coma; myoclonus; seizures; chorea; ataxia
(2)specific diagnostic testing: EEG demonstrates reduced background activity with generalized slow wave complexes occurring regularly every 5–10 seconds (Fig. 11–4)
(3)treatment: intrathecal systemic interferon may be helpful
(4)prognosis: death within a few years
c.general diagnosis: high measles antibody titres in the cerebrospinal fluid; neurological infections do not involve pleocytosis
d.general treatment: prophylaxis with the measles vaccine given routinely to infants
7.Human immunodeficiency virus (HIV) (Table 11–3)
Figure 11–4 Subacute sclerosing panencephalitis. (From McKhann GM et al. Q&A Color Review of Clinical Neurology and Neurosurgery. Stuttgart, Germany: Georg Thieme; 2003:19, Fig. 9. Reprinted by permission.)
a.pathophysiology: infects monocytes, T lym-
phocytes, and microglia by binding to CD4 and a chemokine receptor with the viral gp120 glycoprotein; direct infection of neurons and oligodendroglia does not appear to occur, despite neuron apoptosis and myelin loss that is likely due to the actions of proinflammatory cytokines
i.histology: reactive gliosis diffusely distributed throughout the brain parenchyma
b.general diagnostic testing: blood HIV ELISA with confirmatory Western blot, which may take 4–8 weeks from the time of infection to turn positive
i.asymptomatic cerebrospinal fluid abnormalities occur throughout the course of HIV infection, typically protein 45 but 100, lymphocytic pleocytosis 25, and presence of anti-HIV IgG
c.treatment of HIV infection: HAART, which is a combination of three or more antiretroviral medications
i.side effects of antiretroviral medications include neuropathy (didanosine, zalcitabine, stavudine), myopathy (zidovudine), and oral paresthesias (ritonavir)
d.direct effects of HIV infection
i.HIV encephalitis/AIDS dementia complex—occurs in 1% with CD4
500, 7% with CD4 100
(1)pathophysiology: perivascular inflammation with multinucleated giant cells and reactive gliosis that may ultimately produce diffuse cerebral atrophy
(2)symptoms: develop over months
Table 11–3 Manifestations of Human Immunodeficiency Virus (HIV) Infection Related to CD4 Count
CD4 |
Direct effects of HIV |
Opportunistic |
|
|
|
500 |
Distal sensory polyneuropathy |
|
200–500 |
Dementia |
VZV radiculitis (shingles) |
100–200 |
Myelopathy |
Progressive multifocal leukoencephalopathy |
100 |
|
Toxoplasma, Cryptococcus |
50 |
|
Lymphoma, CMV |
|
|
|
Abbreviations: CMV, cytomegalovirus, VZV, varicella-zoster virus.
(a)subcortical dementia: impaired concentration, memory loss, apathy
(b)psychosis (10%)
(c)bradykinesia; hyperreflexia and spasticity
(3)diagnostic testing
(a)neuroimaging: MRI demonstrates diffuse high white matter signal on T2; late in the disease diffuse atrophy becomes apparent
(b)cerebrospinal fluid demonstrates increased protein, lymphocytosis, and elevated markers of lymphocyte and macrophage activation (e.g., 2 microglobulin, quinolinic acid)
(4)treatment: high-dose zidovudine, which can be a component of HAART
ii.vacuolar myelopathy: symptoms include spastic weakness, loss of large-fiber sensation (particularly proprioception), and incontinence developing over a period of months, caused by degeneration of posterior and lateral spinal cord columns
(1)unlikely caused directly by HIV infection, instead may be related to impairment of vitamin B12 utilization
iii.distal sensory polyneuropathy: symmetric sensory motor, rarely painful (Box 11.11)
(1)epidemiology: develops in 30% of AIDS patients
iv.HIV myopathy: can develop at any CD4 count; histologically is a poorly defined condition because of coincident myopathy from antiretroviral medications (particularly zidovudine) and from cachexia
(1)symptoms: proximal distal weakness, muscle soreness
(2)diagnostic testing: normal or increased baseline CK that is greatly increased by exercise
v.stroke: may be caused by HIV vasculitis, induced protein C and S deficiency, antithrombin III deficiency, or antiphospholipid antibody syndrome
(1)vasculitis can also be caused by cytomegalovirus, varicella, tuberculosis, or syphilis coinfection
e. opportunistic diseases with focal findings (Box 11.12)
i.toxoplasmosis: caused by Toxoplasma carinii (see p. 264)
(1)epidemiology: develops in 10% of AIDS patients
(2)symptoms: focal symptoms and encephalopathy fever headache developing over days to weeks
(3)diagnostic testing
(a)neuroimaging: usually exhibits multiple ring-enhancing lesions with mass effect, most commonly at the gray–white junction or basal ganglia
(i)70% of cases have multiple lesions
(ii)negative Toxoplasma IgG titers suggests the lesions are lymphoma
(iii)repeat neuroimaging after antibiotic treatment for 10 days to determine if the lesions have been reduced in size; avoid glucocorticoids unless the mass is inducing herniation as they will reduce both infectious and cancerous lesions
(4)treatment
(a)acute: pyrimethamine sulfadiazine or clindamycin folate
(b)prophylactic: TMP-SMX or dapsone
(5)prognosis: 90% of patients respond to treatment
Box 11.11
Neuropathy from antiretroviral treatments is painful unlike that directly from HIV infection.
Box 11.12
Brain Lesion Management in AIDS
1.Acute empiric treatment for toxoplasmosis except in cases of negative serologic studies
Evaluate for radiographic shrinkage within 10 days
Continue toxoplasmosis treatment indefinitely if the lesions are shrinking
2.Lesions with negative toxoplasmosis serology are an indication for brain biopsy
3.SPECT or PET suggests primary CNS lymphoma, which still requires brain biopsy confirmation
4.Immediately brain biopsy in children with AIDS and lesions
Viruses
261
11 Infections of the Nervous System
ii.primary CNS lymphoma
(1)epidemiology: develops in 5% of AIDS patients
(2)histology: lymphoma is of the non-Hodgkin B-cell type and it always is related to transformation by the Epstein-Barr virus
(3)symptoms: focal neurological injury (usually multiple), encephalopathy, and headache developing over weeks; symptoms are less severe than those of toxoplasmosis
(4)diagnostic testing
(a)neuroimaging: usually one or a few ring-enhancing lesions with mass effect, most commonly in the periventricular region or corpus callosum; lesions do not shrink after 10 days of antibiotics
(b)thallium201 SPECT or fluoro-2-deoxyglucose (FDG)- PET scanning shows label uptake
(c)cerebrospinal fluid cytology is usually negative, but markers of lymphocyte activity ( 2-microglobulin, lactate dehydrogenase) are usually increased
(d)tissue biopsy is ultimately required for diagnosis
(5)treatment: irradiation, particularly with the gamma knife; chemotherapy cannot usually be tolerated
(6)prognosis: 1-month survival untreated; 6-month survival with conformal radiation therapy (XRT)
iii.progressive multifocal leukoencephalopathy (see p. 259) caused by the JC virus, occurring in 5% of AIDS patients
f. opportunistic diseases with minimal focal findings
i.cytomegalovirus (see p. 257)
ii.meningitis, caused by
(1)Cryptococcus (see p. 263) occurs in 10% of AIDS patients, usually with CD4 200; 30% acute mortality; outcome is proportionate to Cryptococcus antigen titer and low level of CSF pleocytosis
(2)syphilis (see p. 254): rate of neurosyphilis is not affected by HIV infection, although it generally develops faster; cerebrospinal fluid examination for syphilis is required even without clinical evidence of neurosyphilis
(3)tuberculosis (see p. 252): in HIV infected patients, there is an increased risk of developing tuberculous meningitis (10% overall incidence) but there does not appear to be a worse outcome from the disease
(4)bacteria: Listeria monocytogenes, Gram-negative bacilli
(5)cancer (carcinomatous meningitis, see p. 131), particularly from systemic lymphoma
VI. Fungus
1.General fungal pathophysiology: most are initially respiratory infectious processes that enter the CNS via hematogenous spread
2.General symptoms: all can present as acute, subacute, or chronic meningitis; focal neurological
Figure 11–5 Aspergillus centered about a small parenchymal vessel. Courtesy of Dr. C. Yamada.
262
signs can be indicative of abscess formation, but may also be part of subacute-chronic meningitis (e.g., cranial nerve palsies)
a.vision loss may occur from an elevated intracranial pressure
3.General diagnosis: fungal stain is positive in 50% or less of cases, depending upon the fungus
4.Types of fungi
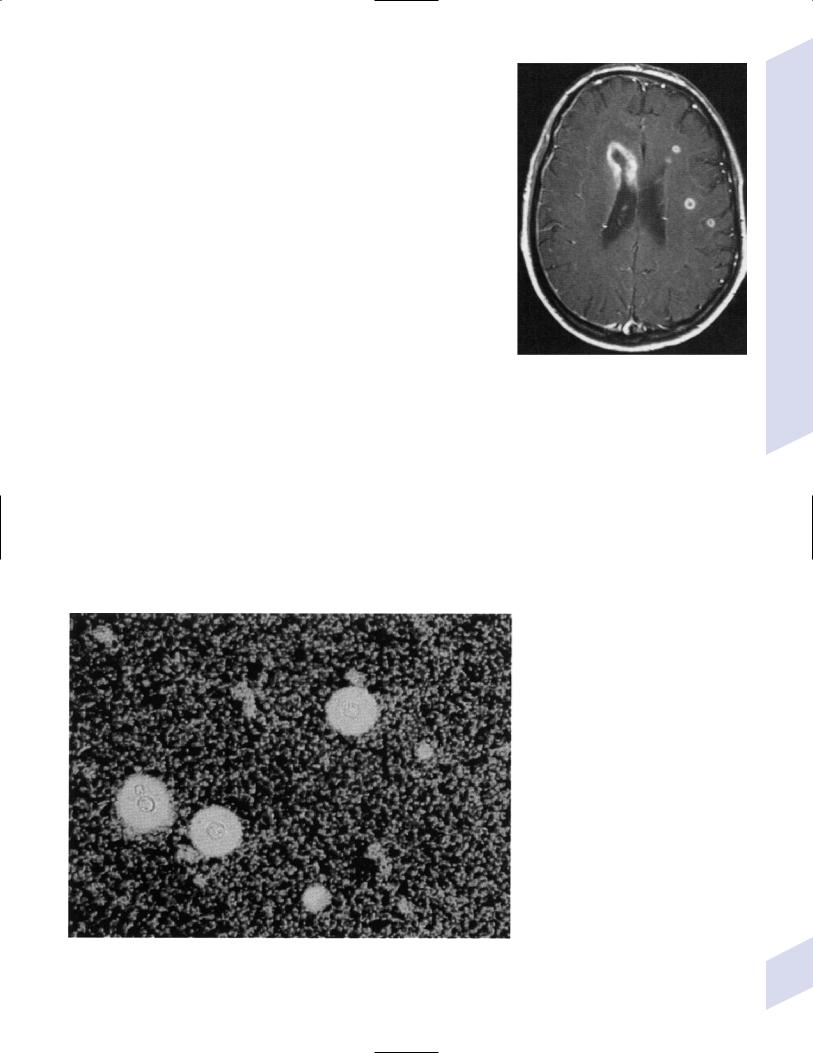
a.Aspergillus—invades blood vessels in the brain (Fig. 11–5), leading to a necrotizing vasculitis, thrombosis, and/or granulomatous inflammation; forms abscesses more commonly than meningitis, particularly around the ependymal region (Fig. 11–6)
i.epidemiology: common in postsurgical/post-head trauma patients
ii.specific diagnostic testing: Aspergillus PCR
b.Cryptococcus—found in bird droppings and soil, which is subsequently inhaled; can also directly colonize human skin
i.epidemiology: can infect the immunocompetent, but more commonly infects HIV patients and chronic glucocorticoid users
ii.causes meningitic that form involves granulomas with multinucleated giant cells 5-mm diameter, similar to tubercular meningitis
(1)50% of cases develop small groups of intraparenchymal cysts that represent cryptococcal abscesses {cryptococcoma}
iii.specific diagnostic testing
Figure 11–6 Cerebral aspergilliosis with cerebral and ependymal lesions on MRI. (From McKhann GM et al. Q&A Color Review of Clinical Neurology and Neurosurgery. Stuttgart, Germany: Georg Thieme; 2003:115, Fig. 112. Reprinted by permission.)
(1)Cryptococcus antigen latex agglutination test is positive in the cerebrospinal fluid of 90% of meningitis cases; in HIV patients, a negative serum cryptococcal antigen testing is sufficient to rule out the diagnosis of Cryptococcus meningitis
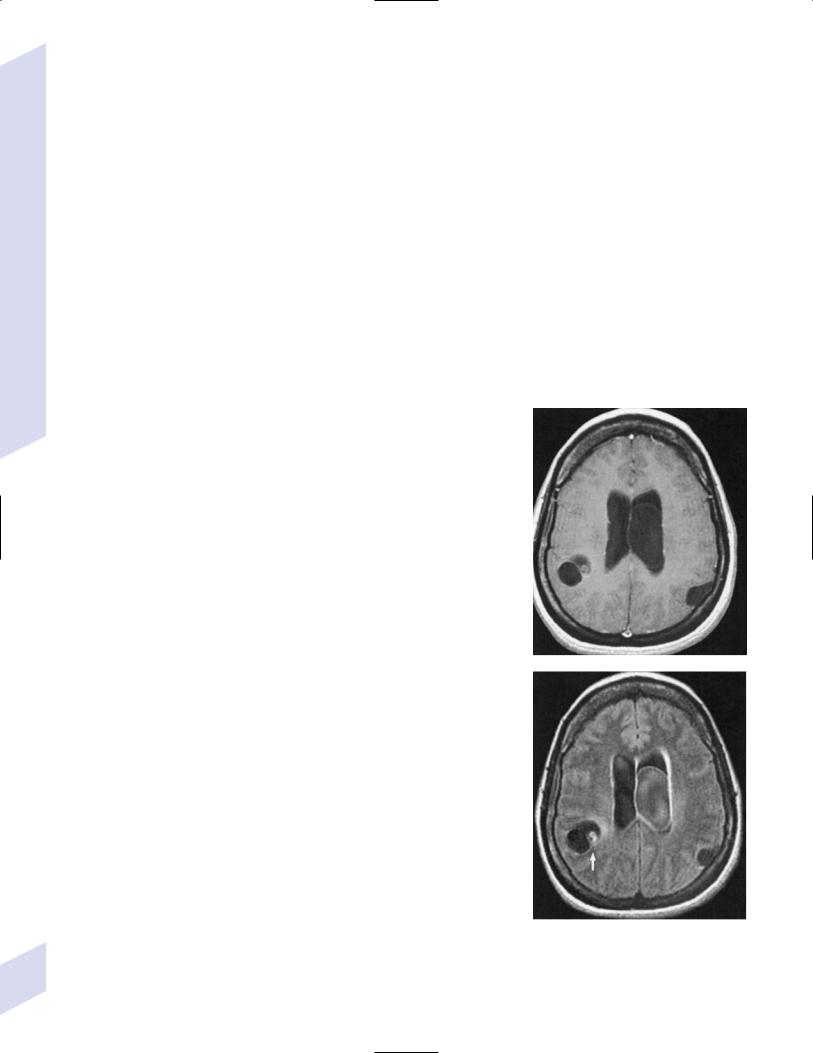
(2) India ink stain may demonstrate encapsulated organisms (Fig. 11–7)
c.Coccidioides sp., Histoplasma sp.: cause chronic meningitis
Figure 11–7 Encapsulated Cryptococcus. (From Citow JS et al. Neuropathology and neuroradiology: a review. Stuttgart, Germany: Georg Thieme; 2001:28, Fig. 29. Reprinted by permission.)
Fungus
263
11 Infections of the Nervous System
264
VII. Parasites
1.Protozoans
a.amebiasis: cause hemorrhagic encephalitis
i.Acanthamoeba/Balamuthia—infects the lower respiratory tract and skin
ii.Naegleria—initially infects the nasal cavity, then spreads to the brain via the cribriform plate
iii.Entamoeba—transmission is human to human via reservoirs of food or water; commonly infects the GI tract or liver
b.toxoplasmosis (Toxoplasma carinii)—ingested cysts develop into larva in the intestines and disseminate hematogenously to the brain (causing encephalitis and focal mass lesions), eye (causing chorioretinitis), heart and muscle; can be transmitted from reservoir animals (birds, cats) directly or across the placenta
i.diagnostic testing: Toxoplasma antibodies or PCR on cerebrospinal fluid
ii.treatment: pyrimethamine sulfadiazine or clindamycin
(1)prevent fetal infection by avoiding risk factors (e.g., cleaning cat boxes) in seronegative pregnant women, particularly during the first trimester when the complications of infection to the neonate are most severe
(2)treat acutely infected pregnant women with sulfadiazine; may also use pyrimethamine after the first trimester
c.malaria (Plasmodium falciparum)—parasite-infected erythrocytes aggregate and cause capillary and small vein obstruction and vascular congestion, producing the characteristicly swollen “slate gray” brain on autopsy
i.symptoms: develop after a 12-day period of a flu-like syndrome that involves periodic (approximately q.3.day) attacks of shaking chills, high fever, and diaphoresis, as well as hepatosplenomegaly
(1)cerebral malaria: develops in 1% of cases, usually within 2 weeks of the initial infection in adults but may develop as rapidly as 1 day in children
(a)symptoms include encephalopathy, seizures, and meningismus; focal neurological injury is common
ii.diagnostic testing
(1)ring-shaped trophozoites in erythrocytes on blood smear
(2)cerebrospinal fluid demonstrates increased intracranial pressure, elevated protein, and a lymphocytosis 50
(3)neuroimaging demonstrates diffuse cerebral edema
iii.treatment: quinine; artemether for multidrug resistant Plasmodium falciparum; transfusion for severe anemia
(1)do not use glucocorticoids as they worsen outcome
iv.prognosis: cerebral malaria is lethal within 3 days if untreated
(1)postmalaria syndromes
(a)delayed-onset coma, which develops within 2 days of recovery from cerebral malaria; recovery is variable
(b)delayed-onset psychosis
2.Metazoans: Cause cystic mass lesions in the brain parenchyma, ventricles, or subarachnoid spaces; the death of the organism leads to an intense local inflammatory response that can exacerbate symptoms up to 30 years after the initial infection
a.cysticercosis (Taenia solium; pig tapeworm)—acquired by eating poorly cooked pork or by fecal-oral transmission; ingested eggs hatch in GI tract, migrate to brain, eye, and muscle
i.diagnostic testing: Taenia ELISA; neuroimaging demonstrates multiple cystic mass lesions with a visible scolex (Fig. 11–8)
ii.treatment: albendazole or praziquantel
A
B
Figure 11–8 Neurocysticercosis. (A) T1 posgadolinium. (B) FLAIR demonstrating the scolex (arrow). (From McKhann GM et al. Q&A Color Review of Clinical Neurology and Neurosurgery. Stuttgart, Germany: Georg Thieme; 2003:79, Fig. 70b, Fig, 70c. Reprinted by permission.)
b.echinococcosis/hydatid disease (Echinococcus)—acquired by eating poorly cooked meat from several types of animals or by fecal-oral transmission; ingested eggs hatch in GI tract, migrate to brain, spinal cord {hydatid Pott’s disease}, liver, and lung
i.diagnostic testing: neuroimaging demonstrates parasite cysts
ii.treatment: surgery after cyst reduction with alben-
dazole; if unresectable, use prolonged albendazole |
|
treatment |
A |
|
VIII. Prion Diseases
1.General pathophysiology: caused by a toxic gain-of-function that is associated with a conformational change in the prion protein (PrP), which is a membrane-bound glycoprotein that may be involved in copper metabolism
a.abnormal PrP is resistant to denaturation and proteases, and can induce the toxic conformational change in native PrP
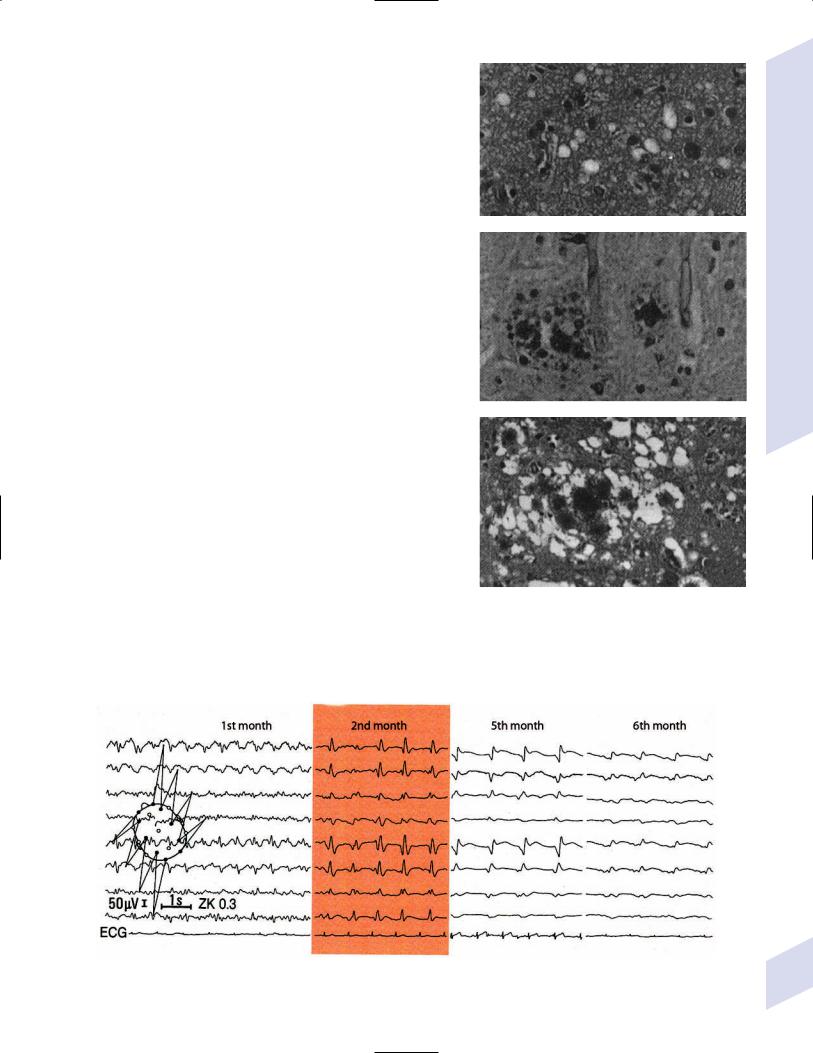
b.general histology: prion diseases involve spongiform degeneration of the gray matter that is caused by the loss of neurons (Fig. 11–9); affected brain parenchyma does not have inflammatory changes but may have a reactive gliosis, depending upon the specific prion disease
i.spongiform changes are not unique to prion diseases, and can be observed in Alzheimer’s, Pick’s, and Lewy body dementias, and in certain aminoacidopathies
2.Types of prion diseases
a.idiopathic prion diseases
i.sporadic Creutzfeldt-Jakob (sCJD) disease—accounts for 80% of CJD; typically occurs in older patients (average age of onset 65 years of age)
(1)symptoms: rapidly progressive myoclonus and dementia; average 4-month duration of symptoms at time of diagnosis
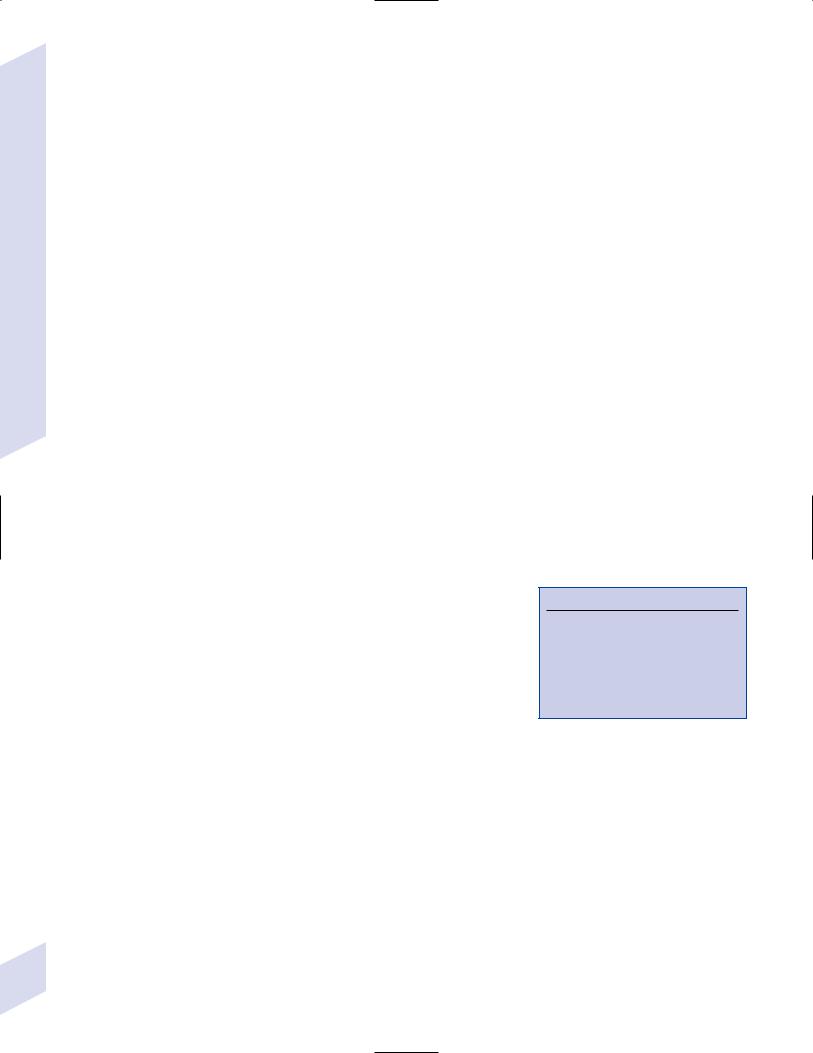
(2)diagnostic testing: EEG demonstrates periodic sharp waves (Fig. 11–10)
B
C
Figure 11–9 (A) Spongiform vacuolation. (B) Multicentric plaques of Gerstmann-Straussler-Scheinker disease. (C) florid plaques of vCJD. (From Mastrianni JA, Roos RP. The prion diseases. Semin Neurol 2000,20:343, Fig. 3a, Fig, 3c, Fig, 3d. Reprinted by permission.)
Prion Diseases
Figure 11–10 Development of EEG abnormalities in Creutzfeldt-Jakob disease. Within 2 months of symptomatic onset, periodic activity is evident |
265 |
and it becomes dominant within 1 month. Gradual attenuation develops thereafter. (From Mumenthaler M, Neurology. 3rd ed. Stuttgart, Germany: |
Georg Thieme; 1990:170, Fig. 1.23. Reprinted by permission.)
b. |
familial/inherited prion diseases |
|||
|
i. inherited Creutzfeldt-Jakob disease—accounts for 15% of all CJD; |
|||
|
symptomatic onset is typically less than 60 years of age, but there is |
|||
|
still an increasing incidence with advanced age |
|||
|
(1) |
pathophysiology: caused by mutation of the PrP gene on chromo- |
||
|
|
some 20; exhibits autosomal dominant inheritance |
||
|
(2) |
symptoms: as per sCJD |
||
System |
(3) |
diagnostic testing: as per sCJD |
||
ii. fatal familial insomnia |
||||
|
||||
|
(1) pathophysiology: degeneration occurs predominantly in the |
|||
Nervous |
|
anterior and mediodorsal thalamus with pronounced gliosis and |
||
|
minimal spongiform degeneration |
|||
|
|
|||
|
(2) |
symptoms: onset between 30–40 years of age |
||
the |
|
(a) |
insomnia, fatigue |
|
|
(b) |
anorexia |
||
of |
|
(c) |
hallucinations; stupor progressing to coma |
|
Infections |
|
|||
|
(d) |
ataxia, dysarthria |
||
|
|
|||
|
|
(e) |
sympathetic hyperactivity |
|
|
(3) |
diagnostic testing: cerebrospinal fluid is reliably negative for |
||
11 |
|
14-3-3 protein, and neuroimaging is normal |
||
|
(a) |
polysomnography demonstrates absent rapid eye movement |
||
|
|
|||
|
|
|
(REM) and slow-wave sleep stages |
|
|
|
(b) |
PET scan shows reduced blood flow in thalamus |
|
|
iii. Gerstmann-Straussler-Scheinker disease |
|||
|
(1) histology: Alzheimer-like amyloid plaques and kuru-type plaques |
|||
|
|
in cerebellum cerebrum, brainstem; relatively little spongiform |
||
|
|
degeneration |
||
|
(2) |
symptoms: onset between 50–60 years of age; symptoms are |
||
|
|
progressive over a period of years |
||
|
|
(a) |
limb and trunk ataxia |
|
|
|
(b) |
dementia, delirium |
|
c. infectious prion diseases (Box 11.13) |
||||
|
i. variant Creutzfeldt-Jakob (vCJD) disease/transmissible spongiform |
|||
|
encephalopathy/mad cow disease |
|||
|
(1) |
specific histology: spongiform changes occur in the basal ganglia |
||
|
|
and thalamus; PrP aggregates into dense cores with halos in the |
||
|
|
cerebrum and cerebellum that are surrounded by spongiform |
||
|
|
change {florid plaques} (Fig. 11–9C) |
||
|
(2) |
symptoms: onset between 20–30 years of age, with a relatively |
||
|
|
slow course compared with other CJD subtypes |
||
|
|
(a) |
psychiatric disorders: anxiety, insomnia |
|
|
|
(b) |
dysesthesias |
|
|
|
(c) |
gait ataxia, dysarthria, tremor; chorea; myoclonus |
|
|
|
(d) |
dementia |
|
|
(3) |
diagnostic testing |
||
|
|
(a) |
EEG does not demonstrate periodic complexes as it does in |
|
|
|
|
sporadic or inherited CJD |
|
|
|
(b) |
cerebrospinal fluid demonstrates 14–3-3 protein in 50% of |
|
|
|
|
cases |
|
|
|
(c) |
neuroimaging: increased T2 signal in posterior thalamus |
|
|
|
|
(75%) |
|
d. iatrogenic prion diseases: rarely causes of CJD include contaminated |
||||
266 |
dural grafts, cadaveric human growth hormone therapy, or surgical |
|||
instruments |
|
|||
Box 11.13
Kuru
Pathophysiology: Transmissible only by cannibalism, occurs only in New Guinea
Histology: Kuru-type plaques in cerebellum
Symptoms: limb and truncal ataxia; dementia
Table 11–4 Toxin Syndromes
|
Toxin pathophysiology |
Route |
Key symptoms |
Treatment |
|
|
|
|
|
Botulism |
Cleaves SNAP-25, syntaxin, |
Contaminated food, |
Flaccid paralysis & hyporeflexia; |
Emetics, antiserum, |
(Clostridium |
or synaptobrevin; inhibits |
wound, GI colonization |
ophthalmoparesis & unreactive |
wound debridement, |
botulinum) |
presynaptic acetylcholine |
(infants), inhalation |
pupils |
penicillin |
|
release from motoneurons |
(bioweapons) |
|
|
|
|
|
|
|
Tetanus |
Inhibits GABA & glycine |
Wound, including |
Painful muscle spasms that are |
Anti-tetanus IgG, wound |
(Clostridium |
release from pre- |
umbilical stump |
inducible by startle; |
debridement, |
tetani) |
motoneuron inhibitory |
|
hyperreflexia; sympathetic |
metronidazole, penicillin |
|
interneuron |
|
hyperactivity |
|
|
|
|
|
|
Diphtheria |
Inhibits protein synthesis |
Skin infection; pharyngitis, |
Local sensory loss and |
Diphtheria antitoxin, |
(Corynebacterium |
|
tonsillitis, sinusitis |
weakness (skin infection); |
erythromycin, penicillin |
diphtheriae) |
|
|
sensorimotor polyneuropathy |
|
|
|
|
involving vagus and phrenic |
|
|
|
|
nerves |
|
|
|
|
|
|
Tick paralysis |
Inhibits presynaptic |
Tick bite |
Ataxia, flaccid paralysis (like |
Remove tick |
(holocyclotoxin of |
acetylcholine release |
|
GBS), diffuse paresthesias |
|
Ixodes ticks) |
|
|
|
|
|
|
|
|
|
Abbreviations: GI, gastrointestinal; IgG, immunoglobulin G; GBS, Guillain-Barre syndrome; SNAP, sensory nerve action potential.
IX. Toxin Syndromes
1.Major neurotoxins (Table 11–4)
2.Other neurotoxins (Table 11–5)
X. Idiopathic Infection-Like Conditions
1.Idiopathic hypertrophic pachymeningitis (Box 11.14)
a.histology: infiltration of meninges by mature lymphocytes and histiocytes, rarely with granuloma formation
b.symptoms: severe headache (90%); vision loss (60%), papilledema; ataxia; seizures
c.diagnostic testing
i.neuroimaging demonstrates dural enhancement involving nodularity and cerebral edema in the proximal brain parenchyma
Table 11–5 Other Neurotoxins
Box 11.14
Unexpected Causes of Dural Enhancement
Infections: Spirochetes, mycobacteria, fungi, parasites
Autoimmune disorders: Wegener’s granulomatosis, sarcoidosis, Behçet’s syndrome, Sjögren’s syndrome, rheumatoid arthritis
Carcinomatous meningitis Spontaneous intracranial hypotension En plague: Meningioma
Toxin |
Source |
Action |
Symptoms |
|
|
|
|
|
|
Bungarotoxin |
Snakes |
Nicotinic ACh receptor antagonist; |
Paralysis, including respiratory muscles |
|
Alpha |
|
|
inhibits presynaptic ACh release |
|
Beta |
|
|
|
|
|
|
|
|
|
Latrotoxin |
Black Widow spider |
Binds presynaptic neurexin, causing increased |
Local pain, muscle fibrillation; generalized |
|
|
|
|
neurotransmitter release |
muscle cramping; hypertension |
|
|
|
|
|
Domoate* |
Shellfish |
Kainate NMDA receptor agonist |
Nausea, diarrhea, abdominal pain; somnolence, |
|
|
|
|
|
amnesia, mutism, seizures, myoclonus |
|
|
|
|
|
Saxitoxin,* |
Shellfish |
Voltage-gated sodium channel antagonists |
Nausea, diarrhea, abdominal pain; |
|
|
|
|
|
paresthesias; progressive weakness with |
tetrototoxin* |
Pufferfish, newts |
|
||
|
oropharyngeal involvement; hypotension |
|||
|
|
|
|
|
Brevitoxin |
“Red tide” shellfish |
Voltage-gated sodium channel agonists |
Nausea, diarrhea; paresthesias |
|
|
|
|
|
|
Ciguatoxin,* |
Large fish |
|
Diffuse myalgias, paresthesias; inverted |
|
scaritoxin* |
|
|
|
temperature sensation (e.g., cold feels hot) |
|
|
|
|
|
*Produced by single-cell microorganisms eaten by the larger animal. Abbreviations: NMDA, nicotimide adenine dinucleotide.
Idiopathic Infection-Like Conditions
267

11 Infections of the Nervous System
ii.cerebrospinal fluid demonstrates a lymphocyte pleocytosis and increased protein
d.treatment: glucocorticoids
2.Mollaret’s meningitis
a.pathophysiology: possibly related to recurrent bouts of limited herpes virus reactivation, but generally no pathogen can be identified
b.symptoms: several days of meningitis-like symptoms that spontaneously resolve
c.diagnostic testing: cerebrospinal fluid contains various types of leukocytes and large endothelium-like cells {Mollaret’s cells}; Mollaret’s cells usually are observable only at the very beginning of an attack
d.treatment: none specific
3.Vogt-Koyanagi-Harada syndrome
a.pathophysiology: possibly an autoimmune reaction against retinal photoreceptor antigens; associated with (HLA)-DR
b.symptoms: a self-limited episode of meningitis and iritis followed weeks later by hair loss, focal loss of hair pigmentation {poliosis}, and vitiligo; meningitis episode can involve focal neurological injury, and vision loss can occur from iritis
c.diagnostic testing: cerebrospinal fluid has melanin-containing macrophages
d.treatment: glucocorticoids
268
12
Developmental and Metabolic
Diseases of the Nervous System
Note: Significant diseases are indicated in bold and syndromes in italics.
I. Disorders of Embryogenesis
A. Malformations
1. Cortical malformations—general symptoms include medication-refractory
seizures, mental retardation, and focal neurological abnormalities |
|
|
|
|
|||
a. |
heterotopia—formed by either the abnormal proliferation or migration of |
|
|
|
|||
|
neurons; neurons are generally misshapen and incorrectly organized |
|
|
|
|
||
|
i. |
subtypes |
|
|
|
|
|
|
|
(1) focal cortical dysplasia—a gray matter connection between the |
|
|
|
||
|
|
cortex and the periventricular zone formed by neuroblasts that |
|
|
|
||
|
|
have incompletely migrated toward the cortex |
|
|
|
|
|
|
|
(2) periventricular heterotopia—clumps of neurons that failed to |
|
|
|
||
|
|
migrate to the cortex (or move at all) and instead reside in the |
|
|
|
||
|
|
periventricular white matter (Fig. 12–1) |
|
|
|
|
|
|
|
|
|
Box 12.1 |
|||
|
|
(a) some cases are related to X-linked mutations in the filamin A |
|
||||
|
|
|
Laminar heterotopia and lissencephaly are |
|
|||
|
|
gene, the protein of which regulates cytoskeletal actin filament |
|
||||
|
|
|
both associated with mutations of the |
||||
|
|
formation |
|
|
|||
|
|
|
|
doublecortin gene (X-linked) or LIS-1 gene |
|||
|
|
(3) laminar heterotopia: waves of neurons fail to migrate the entire |
|
||||
|
|
|
(autosomal dominant). |
||||
|
|
distance to the cortex and instead form a band underneath |
|
|
|
|
|
|
|
|
|
|
|
||
|
|
the cortex in the white matter (Box 12.1) |
|
|
|
|
|
b. |
lissencephaly—diffuse neuronal migration failure that produces only |
|
|
|
|
||
|
a few enlarged gyri {pachygyria} |
|
|
|
|
||
c. |
polymicrogyria—small cortical gyri with fusion and/or loss of the un- |
|
|
|
|
||
|
derlying neuronal layers, often occurring in a regional distribution |
|
|
|
|
||
|
(e.g., peri-Sylvian) |
|
|
|
|
||
d. |
schizencephaly—clefts in the cortex that penetrate to the ventricular |
|
|
|
|
||
|
system occurring in areas of polymicrogyria; the clefts are lined by |
|
|
|
|
||
|
cortical neurons, unlike porencephaly (Fig. 12–2) |
|
|
|
|
||
|
i. the severity of symptoms is related to size of the cleft and a |
|
|
|
|
||
|
|
patent cleft opening |
|
|
|
|
|
e. |
porencephaly—cortical clefts that can penetrate to the ventricular |
|
|
|
|
||
|
system, and that have openings that are surrounded by abnormal |
|
|
|
|
||
|
gyri (often polymicrogyria); unlike schizencephaly, clefts are usu- |
|
|
|
|
||
|
ally bilateral and symmetric, and gray matter does not extend into |
|
|
|
|
||
|
the cleft |
|
|
|
|
||
|
i. |
clefts likely represent tissue loss from in utero injuries |
|
|
|
|
|
|
ii. growth of the clefts due to fluid retention may compress ven- |
|
|
|
|
||
|
|
tricular flow, causing hydrocephalus |
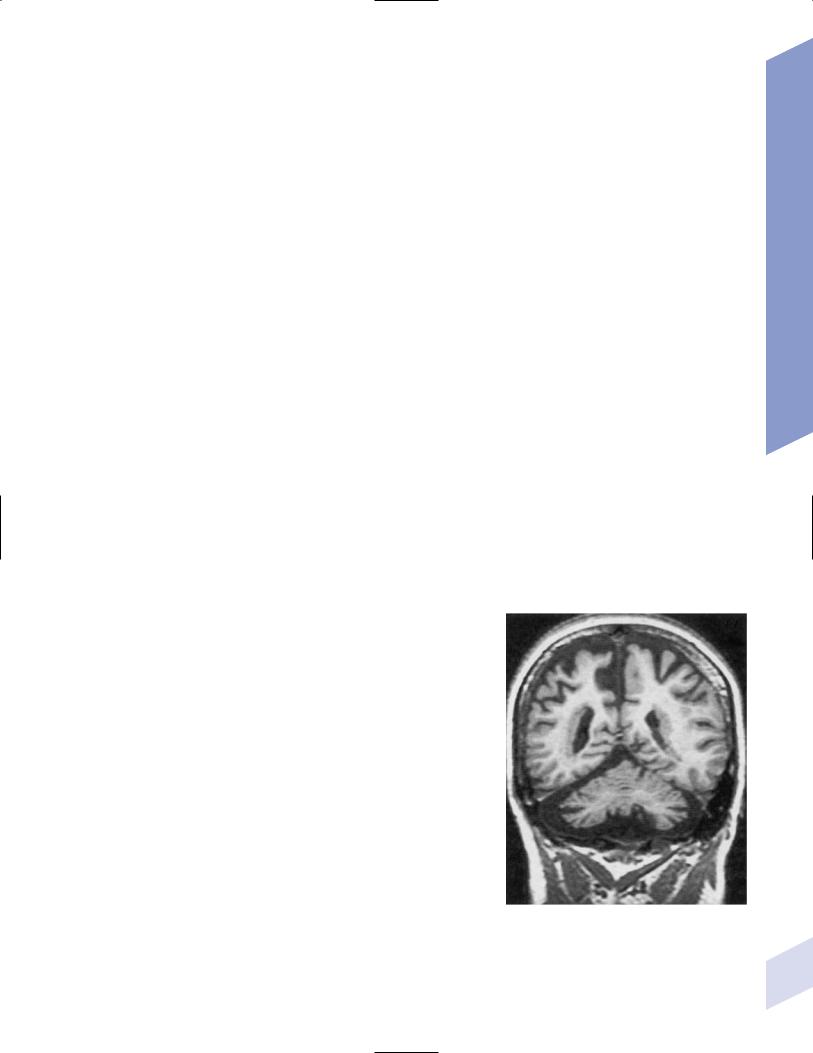
Figure 12–1 Bilateral periventricular nodular het- |
||||
f. |
holoprosencephaly—failure of prosencephalon development, producing |
||||||
erotopias located on the lateral surface of the ven- |
|||||||
|
a smooth cortical surface without gyri or sulci |
||||||
|
tricles. (From McKhann GM et al. Q&A Color Review |
||||||
|
i. |
related to |
of Clinical Neurology and Neurosurgery. Stuttgart, |
||||
|
Germany: Georg Thieme; 2003:73, Fig. 64. Reprinted |
||||||
|
|
(1) maternal diabetes |
|||||
|
|
by permission.) |
|||||
Disorders of Embryogenesis
269

Developmental and Metabolic Diseases of the |
Nervous System |
12 |
|
(2) |
mutation in the sonic hedgehog gene (10% of cases) (Box 12.2), |
|
Box 12.2 |
|
|
which encodes a secreted intracellular signaling molecule; under |
|
||
|
|
|
|
|
|
|
|
|
|
|
expression of sonic hedgehog at the rostral end of the neural tube |
|
Other mutations of the sonic hedgehog |
|
|
leads to incomplete development |
|
gene cause cerebellar hypoplasia and/or |
|
(3) |
mutations in Zic-2, a transcription regulatory protein |
|
pituitary gland abnormalities and VACTERL |
|
|
syndrome. |
|||
|
|
|
||
ii.subtypes
(1)alobar holoprosencephaly: no divisions develop between the hemispheres or lobes; associated with a single underlying ventricle, agenesis of the septum and corpus callosum, and olfactory and optic nerve hypoplasia
(2)semilobar prosencephaly: failure of the anterior interhemispheric division allows for fusion of the anterior cortices with significant hypoplasia of the corpus callosum and olfactory nerves; a posterior interhemispheric division exists and the basic lobar structure is otherwise preserved
(a)septo-optic dysplasia—a variant of semilobar prosencephaly; specific features include hypothalamic hamartomas causing panhypopituitarism, agenesis of the cerebellar vermis and fusion of the dentate nuclei causing ataxia (Fig. 12–3).
(3)lobar prosencephaly: interhemispheric connections occurring between frontal poles and the cingulate cortices through an oversized indusium griseum (a vestigial gray matter band normally located on top of the corpus callosum); limited hypoplasia of the corpus callosum
g.hydranencephaly—reduction of the cerebral cortex to a flat, thin layer, often sparing the basal cortex and hippocampus but severely disrupting the thalamus and basal ganglia; caused by destructive processes (which produce microcephaly at birth) or by hydrocephalus
(which produces macrocephaly at birth) |
Figure 12–2 Schizencephaly. (From Citow JS et al. Neuropathol- |
|
ogy and Neuroradiology: A Review. Thieme; Stuttgart, Germany: |
||
|
||
|
Georg Thieme; 2001:11, Fig. 9. Reprinted by permission.) |
A B
Figure 12–3 Septo-optic dysplasia with absent septum pellucidum between the two frontal horns
(A) and thin optic nerves (B). (From Citow JS et al. Neuropathology and Neuroradiology: A Review. Thieme; Stuttgart, Germany: Georg Thieme; 2001:10, Fig. 7A. Reprinted by permission.)
270

i.generally fatal within a few days due to hypothalamic dysfunction
2.Subcortical malformations
a.agenesis of the corpus callosum—caused by complete or partial failure of the extension of the developing corpus callosum posteriorly from the lamina terminalis; fibers that cannot cross in the corpus callosum run ipsilaterally in a rostrocaudal direction {bundle of Probst} (Box 12.3)
i.other commissures are enlarged
ii.occurs in isolation in 2% of population
iii.symptoms: generally asymptomatic without other malformations, but may exhibit
(1)learning disabilities or mental retardation
(2)abnormally wide-spaced eyes {hypertelorism}
(3)exotropia with inability to converge
3.Abnormalities of cerebrospinal fluid and the ventricular system
a.symptoms of hydrocephalus include irritability, bulging fontanelles, prominent scalp veins, false-localizing CN VI palsies, Parinaud’s syndrome, hyperreflexivity, and irregular respiration
b.diagnosis requires a head circumference 2 standard deviations for gestational age or an increase in head circumference 2 standard deviations during first year of life; diagnosis does not require elevated intracranial pressure, which only occurs after fusion of the cranial sutures
c.pathophysiology: acquired pediatric hydrocephalus conditions
i.noncommunicating: aqueduct stenosis caused by intrauterine infection, hemorrhage, trauma, or tumor
ii.communicating: following subarachnoid hemorrhage or meningitis
d.pathophysiology: syndromal hydrocephalus conditions
i.trisomy 9, 13, 18
ii.VACTERL syndrome with hydrocephalus (Box 12.4)
iii.Hunter’s and Hurler’s syndromes
iv.Walker-Warburg syndrome (also has congenital muscular dystrophy, lissencephaly, Dandy-Walker malformation, and eye malformations)
v.craniosynostosis syndromes: all involve premature closure of skull sutures leading to skull deformation, and have autosomal dominant and sporadic forms
(1)simple craniosynostosis: early closure of one or a few sutures that does not cause hydrocephalus or other symptoms
(2)complex craniosynostosis
(a)Crouzon’s syndrome—other symptoms include multiple cranial nerve palsies caused by jugular foramen narrowing; no mental retardation
(b)Apert’s syndrome—other symptoms include facial deformity, syndactyly, short upper extremities, and mental retardation
vi.X-linked hydrocephalus syndromes: both are caused by mutations in L1CAM, a cell adhesion molecule (Box 12.5)
(1)mental retardation, aphasia, shuffling gait, adducted thumbs (MASA) disorder
(2)X-linked aqueduct stenosis—also exhibits adducted thumbs, agenesis of the corpus callosum, brainstem malformations, and hypoplastic corticospinal tracts
4.Cerebellar and brainstem malformations
a.Chiari malformations
Box 12.3
Aicardi’s syndrome
Pathophysiology—X-linked inherited agenesis of corpus callosum and anterior commissure, and polymicrogyria
Symptoms—Mental retardation; myoclonic epilepsy; chorioretinal abnormalities; vertebral abnormalities
Box 12.4
VACTERL syndrome
Vertebral anomalies; anal atresia; cardiac defects; tracheoesophageal fistula; renal abnormalities; l imb abnormalities. Some cases can be caused by mutation in the sonic hedgehog gene, like holoprosencephaly.
Box 12.5
L1CAM is also mutated in X-linked hereditary spastic paraparesis.
Disorders of Embryogenesis
271
Developmental and Metabolic Diseases of the |
Nervous System |
12 |
|
272
Table 12–1 |
Chiari Malformations |
|
|
Chiari I malformation |
Chiari II malformation |
|
|
|
Symptoms |
Onset in young adulthood |
Onset in infancy |
|
Head, neck, or upper back |
|
pain; extremity spastic |
|
weakness distal atrophy; |
|
extremity sensory loss; |
|
gait ataxia; diplopia; |
|
downbeat nystagmus |
Cerebellum |
Downward herniation of |
|
cerebellar tonsils; |
|
elongated, peg-like tonsils |
Brainstem |
Normal |
Spine and |
Syringomyelia, scoliosis |
spinal cord |
|
Ventricular |
Hydrocephalus due to |
system |
adhesions occluding 4th |
|
ventricle foramen ( 5%) |
Skull |
Minor abnormalities of |
|
basilar skull |
Supratentorial |
Normal |
structures |
|
Diagnostic |
Neuroimaging: Descent of |
testing |
one tonsil 5 mm below |
|
foramen magnum, or both |
|
tonsils 3–5 mm below |
|
foramen magnum |
Treatment and |
Craniocervical |
prognosis |
decompression |
|
Good outcome with pain or |
|
cerebellar injury; poor out- |
|
come with central cord injury |
Dysphagia: cyanosis during feeding, regurgitation, aspiration; apnea, stridor; upper extremity weakness; opisthotonos; nystagmus, particularly downbeat
Downward herniation of cerebellar vermis; dysplastic, hypomyelinated cerebellar folia
Herniation of medulla into the spinal canal; buckling of medulla {Z deformity}, tectal beaking; stretched and atrophic lower cranial nerves
Myelomeningocele; syringomyelia, scoliosis
Hydrocephalus from cerebral aqueduct stenosis; big occipital horns of lateral ventricles. {colpocephaly}
Enlarged foramen magnum; multifocal skull defects {luckenschadel} (See figure at right.); platybasia/basilar impression
Hypoplastic and irregular tentorium and falx {Chinese lettering sign}; enlarged thalamic massa intermedia, absent septum; agenesis of corpus callosum
Neuroimaging: For aforementioned abnormalities; skull films demonstrate luckenschadel
May need ventricular shunting
Craniocervical decompression
Older patients (i.e., children) tend to improve after surgery; unclear if decompression alters course in infants
Luckenschadel. (From Citow JS et al. Neuropathology and Neuroradiology: A Review. Thieme; Stuttgart, Germany: Georg Thieme; 2001:14, Fig. 11, Fig. 13A. Reprinted by permission.)
i.Chiari I and II malformations (Table 12–1) (Fig. 12–4)
ii.other Chiari malformations
(1)Chiari III malformation: a very rare malformation characterized by herniation of an abnormal brain stem and cerebellum into a low occipital/high cervical encephalocele; associated with hydrocephalus
(2)Chiari IV malformation: aplasia or hypoplasia of the cerebellum, therefore not a true Chiari malformation due to the lack of any hindbrain herniation
(3)Chiari 0 malformation: syringohydromyelia not associated with hindbrain herniation or other apparent etiology, likely due to functional disturbances of cerebrospinal fluid flow at the foramen magnum
b.Dandy-Walker malformation (Box 12.6)
i.pathophysiology: atresia of the foramen of Luschka and Magendie together with agenesis of the cerebellar vermis creates a fluid space that communicates with the subarachnoid space (Fig. 12–5)
(1)associated abnormalities include agenesis of the corpus callosum (20%) and occipital encephalocele (10%); rarely associated with true hydrocephalus
(a)may be associated with cleft palate, ocular abnormalities (iris defects {coloboma}, microphthalmia), and cardiac structural defects
Box 12.6
Dandy-Walker malformations look similar to posterior fossa subarachnoid cysts, which do not communicate with the subarachnoid space as does the Dandy-Walker malformation.
A B
Figure 12–4 Chiari I malformation with syrinx (A), Chiari II malformation with mesencephalic malformation (“tectal beaking”), low-lying torcula (arrow), medullary/cervical spinal cord buckling, and descent of the
cerebellar vermis. (From Citow JS et al. Neuropathology and Neuroradiology: A Review. Thieme; Stuttgart, Germany: Georg Thieme; 2001:13, Fig. 11, Fig. 12. Reprinted by permission.)
Figure 12–5 Dandy-Walker malformation demonstrating a hypoplastic cerebellar vermis, laterally displaced cerebellar hemispheres, retrocerebellar cyst, and hydrocephalus. (From Mori K. Anomalies of the Central Nervous System. Thieme; Stuttgart, Germany: Georg Thieme; 1985:92, Fig. 6–12A. Reprinted by permission.)
Disorders of Embryogenesis
273
Developmental and Metabolic Diseases of the |
Nervous System |
12 |
|
274
ii.symptoms: mental retardation; ataxia; spasticity; seizures (15%)
iii.treatment: if symptomatic, shunt the posterior fossa fluid space; ventricular shunts in absence of posterior fossa shunt may cause upward herniation
5.Spinal malformations
a. spina bifida: a neurulation abnormality
i.subtypes: all are most common in lumbosacral region and may be associated with an overlying tuft of hair
(1)spina bifida occulta: incidental defect in the dorsal bony vertebra; occurs in 20% of the general population
(2)meningocele: a meningeal outpouching that does not contain neural tissues and is well-covered by skin; has minimal association with brain malformations
(a)treatment: early surgical closure
(3)myelomeningocele: failure of the posterior neuropore closure; meningeal outpouching includes neural tissues and is covered by minimal skin elements (Box 12.7)
(a)always associated with a type II Chiari malformation, and may have hydrocephalus from aqueduct stenosis (80%)
(b)treatment: early surgical closure, although this is unlikely to improve the patient’s quality of life
ii.diagnostic testing: prenatal diagnosis with elevated amniotic fluid-fetoprotein and ultrasound
iii.treatment: prenatal folate supplementation reduces the incidence of spina bifida
b.tethered spinal cord
i.pathophysiology: stretching of the spinal cord by an unusually strong filum terminalis or lipoma, causing ischemia; associated with spina bifida occulta in 90% of cases, and the tethered cord usually accounts for deterioration in such patients who have back pain (deterioration without back pain is usually due to syringomyelia)
(1)can be associated with chromosomal abnormalities (trisomy 13, 18; DiGeorge syndrome)
ii.symptoms: back pain; lower extremity weakness and sensory loss; lower extremity atrophy and/or foot deformities; bowel/bladder dysfunction; progressive kyphoscoliosis; delayed walking; constipation (most common presenting sign in children); imperforate anus and other anal anomalies
iii.treatment: surgical transection of the filum; removal of any lipoma
c.caudal regression syndrome/sacral agenesis
Box 12.7
Other Neurulation Abnormalities
Anencephaly—Failure of anterior neuropore closure; extends caudally as failure of spinal cord neurulation {rachischisis}
Encephalocele—Unilateral cortical protrusion through a midline skull defect (occipital frontal)
i.pathophysiology: degeneration of the coccyx and sacral vertebrae, and of the sacral spinal cord that causes atrophy of the lower extremities and malformations of the rectum and genitourinary system; caused by decreased expression of the sonic hedgehog protein at the caudal end of neural tube
(1)also associated with maternal diabetes mellitus
6.Vein of Galen malformations: Caused by direct arterial connections into the vein of Galen
a. subtypes
i.neonatal onset: the shunt involves a significant proportion of the cardiac output, therefore it presents as high-output congestive heart failure
ii.infantile onset: presents with hydrocephalus, likely due to obstruction of the aqueduct of Sylvius by the enlarged vein of Galen
Figure 12–6 Lisch nodules. (From McKhann GM et al. Q&A Color Review of Clinical Neurology and Neurosurgery. Stuttgart, Germany: Georg Thieme; 2003:93, Fig. 87a. Reprinted by permission.)

iii.childhood onset: presents as mental retardation, seizures, and/or focal neurological injury caused by arterial steal
iv.adult onset: presents as hemorrhagic infarction
b. treatment: endovascular occlusion of the arterial connections
II. Neurocutaneous Syndromes/Phakomatoses
A. Neurofibromatosis Type 1/von Recklinghausen’s Disease
1.Pathophysiology: caused by mutation of NF-1 gene on chromosome 17, which encodes neurofibromin protein that is likely a GTPase-activating protein; somatic mosaicism may cause localization of abnormalities to one part of the body
a.histology
i.subcutaneous neurofibromas develop along peripheral nerves; neurofibromas and/or schwannomas can develop on cranial nerves, but not as frequently as in neurofibromatosis type 2
ii.gliomas (usually juvenile pilocytic astrocytomas) develop on the optic nerve (25%) of in the brain parenchyma (5%)
iii.pigmented hamartomas or the iris {Lisch nodules (Fig. 12–6)} or retina
2.Symptoms: onset by 5 years of age (Box 12.8)
a.neurological symptoms
i.mental retardation (40%), seizures (10%)
ii.painful peripheral neuropathies or radiculopathies from subcutaneous neurofibromas
b.eye: abnormalities are asymptomatic
c.skin: café-au-lait spots (Fig. 12–7); axillary freckling
d.hypertension from pheochromocytoma and/or renal artery stenosis
3.Diagnostic testing
a.neuroimaging: MRI demonstrates areas of non-enhancing increased T2 and diffusion-weighted signal {unidentified bright objects (UBOs)} that are located commonly in the basal ganglia, brainstem, and cerebellum (Fig. 12–8)
i.UBOs likely represent areas of glial proliferation, and they are frequently sites for glioma development; 40% will spontaneous resolve, but persistence of UBOs into adulthood is associated with mental retardation
Figure 12–7 Café-au-lait spots. (From Panteliadis CP, Darras BT. Encyclopaedia of Paediatric Neurology: Theory and Practice, 2nd ed. Stuttgart, Germany: Georg Thieme; 1999:491, Fig. 25–1. Reprinted by permission.)
Box 12.8
Diagnostic Criteria for
Neurofibromatosis (NF) Type 1
Requires two or more of the following:
At least six café-au-lait spots that are5 mm before puberty or 15 mm after puberty
Multiple neurofibromas or one plexiform neurofibroma
Multiple Lisch nodules
Axillary freckling
Optic glioma
Bony lesions: thinning of long bones, pseudoarthrosis
A family history of NF type 1
b. genetic abnormalities of NF-1 gene are detected in only 70%
4.Treatment: None specific
B.Neurofibromatosis Type 2
1.Pathophysiology: mutation of NF-2 gene on chromosome 22, encodes merlin/ schwannomin protein, which acts as a tumor suppressor (Box 12.9)
a.histology: multiple schwannomas, classically bilateral on CN/VIII but also on peripheral nerves; frequently associated with ependymomas meningiomas and astrocytomas
2.Symptoms: typically develop around 20 years of age (Box 12.10)
a.neurological symptoms: vertigo, hearing loss, tinnitus from CN/VIII schwannomas (usually develop on the vestibular portion); schwannomas neurofibromas on peripheral nerves may cause painful peripheral neuropathies
i.may develop facial weakness from compression of CN/VII in the internal acoustic meatus
b.skin: as per neurofibromatosis type 1 but less prominent; café-au-lait spots and subcutaneous neurofibromas are rare
c.eye: rare Lisch nodule
Box 12.9
Other mutations of merlin/schwannomin produce sporadic low-grade meningiomas
Box 12.10
Diagnostic Criteria for Neurofibromatosis Type 2: Requires All Three of the Following
Bilateral CN VIII tumors
Family history of neurofibromatosis type 2
One family member with a CN VIII tumor or two other nervous system tumors (neurofibroma, schwannoma, meningioma, glioma)
Neurocutaneous Syndromes/Phakomatoses
275

Developmental and Metabolic Diseases of the |
Nervous System |
12 |
|
3.Diagnostic testing: neuroimaging with MRI; genetic testing
4.Treatment: none specific
C. Tuberous Sclerosis
1.Pathophysiology
a.caused by autosomal dominant mutations in the
i.TSC-1 gene (chromosome 9): encodes the hamartin protein, which functions in linking the cell membrane to the cytoskeleton
ii.TSC-2 gene (chromosome 16): encodes the tuberin protein, which is similar to GTPase-activating proteins and interacts with hamartin
b.TCS1 mutations may be more likely to cause familial disease and have a milder course, but otherwise there is no obvious phenotype difference between the two genetic subtypes
2.Histology
a.nervous system: cortical hamartomas {tubers}; focal cortical hypoplasia; subependymal heterotopias that are generally calcified; subependymal giant cell astrocytomas
b.eye: hamartomas, retinal depigmentation
c.cardiac, pulmonary, and renal hamartomas
d.vasculature: degeneration of the tunica media promotes aneurysm formation
3.Symptoms
a.neurological symptoms
i.mental retardation ( 50%)
ii.seizures (90%): may occur in the absence of mental retardation (Box 12.11)
iii.autism and behavioral disorders (psychosis, attention deficit– hyperactivity disorder)
b.skin
i.ash-leaf patches (Fig. 12–9): nonspecific for the phakomatoses
Figure 12–8 Unidentified bright objects (arrow) of neurofibromatosis type 1. (From Raininko R et al. Non-neoplastic brain abnormalities on MRI in children and adolescents with neurofibromatosis type 1. Neuropediatrics, 2001, 32: 226, Fig. 1b. Reprinted by permission.)
Box 12.11
Tuberous sclerosis is the most common cause of infantile spasms with a known etiology.
A
Figure 12–9 An ash-leaf patch (A). In a fair-skinned child (B), ash-leaf patches may be difficult to observe directly (left) and may require a Wood’s lamp (right). (Panel A, from Panteliadis CP, Darras BT. Encyclopaedia of Paediatric Neurology: Theory and Practice, 2nd ed. Stuttgart,
B
Germany: Georg Thieme; 1999:497, Fig. 25–4. Reprinted by permission. Panel B, from Kurlemann G, Schuierer G. Ash-leaf spots in tuberous sclerosis. New Eng J Med, 1998, 338:1887. Copyright 1998 Massachusetts Medical Society. All rights reserved.)
276
Figure 12–10 Ungual fibromas. (From McKhann GM |
Figure 12–11 Shagreen patch. (From Thappa DM et al. Giant sha- |
et al. Q&A Color Review of Clinical Neurology and Neu- |
|
rosurgery. Stuttgart, Germany: Georg Thieme; 2003: |
green patch associated with spina bifida occulta in tuberous sclerosis. |
177, Fig. 177a. Reprinted by permission.) |
Pediatric Dermatol 2003, 453–454. Reprinted by permission.) |
ii.ungual fibromas (Fig. 12–10): nodular fibromas that develop under the nails, particularly after trauma
iii.shagreen patch (Fig. 12–11): raised, leather-like skin usually on the back; usually develops in childhood, late in the disease
iv.adenoma sebaceum/facial angiofibromas (Fig. 12–12): specific for tuberous sclerosis
v.poliosis: white patches of hair on the forehead or eyebrows
c.eye: abnormalities are asymptomatic
d.cardiac: occasionally rhabdomyomas cause congestive heart failure, rarely arrhythmias
4.Diagnostic testing: neuroimaging
a.MRI demonstrates cortical tubers (in neonates, ↑ T1 and ↓ T2; in children, ↓ T1 and ↑ T2) and poorly defined underlying white matter abnormalities (Fig. 12–13)
Figure 12–12 Adenoma sebaceum. (From McKhann GM et al. Q&A Color Review of Clinical Neurology and Neurosurgery. Stuttgart, Germany: Georg Thieme; 2003:177, Fig. 177b. Reprinted by permission.)
b.computed tomography (CT) demonstrates intracranial calcifications, particularly of the subependymal nodules
c.angiography can demonstrate intracranial and peripheral aneurysms
5.Treatment: None specific
D. Sturge-Weber Syndrome
1.Pathophysiology: no known genetic cause; occurs sporadically
2.Histology: angiomas of the face and the underlying meninges and brain that cause atrophy and dystrophic calcification of the brain
a.intracranial angiomas drain into a dilated subependymal venous plexus; cortical veins are often thrombosed, leading to poor filling of the superior sagittal sinus
b.angioma distribution is variable, and isolated face or intracranial angiomas can occur
3.Symptoms
a.neurological symptoms
Figure 12–13 Subependymal nodules in tuberous sclerosis. (From McKhann GM et al. Q&A Color Review of Clinical Neurology and Neurosurgery. Stuttgart, Germany: Georg Thieme; 2003:75, Fig. 66b. Reprinted by permission.)
i.mental retardation (70%), particularly in patients with bilateral intracranial angiomas
Neurocutaneous Syndromes/Phakomatoses
277
Developmental and Metabolic Diseases of the |
Nervous System |
12 |
|
ii.seizures (90%), generally focal motor or generalized tonic-clonic; early onset of seizures increases the risk for mental retardation
iii.focal neurological deficits: typically develop acutely after the onset of seizures and suggest ischemic stroke
iv.megacephaly with hydrocephalus
b.skin: angioma {port-wine stain} typically is in the CN V1 distribution but it may extend downwards on neck, trunk, and upper extremity; may occur bilaterally (Fig. 12–14)
c.eye: glaucoma when the angioma involves the choroid
4.Diagnostic testing
a.contrast MRI and MR angiography (MRA) demonstrate intracranial angiomas; confirm with angiography
b.CT: demonstrates dystrophic calcification
c.skull plain films: classic “tram-track” calcifications develop over decades, and are rarely present at the time of symptomatic onset (Fig. 12–15)
5.Treatment: Complete seizure control with antiepileptic medications in 50%; may require resection of seizure foci or hemispherectomy
Figure 12–14 Port-wine stain. (From Panteliadis CP, Darras BT. Encyclopaedia of Paediatric Neurology: Theory and Practice, 2nd ed. Stuttgart, Germany: Georg Thieme; 1999: 501, Fig. 25–8. Reprinted by permission.)
E. von Hippel-Lindau Syndrome
1.Pathophysiology: autosomal dominant mutations in the VHL gene, the protein of which acts as tumor suppressor by inhibiting endothelial-derived growth factor
2.Histology: multiple hemangioblastomas of the eye and brain
a.in the brain, hemangioblastomas are found in the cerebellum spinal cord (cervical and conus regions) or medulla
i.supratentorial hemangioblastomas occur in 5%
ii.hemangioblastomas in the spinal cord can cause syrinx formation, and in the medulla they may cause syringobulbia
iii.hemangioblastomas are slow growing, so symptoms increase with age
b.renal and pancreatic cysts
c.tumors: renal carcinoma, pheochromocytoma (20%)
3.Symptoms
a.neurological symptoms: usually minimal unless a hemangioblastoma hemorrhages
b.eye: usually asymptomatic unless the hemangioblastoma is large and centrally located, or unless it hemorrhages
c.episodic hypertension from pheochromocytoma with associated flushing and diaphoresis; chronic hypertension from renal cysts
d.pancreatitis from bile duct obstruction due to multiple pancreatic cysts
4.Diagnostic testing: contrast MRI to identify hemangioblastomas only in patients who are symptomatic; confirm with angiography
Figure 12–15 Tram-track calcifications of advanced Sturge-Weber syndrome. (From Citow JS et al. Neuropathology and Neuroradiology: A Review. Thieme; Stuttgart, Germany: Georg Thieme; 2001:123, Fig. 180B, Fig. 12. Reprinted by permission.)
III. Aminoacidopathies (Box 12.12)
1.Maple syrup urine disease
a.pathophysiology: caused by autosomal recessive mutations of the branchedchain ketoacid dehydrogenase (BCKD) complex, which also decarboxylates branched chain amino acids (isoleucine, valine, leucine); BCKD requires thiamine cofactor, which accounts for a thiamine-responsive variant of the disease
i.histology: immature central nervous system myelination {dysmyelination}, heterotopias, diffuse spongy degeneration, and failure to develop all cortical layers
278 b. symptoms
Box 12.12
Diseases with Hyperammonemia
Aminoacidopathies—Glycine encephalopathy
Disorders of urea metabolism—Argininosuc- cinate lyase deficiency; argininosuccinate synthase deficiency; arginase deficiency
Disorders of organic acids—Isovaleric acidemia; propionic acidemia; methylmalonic acidemia
Toxicities—Valproate use (the most common cause)
i.neonatal onset (severe enzyme deficiency): normal at birth, but symptoms develop within days of starting a protein diet
(1)general symptoms: ketoacidosis and hypoglycemia
(2)neurological symptoms: hypotonia; lethargy; apnea, respiratory failure; seizures and coma, usually leading to death or mental retardation in the survivors
ii.infant onset (moderate enzyme deficiency): symptoms usually develop after a catabolic state (e.g., febrile illness)
(1)general symptoms: mild ketoacidosis
(2)neurological symptoms: ataxic gait; mental retardation
c.diagnostic testing
i.urine dinitrophenylhydrazine chromogenic test for amino acids
ii.elevated serum branched-chain amino acid levels
iii.neuroimaging: demonstrates areas of abnormal subcortical white matter signal at baseline, and cerebral edema that develops after a protein meal
d.treatment
i.thiamine
ii.low branched-chain amino acid diet; protein-free diet during acute exacerbations
iii.hemodialysis for severe ketoacidosis
2.Phenylketonuria
a.pathophysiology: autosomal recessive deficiency of phenylalanine hydroxylase prevents conversion of phenylalanine to tyrosine; high levels of phenylalanine are directly toxic to glia more so than neurons
i.phenylalanine hydroxylase requires tetrahydrobiopterin cofactor (Box 12.13)
ii.histology: as per maple syrup urine disease
b.symptoms: develop 2–3 months after birth
i.general symptoms: musty body odor; dry, scaling hypopigmented skin; small stature; patients are always blonde and blue-eyed
ii.neurological symptoms: microcephaly with failure to acquire developmental milestones and mental retardation; hyperactivity, irritability; tremor; spasticity; infantile spasms progressing to generalized tonic-clonic seizures
c.diagnostic testing: screening blood test for blood phenylalanine taken 24 hours after protein the first feeding; if positive, phenylketonuria is confirmed by demonstrating normal urine tyrosine and tetrahydrobiopterin levels
d.treatment: phenylalanine-restricted diet beginning by 3 weeks of age
i.diet restrictions must be carefully monitored by a nutritionist and should be continued at least through adolescence; it is unclear if a normal diet can ever be safely instituted
e.prognosis: dietary management limits mental retardation
3.Hartnup’s disease
a.pathophysiology: abnormal function of the neutral amino acid transport protein impairs intestinal absorption and renal reabsorption of neutral amino acids, particularly tryptophan
b.symptoms: develop in childhood, and are unrelated to diet
i.general symptoms: photosensitive dermatitis
ii.neurological symptoms: headache; depression, psychosis; gait ataxia
c.diagnostic testing: increased urine excretion of tryptophan and other neutral amino acids, which is constant and does not correlate with symptom severity
d.treatment: nicotinamide and tryptophan supplementation
Aminoacidopathies
Box 12.13
Tetrahydrobioprotein Methylenetetrahydrofolate (used in conversion of homocysteine to methionine)
279

Developmental and Metabolic Diseases of the |
Nervous System |
12 |
|
280
4.Glycine encephalopathy
a.pathophysiology: caused by an unknown defect in glycine metabolism that leads to glycine accumulation
b.symptoms: irritability, hiccupping, lethargy, and hypotonia developing within a few hours of birth; myoclonic seizures develop thereafter
i.no ketoacidosis, as in some disorders of organic acids (i.e., isovaleric acidemia, propionic acidemia)
c.diagnostic testing
i.EEG: shows a burst-suppression pattern progressing to hypsarrhythmia (see p. 92)
ii.increased serum and cerebrospinal fluid glycine level without hyperammonemia or organic acidemia (Box 12.14)
d.treatment
i.hemodialysis for severe encephalopathy
ii.diazepam benzoate for seizures; avoid valproate
e.prognosis: death within a few weeks of birth
5.Aromatic amino acid/DOPA decarboxylase deficiency
a.pathophysiology: mutations in aromatic amino acid/dihydroxyphenylalanine (DOPA) decarboxylase prevents L-DOPA conversion to dopamine (Box 12.15), and 5-hydroxytryptophan conversion to serotonin
b.symptoms: onset 2 years of age; symptoms become worse throughout the day and improve after sleep (Box 12.16)
i.axial hypotonia with limb hypertonia; dystonia; startle myoclonus
ii.autonomic dysfunction: diaphoresis, temperature instability, ptosis, miosis, episodic hypoglycemia
iii.mental retardation; failure to achieve motor milestones
iv.disturbed sleep–wake patterns
c.treatment: vitamin B6/pyridoxine (cofactor for the decarboxylase); monoamine oxidase (MAO) inhibitors; dopaminergic agonists (pergolide, bromocriptine); melatonin for disturbed sleep
6.Canavan’s disease/spongy degeneration of infancy: see p. 115
7.Homocysteinuria: see p. 72
IV. Disorders of Organic Acids
1.Isovaleric acidemia
a.pathophysiology: caused by autosomal recessive deficiency of isovalerylCoA dehydrogenase that leads to accumulation of isovaleric acid (a fatty acid); normally this enzyme is active in a multistep pathway converting leucine to acetoacetate
b.symptoms: lethargy and protracted vomiting developing within a few days of birth; exhibits ketoacidosis, unlike glycine encephalopathy
c.diagnostic testing: increased isovaleryl-lysine levels, which causes the urine to smell like sweaty feet and the patient to smell like stale sweat
d.treatment: dietary protein restriction, particularly leucine; L-carnitine and glycine supplementation
e.prognosis: 60% 3-week mortality
2.Propionic acidemia
a.pathophysiology: caused by autosomal recessive deficiency of propionylCoA carboxylase; enzymatic deficiency prevents the conversion of propionic acid to methylmalonyl-CoA (Fig. 12–16)
b.symptoms: lethargy, hypotonia, and dehydration developing within days to months of birth
c.diagnostic testing
Box 12.14
Increased glycine and ketoacidosis occurs in propionic academia and isovaleric acidemia.
Box 12.15
Dopamine is formed by tyrosine hydroxylase, which is mutated in Segawa’s syndrome.
Box 12.16
Worsening of symptoms during the day is similar to Segawa’s syndrome.
i.hyperammonemia; ketoacidosis (unlike glycine encephalopathy)
ii.increased serum glycine and propionate levels; increased urine glycine, methylcitrate, and -hydroxypropionate
iii.increased methylcitrate levels in the amniotic fluid for prenatal diagnosis
d.treatment
i.hemodialysis for severe encephalopathy
ii. dietary protein restriction
1.5 g/kg/d
iii.L-carnitine to reduce ketoge-
nesis |
Figure 12–16 Biosynthesis of methylmalonyl-CoA. |
iv.biotin supplementation (an en-
zymatic cofactor for propionyl-CoA carboxylase)
3.Methylmalonic acidemia
a.pathophysiology: caused by autosomal recessive deficiency of methylmalonylCoA mutase, which leads to accumulation of propionyl-CoA, propionic acid, and methylmalonic acid
b.symptoms: lethargy, vomiting, dehydration, and hypotonia developing after the initiation of protein feeding
c.diagnostic testing: hyperammonemia; increased serum glycine and methylmalonic acid; increased urine methylmalonic acid
d.treatment
i.dietary protein restriction 1.5 g/kg/d
ii.vitamin B12 supplementation (a cofactor for methylmalonyl-CoA mutase)
iii.L-carnitine to reduce ketogenesis
e.prognosis: majority die within 2 months; survivors have episodic metabolic acidosis and mental retardation
V. Disorders of Urea Metabolism
1.Pathophysiology: all exhibit autosomal recessive inheritance except for ornithine transcarbamylase deficiency, which is X-linked (Fig. 12–17)
2.General symptoms: vomiting, lethargy, and hypotonia beginning within 24 hours of birth, due to ammonia toxicity; coma develops if ammonia 300 g/dL, and seizures if 500 g/dL
3.Specific symptoms
a.carbamyl phosphate synthetase deficiency—abnormal eye movements
b.ornithine transcarbamylase deficiency—respiratory alkalosis
c.argininosuccinate synthase deficiency/citrullinemia—hepatomegaly; ataxia
d.arginase deficiency—spastic diplegia
e.argininosuccinate lyase deficiency—brittle hair associated with small nodules along the hair shaft {trichorrhexis nodosa}
4.Diagnostic testing
a.hyperammonemia without organic aciduria that is greatly increased after a protein meal
b.elevated orotic acid levels, except in carbamyl phosphate synthetase deficiency
c.elevated serum levels of amino acids and orotic acid, and urine amino acids
Disorders of Urea Metabolism
281
Developmental and Metabolic Diseases of the |
Nervous System |
12 |
|
Figure 12–17 The urea cycle.
5.Treatment
a.hemodialysis for severe hyperammonemia
b.dietary nitrogen intake 2 g/kg/d
c.benzoate, phenylacetate, and arginine supplementation, for both acute hyperammonemia and chronic treatment
VI. Lysosomal Storage Disorders
A. Disorders of Peroxisomes
1.Refsum’s disease
a.pathophysiology: caused by autosomal recessive deficiency of peroxisomal phytanoyl-CoA hydroxylase that prevents the degradation of phytanic acid
i.histology: diffuse lipid deposits in CNS neurons and glia, with areas of dysmyelination; peripheral nerve axons exhibit myelin onion bulbs, like the Charcot-Marie-Tooth neuropathies
b.symptoms: develop by 20 years of age; a slowly progressive course that is interrupted by frequent remissions and that can be exacerbated by fasting (which releases cellular stores of phytanic acid)
i.general symptoms: cataracts; cardiomyopathy, arrhythmia; dry, scaling skin {ichthyosis}
ii.neurological symptoms
(1)ataxia, nystagmus
(2)retinitis pigmentosa, often presenting as night blindness
(3)distal large-fiber sensorimotor polyneuropathy
(4)sensorineural hearing loss
c.diagnostic testing: increased blood phytanic acid levels, and reduced lowdensity lipoprotein (LDL) and high-density lipoprotein (HDL)
282

d.treatment: dietary restriction of phytanic acid; plasmapheresis (not hemodialysis) during acute exacerbations
2.Zellweger’s syndrome
a.pathophysiology: caused by autosomal recessive mutations in any one of 14 proteins (“peroxins”) that prevent transport of extracellular matrix proteins into peroxisomes for degradation; malfunctional peroxisomes are empty structures {peroxisome ghosts}, and are few in number
i.histology: pachygyria; polymicrogyria; heterotopias, particular in the cerebellum; malformations of the inferior olivary complex
b.symptoms
i.general symptoms: cirrhotic liver failure; facial malformations (micrognathia, low nasal bridge, shallow orbits, ear deformities); cataracts, glaucoma
ii.neurological symptoms
(1)hypotonia, hyporeflexia
(2)sensorineural hearing loss
(3)failure to achieve developmental milestones
(4)seizures
c.diagnostic testing
i.increased levels of pipecolic acid (a very-long chain fatty acid normally metabolized by peroxisomes) in blood and urine
ii.decreased levels of plasmalogens (which are phospholipids synthesized by peroxisomes) in erythrocytes or in cultured amniocytes
d.treatment: mild cases may benefit from docosahexaenoic acid supplementation
e.prognosis: death by 1 year of age in severe cases, or by 3–6 years of age in mild cases
3.Adrenoleukodystrophy: see p. 116
B.Disorders of Mucopolysaccharide Metabolism (Mucopolysaccharidoses)
1.Symptoms: Course facial features; sensorineural hearing loss; behavioral regression beginning after 1 years of age, hepatosplenomegaly; dwarfism
2.Subtypes (Table 12–2)
Table 12–2 Mucopolysaccharidoses
Box 12.17
-galactosidase is also mutated in GM1 gangliosidosis
Disorder |
Enzyme |
Onset |
Accumulates |
Brain involvement |
I-H/Hurler’s syndrome |
Iduronidase (AR) |
I-S/Scheie’s syndrome |
Iduronidase (AR) |
II/Hunter’s syndrome |
Iduronate-2-sulfatase (XR) |
III/Sanfilippo’s syndrome |
(All AR) Heparan sulfatase |
Type A |
Acetylglucosaminidase |
Type B |
Acetyl-CoA |
Type C |
Glucosaminide |
IV/Morquio’s syndrome |
|
Type A |
Acetylgalactosamine |
|
Sulfatase (AR) |
Type B |
-galactosidase (AR) |
|
(Box 12.17) |
1 year of age 2–5 years of age 1–3 years of age 2–4 years of age
1–3 years of age
heparan, dermatan |
|
heparan, dermatan |
|
Heparan, dermatan |
|
Heparan |
|
Keratan
Abbreviations: AR, autosomal-recessive; XR, x-linked recessive.
Lysosomal Storage Disorders
283
Developmental and Metabolic Diseases of the |
Nervous System |
12 |
|
Figure 12–18 The sphingolipid metabolic pathway.
C. Disorders of Sphingolipid Metabolism (Sphingolipidoses)
1.Pathophysiology: caused by mutations of enzymes in the sphingolipid metabolic pathway (Fig. 12–18)
2.General symptoms: onset before 2 years of age; developmental delays and/or mental retardation (Box 12.18)
3.Subtypes (Table 12–3)
4.Treatment: Bone marrow transplantation for metachromatic and globoid leukodystrophies with mild neurological involvement
Box 12.18
Diseases with Cherry-Red Spots—GM-1 & GM-2 gangliosidosis; metachromatic leukodystrophy; Niemann-Pick disease
Table 12–3 Sphingolipidoses
Disorder |
Mutation/accumulates |
Distinguishing features |
|
|
|
Fabry’s disease |
-Galactosidase (XR)/trihexosylceramide |
Cutaneous angiomas, cataracts, renal failure, painful |
|
|
neuropathy, early-onset atherosclerosis |
Farber’s lipogranulomatosis |
Ceramidase (AR)/ceramide |
Multiple subcutaneous nodules, interstitial lung disease, |
|
|
macroglossia |
Gaucher’s disease
GM1 gangliosidosis
GM2 gangliosidosis/ Tay-Sach’s disease
Glucocerebrosidase
(AR); rarely saposin C cofactor/ glucocerebrosides
-Galactosidase (AR); rarely saposin B/GM1 ganglioside
Hexosaminidase A or B (AR); rarely glycoprotein activator/GM2 gangliosides
Type I: visceral; IV enzyme responsive
Type II (infant): neurovisceral; therapy resistant
Type III (child): neurovisceral; IV enzyme responsive
Cherry-red macula spot similar to Hurler’s syndrome, a mucopolysaccharidosis
Tay-Sachs is infantile form
CNS: increased startle reflex, MR, motor regression; cherry-red spot on macula
Niemann-Pick disease type |
Sphingomyelinase (AR)/sphingomyelin |
MR, cachexia, cherry-red spot (50%), opisthotonos, |
A & B |
|
blindness, seizures; liver failure |
(Type C is a dyslipidemia, |
|
Type A: onset 6 months of age |
see p. 297) |
|
Type B: like type A, but no neurological symptoms |
|
|
284 Abbreviations: AR, autosomal recessive; CNS, central nervous system; IV, intravenous; MR, mental retardation; XR, x-linked recessive.

D. Neuronal Ceroid Lipofuscinosis (NCL)
1.General histology: accumulation of autofluorescent lipid pigments (ceroid, lipofuscin) in brain, retina, and gastrointestinal tract that form inclusion bodies; degeneration of retinal photoreceptor and loss of specific layers of cortical neurons, depending upon subtype
a.epidemiology: most common in Scandinavians
2.General symptoms: progressive dementia; progressive vision loss from retinal and optic nerve degeneration; ataxia; myoclonus
3.Subtypes
a.early-infantile NCL: onset 1–2 years of age
i.pathophysiology: caused by mutation in the palmitoyl protein thioesterase (CLN1) gene, the protein of which removes fatty acids from lipoproteins
ii.specific symptoms: hyperactivity; hypotonia
b.late-infantile NCL: onset 2–3 years of age
i.pathophysiology: caused by mutations in the CLN2 gene, which encodes tripeptidyl peptidase that removes N-terminal tripeptides from polypeptides
ii.specific symptoms: seizures; dementia progressing to a vegetative state
c.juvenile NCL: onset 4–9 years of age
i.pathophysiology: caused by mutations in CLN3 gene, which encodes a lysosomal membrane protein of unknown function
ii.specific symptoms: Parkinsonism; tonic-clonic seizures
d.adult NCL: onset 40 years of age
i.pathophysiology: unknown
ii.specific symptoms: seizures; peripheral neuropathy
4.Diagnostic testing
a.reduced palmitoyl protein thioesterase and tripeptidyl peptidase levels (infantile NCLs)
b.neuroimaging: diffuse atrophy, usually cerebral cerebellar; increased T2 signal in the thalami and deep white matter
c.EEG, visual evoked potentials, and electroretinograms exhibit increased responsiveness to photostimulation
d.tissue biopsy of muscle, skin, conjunctiva, or rectum for inclusion bodies
5.Treatment: None specific
Disorders of DNA Repair and Nucleoside Metabolism
VII. Disorders of DNA Repair and Nucleoside Metabolism
1.Xeroderma pigmentosa
a.pathophysiology: caused by abnormalities of DNA repair throughout the genome (i.e., in actively transcribed and inactive regions); the majority of cases with neurological disease have autosomal recessive mutations in DNA helicase (Box 12.19)
i.histology: pronounced neuron loss in the cortex, basal ganglia, and cerebellum causes generalized atrophy; also exhibits anterior horn and dorsal root ganglia neuron loss, and cochlear hair cell atrophy
b.symptoms
i.general symptoms: sun sensitivity causing premature skin aging and an increased incidence of skin cancers
ii.neurological symptoms: microcephaly with progressive dementia; sensorineural hearing loss; spastic weakness that ultimately become areflexic due to a sensorimotor neuropathy; ataxia, dysarthria
c.treatment: avoid sun exposure
2.Cockayne’s syndrome
Box 12.19
Certain mutations in DNA helicase may cause a Cockayne-like syndrome.
285

Developmental and Metabolic Diseases of the |
Nervous System |
12 |
|
a.pathophysiology: caused by abnormalities of the DNA excisional repair system in actively transcribed genes
i.subtypes: both exhibit autosomal recessive inheritance
(1)type I/juvenile-onset (rare): mutation of the CSA gene on chromosome 5
(2)type II/infantile-onset (common): mutation of the CSB gene on chromosome 10
ii.histology: death of oligodendrocytes and Schwann cells; gliosis involves pleomorphic astrocytes that are preneoplastic in appearance
(1)atrophy with calcifications are pronounced in cerebellum cortex or basal ganglia
(2)neurofibrillary tangles and Hirano bodies are present in neurons
(3)premature atherosclerosis with calcification, particularly in the basal ganglia
b.symptoms
i.general symptoms: dwarfism, often with skeletal abnormalities; cachexia; minor skin pigmentation, premature skin aging, and increased cancer risk
ii.neurological symptoms
(1)acquired microcephaly: head circumference is normal at birth but fails to increase; occurs despite normal pressure hydrocephalus
(2)progressive dementia that is not as severe as the microcephaly would suggest
(3)vision loss from cataracts and pigmented retinopathy
c.diagnostic testing
i.neuroimaging: basal ganglia calcifications on CT; meningeal thickening and tigroid leukodystrophy on MRI (Fig. 12–19)
ii.nerve conduction study: demyelinating neuropathy, which is rarely symptomatic (unlike xeroderma pigmentosa)
d.treatment: avoid sun exposure
3.Lesch-Nyhan disease
a.pathophysiology: caused by X-linked mutations in the hypoxan- thine-guanine phosphoribosyl transferase (HGPRT) enzyme, which normally converts hypoxanthine and xanthine into nucleic acids; HGPRT deficiency allows for accumulation of hypoxanthine and xanthine that become converted to uric acid by xanthine oxidase
i.hypoxanthine, xanthine, and uric acid levels are related to nephropathy but not to central nervous system dysfunction
ii.partial HGPRT deficiency may cause only mild mental retardation, gout, and asymptomatic hyperuricemia, as in the female carrier state
b.symptoms: develops by 2–3 years of age
i.general symptoms: gout; renal stones
ii.neurological symptoms
(1)neonatal hypotonia developing into spasticity and opisthotonos
(2)mental retardation with aggressive and self-mutilation behaviors despite sensitivity to pain (e.g., chewing off finger tips)
(3)choreoathetosis, dystonia
c.diagnostic testing: elevated blood uric acid levels
d.treatment: no effective treatment for neurological symptoms
Figure 12–19 Tigroid leukodystrophy of Cockayne’s syndrome. (From Menges-Wenzel E et al. Cockaynesyndrom mit betonter zerebraler symptomatik verlaufsbeobachtung bei zwei schwestern. Klinische Padiatrie 2001, 213:136, Fig. 3a. Reprinted by permission.)
286

VIII. Disorders of Oxidative Phosphorylation
Causing Lactic Acidosis
1.General pathophysiology: caused by enzymatic deficiencies in the Kreb’s cycle or mitochondrial electron transport chain that allow the accumulation of lactate, which is neurotoxic; failure of oxidative phosphorylation may also directly cause cellular death
a.neurons in the basal ganglia (particularly the putamen) are susceptible to lactate
b.histological changes are similar to thiamine deficiency (i.e., Wernicke’s encephalopathy), although the mamillary bodies are spared
2.Major subtypes
a.pyruvate dehydrogenase deficiency—the clinical course is variable and proportionate to the degree of enzyme deficiency
i.specific symptoms: often the lactic acidosis is fatal in the neonatal period; survivors exhibit mental retardation, hypotonia, choreoathetoid cerebral palsy, and a demyelinating polyneuropathy
b.Leigh’s disease (see p. 48), a subset of which is caused by pyruvate dehydrogenase deficiency
3.Diagnostic testing
a.elevated fasting lactate and reduced pyruvate in blood, which may require preceding ingestion of carbohydrates to detect
b.skin and muscle biopsy for identification of enlarged mitochondria
4.Treatment: Multivitamin, L-carnitine, and coenzyme Q10 supplementation; bicarbonate; dietary carbohydrate restriction; dichloroacetic acid for pyruvate dehydrogenase deficiency
IX. Developmental Delays and Regression
A.Normal Developmental Milestones (Table 12–4)
B.Abnormal Development
1.Language delay: causes include
a.hearing impairment: the most common cause of language delay; commonly occurs after intrauterine cytomegalovirus infection, neonatal meningitis, or kernicterus
i.even mild hearing loss can cause language delay if the loss is concentrated in the high-frequency tones
b.bilateral hippocampal sclerosis: usually presents with seizures; one normally functioning medial hippocampal gyrus is necessary for language development
Table 12–4 Normal Developmental Milestones
Skills |
3 months |
6 months |
9 months |
12 months |
15 months |
18 months |
24 months |
|
|
|
|
|
|
|
|
Gross motor |
Sits with |
Sits unsupported |
Stands unaided |
Walks |
Runs |
Climbs steps |
Throws |
|
support |
|
|
|
|
|
|
Fine motor |
Puts hands |
Reaches |
Pincer grasp |
Scribbles |
Stacks two |
Stacks six cubes |
Copies straight |
|
together |
|
|
|
cubes |
|
line |
Social |
Social smile, |
Stranger anxiety |
Indicates desires |
Imitates |
Removes |
Helps clean self |
Puts on clothes |
|
recognizes |
begins |
|
others |
clothes |
|
|
|
parents |
|
|
|
|
|
|
Language |
Laughs |
Monosyllabic |
“Dada” & |
Two words |
Six words |
Combines words |
Picture naming |
|
|
utterances |
“mama” |
|
|
|
|
|
|
|
|
|
|
|
|
Developmental Delays and Regression
287

Developmental and Metabolic Diseases of the |
Nervous System |
12 |
|
288
c.focal lesions in cortical language areas: produce language delay only in children 5 years of age; lesions in children 5 years of age displace language functions to the contralateral hemisphere
d.autism: see p. 290
2.Motor delay
a.cerebral palsy
i.pathophysiology: likely causes are in utero/pre-labor injuries (e.g., twining, in utero stroke, congenital malformations); not related to birth injury or obstetrical procedures
(1)the incidence of cerebral palsy has not changed except for the choreoathetotic subtype, which has been decreased by treatment of hyperbilirubinemia that prevents development of kernicterus
ii.general symptoms: usually identifiable only after 6 months of age; choreoathetotic subtype can be identified only by 12–18 months of age; 20% have coincidental mental retardation
iii.subtypes
(1)paraplegic cerebral palsy—associated with white matter damage high in centrum semiovale (i.e., a periventricular leukomalacia), specifically in the area above and lateral to the caudate nucleus; lesions damage the corticospinal tract fibers issuing from the lower extremity region of the motor cortex more so than upperextremity region
(2)hemiplegic cerebral palsy: likely caused by embolisms from the placenta; 70% involve the left hemisphere because the majority of flow of the fetal circulation through the patent ductus arteriosus goes straight into the left carotid artery
(a)may give the appearance of handedness in neonates (a pathological observation)
(3)quadriplegic cerebral palsy: usually caused by hypoxic-ischemic encephalopathy
(4)choreoathetotic cerebral palsy: caused by injuries to the basal ganglia; historically was due to erythroblastosis fetalis/Rh incompatibility causing kernicterus, but now is usually an idiopathic condition
(a)status marmoratus: a marble-like histological appearance of basal ganglia, due to oligodendrocytes that myelinate astrocytes in the absence of neurons
iv.diagnostic testing
(1)neuroimaging to assess for brain malformations or injury; metabolic and genetic testing should be considered if neuroimaging does not define a structural abnormality
(2)evaluation for a hypercoagulable state
b.hypotonic motor delays
i.general symptoms: all causes involve a loss of postural tone
(1)reduced spontaneous movements
(2)absence of neck and limb flexion when pulling the infant upright {traction response} (Box 12.20)
(3)hip dislocation and pectus excavatum due to regional muscle weakness
(4)joint contractures causing congenital limb deformities {arthrogryposis}
ii.upper motoneuron hypotonic disorders
(1)specific symptoms
(a)seizures
(b)decorticate and/or decerebrate posturing, often induced by passive head movements
Box 12.20
The Traction Response
Not present in premature infants; better than horizontal suspension for hypotonia
(c)persistence of fisting of the hand that encloses the thumb {cortical hand} (which is a normal finding only at 3 months of age); crossing of the thighs {scissoring}; hyperreflexia despite hypotonia
(2)subtypes: from cerebral palsy, leukoencephalopathies, or chromosomal disorders (e.g., Down’s syndrome/trisomy 21); also
(a)Prader-Willi syndrome
(i)pathophysiology: partial deletion of chromosome 15 (70%) or else disomy of the maternal chromosome 15; may involve the necdin gene, which encodes a growth suppressor protein that interacts with p53 cell cycle protein (Box 12.21)
(ii)symptoms: hypotonia, obesity and hyperphagia, mental retardation, hypogonadism and cryptorchidism
iii.lower motoneuron hypotonic disorders: perinatal asphyxia or trauma after breech presentation, or inherited (e.g., the spinal muscular atrophies)
iv.peripheral nerve hypotonic disorders: Charcot-Marie-Tooth neuropathies; Guillain-Barre syndrome; leukodystrophies
v.neuromuscular junction hypotonic disorders: botulism, myasthenia gravis
vi.myopathies and muscular dystrophies
c.ataxic motor delays: spinocerebellar degeneration, Friedreich’s ataxia, ataxia telangiectasia, abetalipoproteinemia, Refsum’s disease, aminoacidopathies
3.Global developmental delay
a.static encephalopathy: caused by
i.cerebral malformations from intrauterine infections, alcohol, lead, drug abuse
ii.genetic abnormalities
(1)chromosomal abnormalities: the most common cause of mental retardation
(a)risk factors: affected family members; parental consanguinity; organomegaly
(b)symptoms suggestive of chromosomal abnormalities
(i)head and neck abnormalities: microphthalmia, slanted eyes, small mandible, small/low-set ears, webbed neck
(ii)limb abnormalities: low-set thumb, polydactyly, radial hypoplasia, rocker-bottom feet
(iii)genitourinary abnormalities: ambiguous genitalia, polycystic kidney
(c)subtypes
(i)Down’s syndrome/trisomy 21—hypotonia, typical facies, Brushfield spots (Box 12.22), flat nape of neck
(ii)trisomy 18: pointed ears, micrognathia, occipital protuberance, narrow pelvis, rocker-bottom feet
(iii)5p monosomy: “cri du chat,” moonlike facies, hypertelorism, microcephaly
(2)focal genetic abnormalities: fragile X syndrome
(a)pathophysiology: caused by a trinucleotide repeat in the FMR1 gene, which encodes a protein that likely functions in translational regulation; exhibits X-linked inheritance, but 80% of males and 30% of females are affected
(b)specific symptoms: autistic-like behavior, long faces, enlarged ears, macroorchidism
iii.intrauterine infections: previously referred to as the “TORCH” infections, now considered the “STaRCH” infections
Box 12.21
Paternal chromosome disomy 15 produces Angelman’s syndrome, which has mental retardation, microcephaly, and myoclonus.
Box 12.22
Brushfield spots
Small white spots around the edges of the iris, caused by stromal hyperplasia centered in areas of hypoplasia
Developmental Delays and Regression
289

Developmental and Metabolic Diseases of the |
Nervous System |
12 |
|
290
(1)syphilis (see p. 254)
(a)symptoms: sensorineural hearing loss, interstitial keratitis, peg-shaped upper incisors {Hutchinson’s triad}
(b)high coincidence with HIV infection
(2)toxoplasmosis (see p. 264)
(3)rubella: symptoms include . . .
(a)growth retardation, cataracts, congenital heart malformations
(b)chorioretinitis, hypotonia, seizures, somnolence from increased intracranial pressure, and sensorineural hearing loss
(4)cytomegalovirus/cytomegalic inclusion disease (see p. 257)
(5)HIV: neonatal infection can also occur via infected breast milk
(a)symptoms: onset between 2 months to 5 years of age
(i)encephalitis leading to failure to thrive with microcephaly
(ii)transverse myelitis
(iii)opportunistic infections, particularly with bacteria
(b)diagnostic testing: neonates of HIV-positive mothers will express HIV antibodies by passive transfer, therefore diagnose with HIV polymerase chain reaction (PCR) or culture
iv.subacute sclerosing panencephalitis (see p. 259)
v.other white matter diseases: adrenoleukodystrophy, Alexander’s disease
vi.other gray matter diseases: neuronal ceroid lipofuscinosis; xeroderma pigmentosum; mitochondrial disorders; spinomuscular atrophies
vii.other disorders: lead poisoning; hypothyroidism
b.autism (Box 12.23)
i.pathophysiology: possibly a first trimester disease process based on the high coincidence of minor physical malformations; not associated with childhood vaccinations
(1)anatomical abnormalities are rare, but can include
(a)ventriculomegaly
(b)decreased cerebellar Purkinje cell number; hypoplasia of cerebellar vermis is not a consistent finding
(c)decreased number of neurons and reduced dendritic arborizations in the subcortical limbic system (amygdala, hippocampus, septum) and the anterior cingulated cortex
(d)polymicrogyria
(e)increased volume of temporal, parietal, and occipital lobes
(2)immunologic abnormalities include a high coincidence of inflammatory bowel disease that suggests immunologic reaction to dietary factors; also associated with HLA-DRB1 allele
(3)genetic factors: multifactorial inheritance; monozygotic dizygotic concordance
(4)psychodynamic abnormalities: parents of autistic children exhibit poor language abilities, decreased social interaction, and a tendency toward routine, but this likely represents a mild, inherited autistic phenotype
ii.epidemiology: 3:1 male preference
iii.symptoms: generally begin 3 years of age; symptoms are more severe in females
(1)diagnostic symptoms
(a)abnormal social interactions: involves at least two of the following
(i)failure to use eye contact, facial expression, body posture, or gestures
Box 12.23
Diseases with autistic features: phenylketonuria; intrauterine infections (rubella, cytomegalovirus (CMV); neurofibromatosis, tuberous sclerosis; Rett’s syndrome; fragile X syndrome
(ii)failure to develop relationships with peers
(iii)abnormal social or emotional responses to other peoples’ emotions
(iv)lack of expression of personal interests
(b)abnormal communication skills: involves at least one of the following
(i)delayed or deficient speech without compensatory gesturing
(ii)failure to initiate or sustain conversation
(iii)stereotyped or repetitive use of words or phrases
(iv)lack of imitative play
(c)stereotyped or repetitive behaviors: involves at least one of the following
(i)preoccupation with specific, nonfunctional interests
(ii)preoccupation with nonfunctional elements of normal objects
(iii)compulsive performance of inappropriate routines
(iv)stereotyped or repetitive motor movements
(2)other symptoms (Box 12.24)
(a)mental retardation (70%)
(b)seizures (30%)
(c)echolalia, dysarthria
(d)decreased pain sensitivity
(e)bulbar affect; hyperactivity, aggressiveness, self-injury
(f)precocious abilities/splinter functions of the idiot savant (rare): photographic memory, mathematical ability, musical skill, early reading ability (may or may not have comprehension)
(g)GI symptoms: nausea, diarrhea, constipation
iv.treatment
(1)language development programs; encourage social behavior
(2)antipsychotics (haloperidol, risperidone) for hyperactivity, repetitive and self-injury behaviors, and irritability
(3)SSRIs for compulsive and repetitive behaviors
v.prognosis
(1)some symptomatic improvement occurs over time, particularly repetitive behaviors; rarely high-functioning autistic children outgrow the diagnosis
(2)majority of autistic patients will require an assisted-living environment
c.Rett’s syndrome
i.pathophysiology: almost all cases are associated with mutation of the MECP2 gene, which encodes a methylated DNA-binding protein that acts as a transcriptional repressor (Box 12.25)
(1)exhibits X-linked recessive inheritance, therefore the disease is most commonly observed in homozygous girls, although it can occur in heterozygous boys
(a)asymptomatic heterozygous females exhibit mosaicism caused by random X chromosome inactivation
(2)histology: reduced dendritic arborizations in the cortex and hippocampus
ii.symptoms: grossly normal development until 6 months of age, although subtle abnormalities of movement and posture are observable and fine motor skills fail to develop
Box 12.24
Asperger’s syndrome
Pathophysiology—Unknown, but occurs frequently in families with autism
Symptoms—As for autism, except that language ability is normal and there is no mental retardation or seizures; children with Asperger’s syndrome exert more effort in making social contacts than those with autism; language development may be delayed, but will ultimately obtain normal levels; thinking often appears illogical
Treatment—As per autism
Box 12.25
Other diseases associated with MECP2 mutations: autism; childhood-onset schizophrenia; an Angelman’s-like syndrome (but not real Angelman’s syndrome)
Developmental Delays and Regression
291
Developmental and Metabolic Diseases of the |
Nervous System |
12 |
|
(1)diagnostic symptoms
(a)motor regression with loss of purposeful hand movements followed by development of repetitive and stereotypic movements
(b)failure to develop further language abilities
(c)gait apraxia, ultimately developing spasticity and muscle wasting
(d)loss of social interaction
(e)failure of normal head growth leading to microcephaly
(2)other symptoms
(a)seizures
(b)irregular breathing pattern when awake, often involving breath holding and apnea; breathing normalizes during sleep
iii.treatment: none specific
iv.prognosis: stabilization of the disease by 20 years of age, with decreasing seizure frequency and improved social and motor function
(1)1% death rate per year, typically from malnourishment or sudden cardiac death
(2)progressive muscle wasting will require use of a wheelchair
292