10 Diseases of the Muscles
10
Diseases of the Muscles
Note: Significant diseases are indicated in bold and syndromes in italics.
I. Muscle Physiology
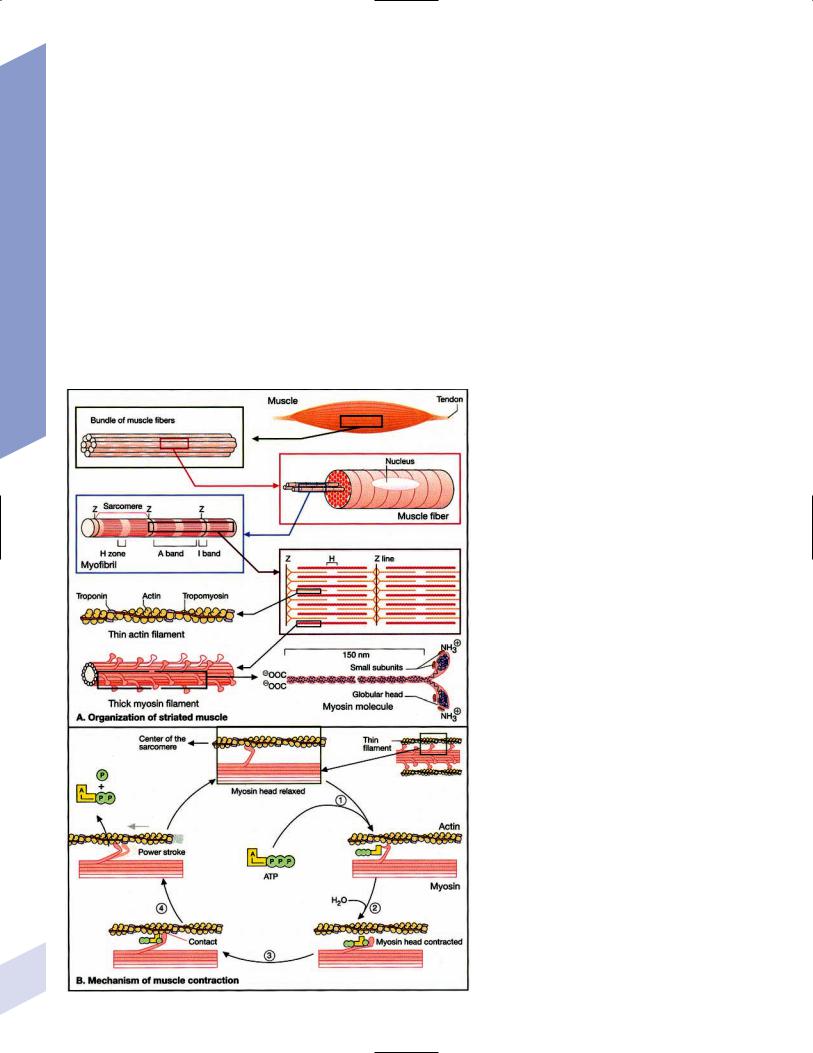
1.Muscle contraction (Fig. 10–1) and neuromuscular transmission (Fig. 10–2)
2.Muscle fibers: distinguished biochemical by the expression of different isozymes of myosin
Figure 10–1 Muscle contraction. (From Koolman J, Rohm 230 KH. Color Atlas of Biochemistry. Stuttgart, Germany:
Georg Thieme; 1996:307. Reprinted by permission.)

Muscle Physiology
Figure 10–2 Neuromuscular transmission. SR smooth endoplasmic reticulum. (From Koolman J, Rohm KH. Color Atlas of Biochemistry. Stuttgart, Germany: Georg Thieme; 1996:309. Reprinted by permission.)
a.slow twitch/type I muscle fibers: characterized by a small diameter, a high myoglobin content, and numerous mitochondria; relies on aerobic metabolism, which is better for tonic contraction
b.fast twitch/type II muscle fibers: characterized by a large diameter, a low myoglobin content, and few mitochondria; relies on anaerobic metabolism, which is better for phasic contraction (Fig. 10–3)
c.super-fast muscle fibers: found in extraocular muscles
d.tonic muscle fibers: found in extraocular muscles, tensor tympani
3.The motor unit: composed of the motoneuron and the muscle fibers it innervates; the muscle fibers for a particular motoneuron are always of the same type, but are not necessarily located near to each other in the muscle
a.motoneurons innervate a greater number of muscle fibers in larger muscles; conversely, motoneurons for the small muscles of face and hands may innervate less than 10 muscle fibers, allowing for fine motor control
Figure 10–3 Normal gastrocnemius muscle. ATPase stain demonstrates fast-twitch (f) and slow-twitch (s) muscle fibers. (From Beltri K et al. Contribution of the distal nerve sheath to nerve and muscle preservation following denervation and sensory protection. J Recon-
struct Microsurg 2005, 21:67, Fig. 8e. Reprinted 231 by permission.)
10 Diseases of the Muscles
b.types of motor units listed in order of recruitment
i.fatigue-resistant slow-twitch motor units: respond to low levels of activation of the motoneuron pool
ii.fatigue-resistant fast-twitch motor units: respond to intermediate levels of activation of the motoneuron pool; produce only half the contraction force of fatigue-resistant slow-twitch motor units
iii.fatigable fast-twitch motor units: respond to the highest levels of activation of the motoneuron pool; generates the largest force during twitch or tetanic contraction
4.Recruitment principle: the force of a muscle contraction is produced by activating an increasingly larger number of motor units in a fixed order beginning with the weakest and progressing to the strongest; this simplifies the modulation of force, because only the magnitude of the response has to be determined, not the identity of the responding motor units
a.the recruitment principle also keeps fatigable fast-twitch motor units quiescent until they are absolutely needed, which is metabolically advantageous
b.the order of activation can be reversed under certain physiological conditions, for example, activation of cutaneous pain receptors that inhibit fatigueresistant slow-twitch motor units while promoting the fast-twitch motor units, thereby allowing rapid force modulation
II. Diagnostic Testing
1.EMG: normal motor unit potentials (MUPs) (Fig. 10–4) a. pathological MUPs
i.myopathic changes on EMG
(1)MUPs tend to exhibit a decreased, not increased, amplitude after myopathic changes, because the muscle fibers are atrophying
(2)contractions of a diseased muscle results in an increased number of MUPs that are firing at normal or slower-than-normal rates
ii.neuropathic changes on EMG: involve a lesser degree of denervation and reinnervation than do myopathic changes
(1)denervation: acutely denervated muscle fibers are electrically
inactive unless they are irritated (e.g., at the time of needle insertion), until the development of fibrillation potentials 3 weeks after injury
(a)fibrillation potentials: spontaneous, regular potentials of chronically denervated muscle fibers
(i)the most accurate indicator of axonal loss in a neuropathic disorder, but fibrillation potentials last only 2 years after denervation before they spontaneously resolve
(ii)presence of fibrillation potentials in the proximal musculature (e.g., paraspinal muscles) indicates a motor root injury (versus peripheral nerve disease), but it is not always present
(2)reinnervation: can be accomplished by regeneration of an injured axon or by collateral sprouting of nearby intact motoneuron terminals
(a)MUPs from collateral sprouts are different from those of normally innervated motor units in that they
(i)have an increased duration
(ii)have an increased amplitude
(iii)have a polyphasic waveform
232
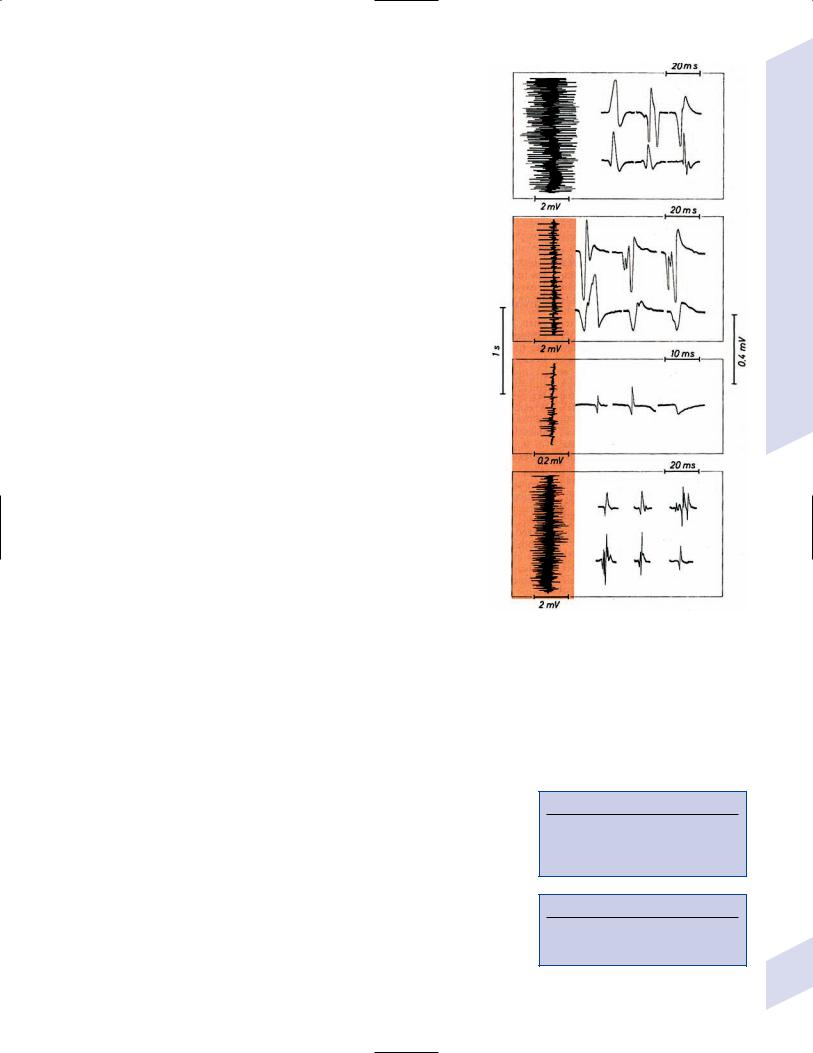
(iv)are of reduced number and are firing at faster than normal rates ( 15 Hz) during muscle contraction (i.e., abnormal recruitment pattern)
iii.abnormalities of insertional activity and irritability on EMG (Fig. 10–4)
(1)reduced insertional activity and irritability are caused by replacement of the muscle by other soft tissues or by physiological contracture (e.g., as in McArdle’s dis-
ease, phosphofructokinase deficiency, paramyotonia |
A |
congenita, or during episodes of the periodic paralysis |
|
disorders) |
|
(2)increased insertional activity and irritability exists in several forms including
(a) |
fibrillation potentials: bior triphasic waveforms |
|
|
|
with an initial negative potential originating |
|
|
|
in the muscle; occurs at 1–30 Hz at a regular |
|
|
|
rhythm |
|
B |
|
(i) caused by lower motoneuron disease, muscu- |
||
|
|
||
|
lar dystrophies, and inflammatory myopathies |
|
|
|
(ii) insertional sharp waves: essentially a fibrilla- |
|
|
|
tion potential caused by the initial irritation |
|
|
|
of a muscle fiber during insertion of the EMG |
|
|
|
needle |
|
|
(b) |
fasciculation potentials: less than 6 Hz poten- |
C |
|
|
tials with an irregular rhythm and variably |
||
|
|
||
|
shaped waveforms caused by pathological |
|
|
|
processes that irritate the lower motoneuron |
|
|
|
anywhere along its course (even within the spinal |
|
|
|
cord, as with amyotrophic lateral |
sclerosis) |
|
|
(Box 10.1) |
|
|
(c) |
myokymic discharges: short bursts of sharp |
|
|
|
waves occurring at regular intervals and that pro- |
|
|
|
duce a rhythmic, undulating muscle contraction; |
|
|
|
occurs at 5–150 Hz |
|
D |
(i)can be caused by most diseases of the muscle Figure 10–4 Normal (A) and abnormal EMG activity. Rein-
(d) myotonic discharges: sustained runs of positiveor |
nervation (B) demonstrates broad, high-amplitude wave- |
|
forms. Complete denervation (C) demonstrates fibrillation |
||
negative-sharp waves (depending upon the site of |
||
potentials (left and middle potentials) and positive sharp |
||
electrode placement) with fluctuant amplitudes, |
waves (right potential). Myopathy (D) exhibits low-amplitude, |
|
producing a tonic muscle contraction; occurs at |
polyphasic waves that are often split. (From Mumenthaler |
|
50–100 Hz (Box 10.2) |
M, Neurology. 3rd ed. Stuttgart, Germany: Georg Thieme; |
|
1990:389, Fig. 10. 2a–d. Reprinted by permission.) |
||
|
(i)neuromyotonic discharges: similar to myotonic discharges, except they occur at 150–300 Hz,
are continuous even during sleep, and are induced by physical stimulation; caused by diseases of the nerve that involve aberrant activity of the most distal portion of the nerve branches (e.g., Isaac’s disease)
(e)complex repetitive discharges: a coordinated burst involving several MUPs induced by a “pacemaker” muscle fiber that drives nearby muscle fibers, producing a stable but abnormal waveform; occurs at 5–100 Hz
b.technical considerations for EMG
i.limb temperature variations: cold prolongs the MUP and increases its amplitude
ii.patient age
(1)neonates: MUPs amplitudes are proportionately small due to immature size of the muscle fibers
Box 10.1
Differential diagnosis of fasciculation potentials—Motoneuron diseases; chronic radiculopathy or neuropathies; cholinesterase use
Box 10.2
Differential diagnosis of acquired myotonia—
Chronic radiculopathy or neuropathy; severe myopathies; drug exposure
Diagnostic Testing
233

10 Diseases of the Muscles
234
(2)elderly: MUP amplitudes are larger than in young adulthood due to compensatory reinnervation caused by age-appropriate loss of motoneurons
2.Muscle biopsy
a.open biopsy allows for multiple sampling sites that may be necessary for muscle diseases that do not involve the whole muscle body (i.e., are patchy, as in the inflammatory myopathies)
b.evaluate both frozen and glutaraldehyde-fixed sections; fixed sections should be prepared by embedding in plastic (for electron microscopy, if necessary) and paraffin
c.technical limitations
i.in cases of chronic muscle diseases, avoid biopsies of severely affected muscles; in cases of relative acute muscle diseases, select severely affected muscles
ii.do not perform within 1 month of an episode of rhabdomyolysis, and avoid muscles that have suffered recent trauma (e.g., EMG testing)
III. Myasthenic Syndromes
1.Myasthenia gravis
a.pathophysiology: caused by antibodies that disrupt the function of postsynaptic nicotinic acetylcholine receptors on striated muscle; these antibodies may be derived from
i.a paraneoplastic syndrome: anti-acetylcholine receptor antibodies block acetylcholine receptor binding and crosslink the receptor proteins causing their internalization and destruction; antibodies target the gamma subunit of the receptors, which are most concentrated in the extraocular muscles
(1)antibodies on the myocytes induce (Box 10.3)
(a)complement membrane attack complexes: damage to the postsynaptic membrane initially causes loss of infoldings of the muscle fiber membranes {T tubules} and disorganization of the muscle end plate with widening of the synaptic cleft, and may ultimately cause muscle fiber death
(b)mild lymphocyte invasion of the muscle, in severe cases
(2)associated with thymoma (15% of cases) or thymic hyperplasia (80% of cases)
(a)B lymphocytes produce the anti-acetylcholine receptor antibodies but require T lymphocytes to make them in the form of IgG, therefore thymectomy changes the antibody expression from IgG to the less damaging IgM form
ii.an autoimmune reaction: antibodies against the muscle specific tyrosine kinase (MuSK) may disrupt acetylcholine receptor aggregation in the muscle end plate by deregulating the function of the intracellular protein agrin
(1)this form of myasthenia gravis is associated with HLA-B8 and –DR3 in young patients, and with other autoimmune disorders (rheumatoid arthritis, lupus, polymyositis, pernicious anemia) or immunosuppression
b.epidemiology: a bimodal prevalence with peaks at 20–30 years of age (female predominance) and 60–80 years of age (male predominance)
(1)myasthenia gravis patients greater than 40 years of age commonly have a thymoma; patients less than 40 years of age are more likely to have thymic hyperplasia
c.symptoms
Box 10.3
Thymoma paraneoplastic syndromes—
Myasthenia gravis; Isaac’s syndrome; cerebellar degeneration (CV-2 paraneoplastic autoantibody); pure red cell aplasia

i.weakness of the extraocular and facial muscles neck (particularly flexion), bulbar, and oropharyngeal muscles proximal upper extremity muscles lower extremity muscles
(1)weakness is typically asymmetric and becomes evident after prolonged use of a muscle (i.e., it is not present with immediate movements)
(a)weakness is often evident as a loss of postural tone, or as tremulousness during prolonged muscle use
(2)the weakness may be specifically limited to the extraocular muscles; the pupils are spared in myasthenia gravis, unlike botulism
ii.muscle soreness
iii.muscle atrophy occurs in only 15% of chronic cases and reflexes are always normal
(1)the tongue often exhibits longitudinal furrows in patients with MuSK antibodies, but this is a poorly understood abnormality that is not true atrophy
d.diagnostic testing
i.edrophonium (Tensilon) test: double-blinded injection of edrophonium (10 mg total in divided doses) produces improvement in a preexisting weakness within 1 minute of injection
(1)best to measure the degree of ptosis, which can be easily quantified
(2)requires a 2-mg test dose to assess tolerability; monitoring for bradycardia
(3)not as accurate as serological tests and EMG, because the response to edrophonium is not specific for myasthenia gravis
ii.serum antibodies
(1)acetylcholine receptor: present in 95% of myasthenia gravis cases
(2)striated muscle: present in 30% of myasthenia gravis cases; more common in older patients
(3)MuSK: present in 65% of acetylcholine receptor-negative patients; patients usually exhibit mild symptoms limited to the extraocular muscles
(4)titan: more common in patients with normal thymus glands
(5)ryanodine-sensitive voltage-gated calcium channels (Box 10.4)
iii.EMG demonstrates increased jitter, at least in facial muscles if not diffusely
iv.nerve conduction studies: repetitive nerve stimulation produces decremental responses
v.muscle biopsy can be used to demonstrate immune deposits in seronegative cases
vi.thymoma screening with CT chest, which always should be performed irrespective of the likelihood of its occurrence
e.treatment (Fig. 10–5)
i.pyridostigmine (Mestinon): better for paraneoplastic myasthenia (i.e., anti-acetylcholine receptor antibody), and is minimally effective in autoimmune myasthenia (i.e., anti-MuSK antibody)
(1)the results of edrophonium testing should not be used to adjust medication doses
ii.immunosuppressants: better for autoimmune myasthenia (Box 10.5)
(1)chronic prednisone, which may cause transient worsening of the weakness at the time of initiation of the therapy
(2)chronic cyclosporine, azathioprine, or mycophenolate in patients who are poorly responsive to glucocorticoids
(3)plasma exchange or intravenous immunoglobulin (IVIg) for severe bulbar and oropharyngeal dysfunction
Figure 10–5 Myasthenia gravis. Significant decrement at rest and postexercise potentiation followed by exhaustion. (From McKhann GM et al. Q&A Color Review of Clinical Neurology and Neurosurgery. Stuttgart, Germany: Georg Thieme; 2003: 131, Fig. 130. Reprinted by permission.)
Box 10.4
The ryanodine-sensitive voltage-gated calcium channel is also mutated in central core myopathy; not the same as the L-type dihydropyridine calcium channel that is mutated in hypokalemic periodic paralysis.
Box 10.5
Myasthenia crisis
Occurs in patients with a baseline respiratory impairment who develop a second respiratory disorder (usually infection); typically defined as a forced vital capacity (FVC) 15 cc/kg
Distinguish from respiratory failure due to acetylcholinesterase inhibitor overdose by challenging the patient with edrophonium after the patient is intubated
Myasthenic Syndromes
235

10 Diseases of the Muscles
236
iii.thymus removal for thymoma or thymic hyperplasia: thymus removal is useful only for paraneoplastic myasthenia and is not usually effective acutely, in severe disease, or in patients with anti-MuSK antibodies
(1)thymomas are generally encapsulated, but 35% may be invasive; thymoma invasion of surrounding tissues may require irradiation and/or chemotherapy
(2)in postthymectomy patients, rising antibody titers may indicate thymoma recurrence
f.prognosis: slow progression of symptoms over months with periods of exacerbation
i.no clear association exists between exacerbations and pregnancy
ii.no association between antibody titers and severity of disease
2.Pediatric myasthenia syndromes
a.transitory neonatal myasthenia—caused by transplacental passive transfer of antibodies from a myasthenic mother to the fetus; occurs in 20% of children of myasthenic mothers
i.symptoms are present at birth, and include hypotonia and poor feeding
ii.treatment: may require plasmapheresis if severe
iii.prognosis: self-limited course usually lasting 2 weeks
b.congenital myasthenia—caused by abnormalities in acetylcholine release, neuromuscular junction acetylcholinesterase function, or postsynaptic acetylcholine receptors; usually exhibits autosomal recessive inheritance
i.symptoms develop at birth, and include ptosis, ocular motor weakness, and facial weakness; limb weakness is mild
ii.treatment: as per adult myasthenia gravis
c.familial infantile myasthenia—associated with polymorphisms on chromosome 17
i.symptoms are present at birth, and include poor feeding and respiratory failure often leading to sudden death
ii.treatment: as per adult myasthenia
iii.prognosis: symptoms generally improve with age
3.Lambert-Eaton myasthenia
a.pathophysiology: a paraneoplastic syndrome caused by antibodies to the P/Q calcium channel on presynaptic motoneuron terminals, thereby reducing acetylcholine release; rarely involves antibodies against the N-type calcium channels
b.subtypes: both occur with approximately equal frequencies
i.Lambert-Eaton myasthenia without associated cancer: average age of onset 50 years of age; often associated with other autoimmune conditions (i.e., myasthenia gravis, pernicious anemia, vitiligo, diabetes, lupus)
ii.Lambert-Eaton myasthenia with associated cancer: average age of onset 60 years of age; classically associated with small-cell lung cancer or rarely associated with leukemia or lymphoma
(1)3% of small-cell lung cancer patients with develop Lambert-Eaton myasthenia
(2)symptomatic onset of myasthenia precedes the diagnosis of cancer in 80%
c.symptoms: usually is subacute at onset, but may be acutely precipitated by exposure to calcium channel blockers, iodinated contrast agents, and muscle paralytic agents
i.weakness in the proximal lower extremities upper extremities oropharyngeal muscles respiratory muscles
(1)weakness improves with brief exercise but then worsens with sustained exercise; similarly, patients are initially hyporeflexic but the reflexes improve with repetition
(2)ocular and facial muscles are generally spared, although 40% will complain of diplopia
ii.autonomic dysfunction (Box 10.6): dry mouth, constipation, impotency
d.diagnostic testing: EMG exhibits transient, incremental compound muscle action potential (CMAP) response to rapid nerve stimulation (20–50 Hz) and a decremental response to slow nerve stimulation (1–5 Hz), similar to botulism; also exhibits small CMAP amplitudes (Box 10.7)
e.treatment: inconsistently responsive to acetylcholinesterase inhibitors
i.Lambert-Eaton myasthenia without associated cancer: chronic immunosuppression
(1)requires careful and frequent evaluation for development of a neoplasm
ii.Lambert-Eaton myasthenia with associated cancer
(1)treatment of neoplasm
(2)3, 4-diaminopyridine; immunosuppression; plasma exchange, IVIg
IV. Congenital Myopathies
1.X-linked myopathies: Mostly symptomatic in males; however, female carriers may develop mild symptoms at later ages due to random inactivation of the X chromosome
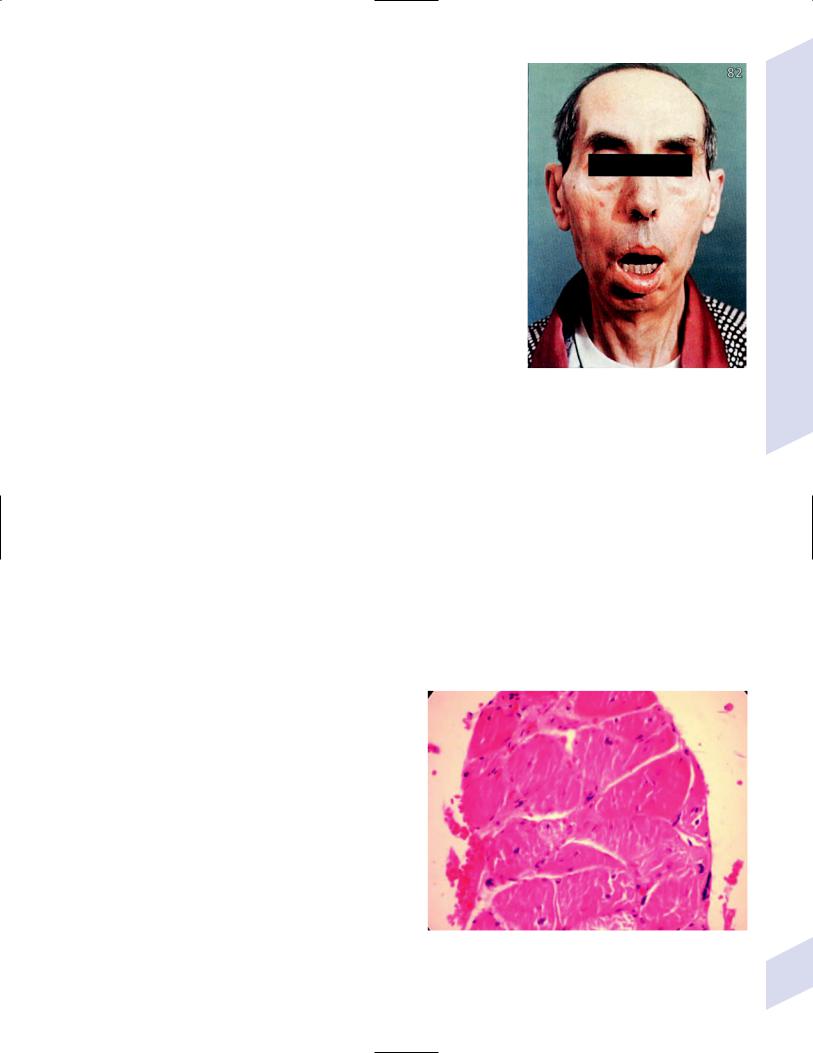
a. Duchenne’s muscular dystrophy
i.pathophysiology: caused by mutation of the dystrophin gene, which encodes a protein that anchors the cytoskeleton to actin in the myocontractile apparatus
(1)the majority of mutations are frameshift; 30% of cases are novel mutations, and 15% of novel mutations are gonadal mosaics
ii.symptoms
Box 10.6
Autonomic dysfunction does not occur with myasthenia gravis.
Box 10.7
CMAP is a summation of multiple MUPs
(1)progressive symmetric weakness beginning at 3–5 years of age as cramping and fatigue of the proximal muscles that progresses to the inability to walk by 13 years of age
(a)the waddling gait with toe walking is due to weakness of the gluteus muscles and shortness of the ankle tendon
(b)proximal muscle weakness necessitates that the patient stands up with the hands pushing on the knees {Gower’s sign}, which is nonspecific for Duchenne’s muscular dystrophy
(2)progressive generalized muscle hypertrophy that is most pronounced in calf muscles
(3)joint contractures, particularly in lower extremities
(4)dilated cardiomyopathy: 60% of cases are symptomatic by 18 years of age
(5)mild mental retardation
iii.diagnostic testing
(1)elevated CK (100-times normal levels) that declines with muscle loss over time
(2)genetic testing for dystrophin mutations
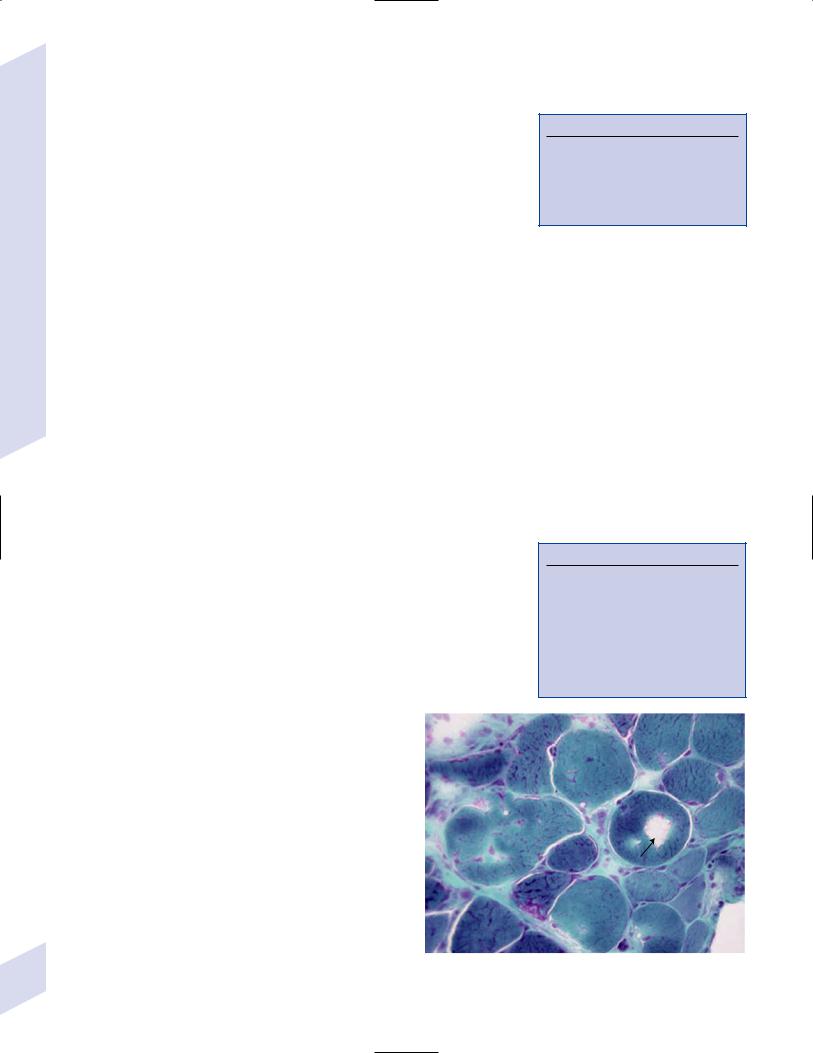
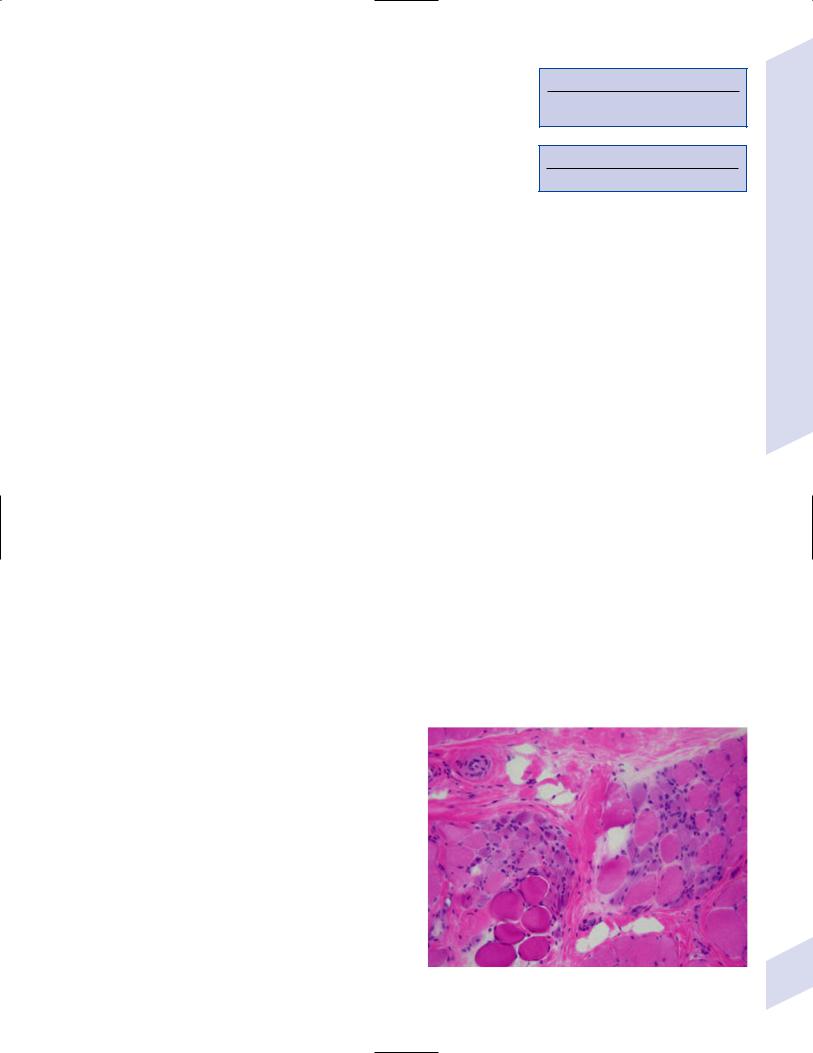
(3)muscle biopsy demonstrates endomysial fibrosis and clumps of variable-sized, irregular muscle fibers in a contracted state but with normal internal muscle architecture (Fig. 10–6)
(a)immunohistochemical analysis demonstrates absent staining for dystrophin
iv.treatment: avoid exercise, which likely worsens muscle injury
Figure 10–6 Duchenne’s muscular dystrophy. Endomysial fibrosis is noted in the region at the top of the figure. Courtesy of Dr. C. Yamada.
Congenital Myopathies
237
10 Diseases of the Muscles