


4
Disorders of Myelination
4 Disorders of Myelination
104
Note: Significant diseases are indicated in bold and syndromes in italics.
I. Multiple Sclerosis (MS)
1.Pathophysiology
a.etiological hypotheses
i.the autoimmune hypothesis: peripheral leukocytes become sensitized to central nervous system myelin antigens by
(1)direct exposure of leukocytes to antigens after breakdown of the blood–brain barrier during a local infection
(2)sensitizing leukocytes to viral antigens that are similar to myelin antigens {molecular mimicry}
ii.the infectious hypothesis: stems from epidemiological studies linking geographical location with disease susceptibility, although no definitive links have been made; it is thought that demyelination is a reaction to chronic viral infection, such as that from
(1)herpes 6 virus infection: evidence of viral infection is present more commonly in patients with MS, although a causative association is not established
(2)Epstein-Barr virus (EBV) infection: pediatric MS patients have twice the rate of EBV exposure than do controls, suggesting an early exposure to the virus; MS patients exhibit higher antibody levels against EBV
(3)other viruses: herpes simplex 1, simian cytomegalovirus, MSassociated retrovirus (HERV-W)
(a)patients with MS have a later mean age of infection with measles, rubella, and mumps, but not a greater overall incidence of infection
(4)Chlamydia pneumoniae—MS patients exhibit greater reactivity to Chlamydia, but equal rates of infection
b.genetics
i.associated with human leukocyte antigen (HLA) A3, -B7, -Dw2, -DQ, and -DR2 subtypes
ii.familial aggregation occurs in 15% of cases; dizygotic twin risk is 5%, similar to the risk of non-twin siblings; monozygotic twin risk is 35%
iii.risk of MS 3% with one affected parent, 30% with two affected parents
c.epidemiology
i.geography: common in northern North America, Europe, Scandinavia, New Zealand, and southern Australia (i.e., maximal distances from the equator)
ii.age: clinical onset occurs almost invariably in patients 60 years of age
(1)risk of developing MS related to geography is acquired by 12 years of age

A |
B |
Figure 4–1 Typical multiple “sclerosis” lesions. (A) T1 non-contrast MRI |
(From Balassy C et al. Long-term MRI observations of childhood-onset |
showing black holes (arrows) within a chronic, inactive lesion. (B) T1 with |
relapsing-remitting multiple sclerosis. Neuropediat 2000;32: 33, Fig. 2a,b. |
contrast MRI demonstrating enhancement of active lesions (arrows). |
Reprinted by permission.) |
iii.sex: for women, relative risk of the relapsing-remitting subtype 3; equal risk for men and women for the primary-progressive subtype
iv.race: relative risk 8% for Whites
2.Histology (Fig. 4–1)
a.plaques: areas of demyelination typically occurring around venules in the white matter
i.acute/active plaques: characterized by T lymphocyte and macrophage infiltration
(1)macrophages are predominant in the core of the plaque and phagocytize myelin debris; reactive astrocytes are also present in the plaque core
(2)T lymphocytes are sensitized to multiple antigens in myelin (i.e., myelin basic protein, proteolipid protein, “P-0” protein, myelinassociated glycoprotein), but the molecule that initiates the inflammatory response is unknown
ii.chronic plaques: hypercellular areas that contain mostly astrocytes, sometimes with a periphery of lipid-containing macrophages
(1)axons are demyelinated but otherwise relatively normal in appearance; loss of axons may approach 50% within the plaque
(a)axon loss is likely a reaction to demyelination and to direct injury from the inflammatory process
(2)most chronic plaques do not contain inflammatory cells, but some have ongoing myelin phagocytosis by macrophages {chronic active plaque}
iii.shadow plaques: areas of demyelination with partial remyelination
(1)oligodendroglia are reduced in the core of plaque but are increased at periphery, which accounts for the limited remyelination that occurs in chronic plaques; most of the oligodendroglia surrounding the plaque are not of a myelinating phenotype
iv.normal appearing white matter (NAWM) that does not contain plaques may show evidence of gliosis
Multiple Sclerosis (MS)
105

4 Disorders of Myelination
106
3.Symptoms (Box 4.1)
a.acute symptoms
i.sensory abnormalities: the most common deficit of MS
(1)subjective sensory complaints include numbness, paresthesias, band-like sensations around the chest or abdomen, saddle anesthesia, itching, and (rarely) radicular pain
(a)Lhermitte’s sign: an electric shock sensation that radiates down the spine and into the limbs upon flexion of the neck; a nonspecific finding that can be seen with many types of cervical spine lesions that involve the dorsal columns (e.g., subacute combined degeneration, tabes dorsalis)
(b)loss of proprioceptive sensation in a limb may produce significant discoordination {useless hand sign of Oppenheim}
(2)objective findings on sensory testing (in order of occurrence)
(a)loss of vibration and position sense
(b)loss of pain and light touch
(c)saddle anesthesia/associated with bowel and bladder hypotonia
ii.vision loss from optic neuritis: most commonly is a centrocecal scotoma (see p. 38), which reduces acuity; commonly is caused by lesions behind optic chiasm {retrobulbar optic neuritis}
(1)vision loss is usually gradual and associated with retroorbital or periorbital pain
(2)vision loss is often associated with a direct afferent pupillary defect {Marcus-Gunn pupil}
(3)papillitis occurs only when the head of the optic nerve is involved
iii.diplopia from ocular motor dysfunction: internuclear ophthalmoplegia impairment of CN VI CN III CN IV function
iv.weakness: acute focal weakness is fairly uncommon except in the primary progressive subtype; slow-developing, progressive weakness is more common
v.cerebellar dysfunction involving ataxia of both the trunk and extremities
vi.syndromes of spinal cord injury (see transverse myelitis below)
vii.bladder and bowel dysfunction: typically includes urinary frequency and urgency with retention, and constipation; sexual dysfunction is also common
viii.epilepsy ( 5%): any type of seizure may occur
b.chronic symptoms: spasticity, appendicular tremor; fatigue; sexual dysfunction; psychiatric disturbance, particularly depression (65%); dementia (5%)
c.paroxysmal symptoms: trigeminal neuralgia; hemifacial spasm; tonic spasms; hemiataxia; paresthesias; dysarthria
i.symptoms last only a few seconds but may recur repeatedly or occur in clusters
ii.paroxysmal symptoms are likely caused by ephaptic neural transmission between demyelinated neural systems that are in direct contact (i.e., a short circuit)
4.Subtypes based on symptomatic progression
a.relapsing-remitting MS (55% of all cases; 85% of cases at the time of presentation)—involves multiple discrete episodes of neurological injury followed by partial recovery
b.secondary progressive MS (30% of all cases)—a progressive course of neurological injury developing after several years of an apparent relapsingremitting course; likelihood of transforming into secondary progressive
subtype from relapsing-remitting subtype increases with disease duration ( 90% likelihood after 25 years of disease)
i.some patients develop progressive neurological dysfunction after a single episode of acute neurological injury (“single progressive MS”)
Box 4.1
MS symptoms are exacerbated or induced by heat, {Uthoff’s phenomenon} menses, and the post-partum period.
MS symptoms are reduced by pregnancy, particularly in the third trimester (due to reduced Th2 leukocytes).

c.primary progressive MS (10% of all cases)—neurological injury that progresses from the onset of the disease and never remits
i.progressive relapsing MS—after a minimum of 1 year of apparent primary progressive MS, the patient develops relapses in addition to the progressive course
d.clinically isolated MS
i.optic/retrobulbar neuritis—as a clinically isolated event, most patients recover to 20/40 acuity but only a third will recover completely and most will have persistent deficits in color vision and/or contrast sensitivity
(1)recurrence occurs in 30%, and often causes a greater impairment of the visual acuity than did the original episode
(2)associated with a 60% 10-year risk of developing MS if the brain magnetic resonance image scan (MRI) is abnormal; only a 10% 10-year risk if the brain MRI is normal
ii.transverse myelitis—symptoms include lower extremity weakness and sensory loss, bowel/bladder dysfunction, back pain, and occasionally spinal shock
(1)does not exhibit a familial pattern
(2)likely due to MS in cases with
(a)asymmetric symptoms (e.g., an incomplete lesion)
(b)involvement of less than two spinal segments
(c)oligoclonal bands in the cerebrospinal fluid
(d)evidence of plaque-like brain lesions on neuroimaging
iii.monophasic brainstem or cerebellar syndromes
5.Diagnostic testing: the Poser criteria for MS and the International Panel on Multiple Sclerosis diagnostic criteria (the McDonald criteria) (Table 4–1)
a.neuroimaging: cognitive and behavioral impairment correlates better with general atrophy than with lesion load
Table 4–1 Diagnostic Criteria for Multiple Sclerosis (MS)
Poser criteria for multiple sclerosis
|
# attacks by |
# exam |
Supportive laboratory |
Category |
history |
findings |
evidence* |
|
|
|
|
Definite MS |
2 |
2 |
None required |
|
2 |
1 |
1 Type |
Probable MS |
2 |
1 |
None |
|
1 |
2 |
None |
|
1 |
1 |
1 Type |
|
|
||
McDonald criteria for definite multiple sclerosis |
|
||
|
|
|
|
# attacks by |
# of physical |
|
|
history |
findings |
Abnormal laboratory test |
|
|
|
|
|
2 |
2 |
None are required for diagnosis, although abnor- |
|
|
|
malities should be found if the various tests are |
|
|
|
performed |
|
2 |
1 |
Barkhof MRI criteria* |
|
|
|
2 MRI lesions abnormal CSF |
|
1 |
2 |
New gad-enhancing lesion within 3 months |
|
1 |
1 |
Barkhof MRI criteria* |
|
|
|
2 MRI lesions abnormal CSF |
|
*Supportive laboratory evidence = typical MRI lesions, asymmetric evoked potentials, abnormal urodynamic testing.
Abbreviations: CSF, cerebrospinal fluid; gad, gadolinium.
Multiple Sclerosis (MS)
107

4 Disorders of Myelination
i.plaques are commonly located radiating outward from the ventricle surface {Dawson’s fingers} or in the corpus callosum; plaques can occur anywhere with oligodendrocyte myelination including the cortical white matter
ii.Barkhof MRI criteria: require at least three of the following four conditions.
(1)one gadolinium-enhancing lesion, or at least nine T2-hyperintense lesions
(2)three periventricular lesions
(3)one infratentorial lesion
(4)one juxtacortical lesion
b.electrophysiological testing
i.visual-evoked responses: 85% abnormal in Poserdefinite MS (see p. 42)
ii.brainstem auditory-evoked responses: 65% abnormal in Poser-definite MS (see p. 50)
iii.somatosensory-evoked responses: 80% abnormal in Poser-definite MS
(1)transcutaneous electrical stimulation of a distal sensory nerve produces electrical po-
tentials that can be measured over the spinal cord and the centroparietal and frontopolar cranial regions
Figure 4–2 Electrodes are placed on the base of the lateral neck inferior to the sternocleidomastoid muscle {Erb’s point} measure the potential generated by the brachial plexus. Somatosensory evoked potential from median nerve stimulation. Potentials are labeled according to positive (P) or negative (N) deflections and their expected delay (in milliseconds). Typically, N11 dorsal columns, N13 potential cervical spinal cord (? cuneate nucleus), N14 and P15 medial lemniscus, N16 and 17 upper brainstem/thalamus, and N20 parietal cortex (for tibial nerve stimulation in a normal person: N34 upper brainstem/thalamus; P37 parietal cortex).
(a)potentials are labeled according to their latencies (in milliseconds) from the stimulus and the polarity of the response
(i)median nerve stimulation in a normal person (Fig. 4–2)
c.cerebrospinal fluid: typical abnormalities include
i.normal or mildly elevated protein
(1)myelin basic protein: very nonspecific; it is no longer used for diagnosis
(2)elevated IgG, usually measured as a fraction of the serum albumin ( 0.27 in 65% of patients with Poser-definite MS versus 3% of controls) or as a fraction of serum IgG {IgG index} ( 0.7 in 80% of patients with Poser-definite MS versus 3% of controls)
(3)oligoclonal bands: detectable in 95% of definite MS but also in 10% of controls
(4)albumin level: a very insensitive measure for MS
ii.leukocytosis 50 in 60% of patients
d.serology: serum anti-myelin antibodies (anti-myelin oligodendroglial glycoprotein and anti-myelin basic protein) may predict second demyelinating events in patients after an initial demyelinating event, although this has not been confirmed
6.Treatment
a.therapy for the relapsing-remitting subtype of MS
i.acute attack treatment: generally reserved for significant neurological impairment
(1)methylprednisolone 1 g IV once q.d. for 3–5 days, or prednisone 1250 mg PO q.d. for 3–5 days; tapering is not required
(a)glucocorticoids increase the rate of recovery, but do not improve overall level of recovered function
108

(b)after an initial demyelinating event, glucocorticoids decrease the likelihood of developing MS over the following 2 years, but they do not reduce the long-term likelihood of developing MS
(c)high-dose oral glucocorticoids are likely to be as efficacious as IV glucocorticoids, although this has not been directly tested
(2)plasmapheresis, although it has no supportive evidence
(a)intravenous immunoglobulin (Ig) has failed to demonstrate any benefit as an adjuvant therapy for glucocorticoids
(3)methotrexate, cladribine, mitoxantrone
(4)combinations of the above therapies
ii.preventative therapy for acute attacks: all are contraindicated during pregnancy, and all have a modest effect on disability in the available short-term follow-up from clinical trials
(1)interferon 1a 30 g IM weekly (qwk.) (Avonex): reduces relapse rate by 18%
(2)interferon 1a 22–44 mg IM q.o.d (Rebif): reduces relapse rate by 30%
(3)interferon 1b 22–44 mg subcutaneously (SQ) 3 times a week (t.i.w.) (Betaseron): reduces relapse rate by 30%
(4)glatiramer acetate (Copaxone) 20 g SQ q.d., if interferons are not tolerated or antibodies against interferons develop; decreases relapse rate by 30% but inconsistent effectiveness against long-term disability
(5)mitoxantrone: used to stabilize patients with rapidly progressive disability or frequent relapses; it exhibits significant cardiotoxicity and is typically given only for a few months
iii.therapy for paroxysmal symptoms: full-dose anticonvulsants, benzodiazepines
b.therapy for progressive subtypes of MS: no therapy has yet proven effective for primary progressive MS; for second progressive MS, or for rapidly developing progressive symptoms, consider
i.interferons, as above
ii.monthly bolus of methylprednisolone
iii.monthly cyclophosphamide treatment
iv.methotrexate, cladribine, mitoxantrone
c.treatment for chronic symptoms
i.spasticity: baclofen, tizanidine, dantrolene, benzodiazepines, botulinum toxin
ii.tremor: anticonvulsants, clonazepam, propranolol, ondansetron, gabapentin; stereotactic thalamotomy in refractory cases
iii.fatigue: amantadine, pemoline, SSRIs, modafinil, 4-aminopyridine, acetylcarnitine
iv.spastic bladder: oxybutynin (Ditropan), propantheline, imipramine
(1)hypotonic bladder is rare
v.psychiatric impairment
(1)depression: SSRIs, amitriptyline, second-generation antidepressants (e.g., venlafaxine, bupropion)
(2)dementia: donepezil may be beneficial
d.treatment for paroxysmal symptoms: full-dose antiepileptics; benzodiazepines; glucocorticoids when resistant to other therapies
Multiple Sclerosis (MS)
109

4 Disorders of Myelination
110
II.Pediatric Multiple Sclerosis/Early-Onset Multiple Sclerosis
1.Pathophysiology
a.pediatric multiple sclerosis has a stronger association with HLA-DR than adult multiple sclerosis but has no gender preference
b.pediatricand adult-onset multiple sclerosis exhibit similar familial patterns
2.Symptoms: as per relapsing-remitting multiple sclerosis; onset 16 years of age but usually continues into adulthood as multiple sclerosis
a.episodes of optic neuritis are typically simultaneous or sequentially bilateral, not isolated unilateral episodes as in adult multiple sclerosis
i.cases with unilateral optic neuritis are more likely to develop into typical multiple sclerosis
b.Devic’s disease-like presentation is extremely rare in children
3.Diagnostic testing: neuroimaging can differentiate the disorder from acute disseminated encephalomyelitis (ADEM). However, neuroimaging and cerebrospinal fluid studies are of uncertain utility in children, therefore establishing the diagnosis of pediatric multiple sclerosis is generally made according to the Poser criteria
4.Treatment
a.acute attack treatment
i.methylprednisolone 20–30 mg/kg/d for 3–5 days, followed by PO prednisone with tapering
ii.IVIg in cases unresponsive to glucocorticoids
b.preventative therapy for acute attacks
i.interferons, adjusting the dose for weight and age
ii.cyclophosphamide
III. Multiple Sclerosis Variants
1.Devic’s disease/neuromyelitis optica
a.histology: inflammatory demyelinating lesions with complete loss of oligodendrocytes and significant leukocyte infiltration that may progress to frank necrosis and cavitation in severe cases (spinal cord optic nerves); inflammation involves gray and white matter of the spinal cord optic nerves, and does not involve much gliosis as does multiple sclerosis
i.lesions exhibit perivascular immunoglobulin and complement deposits
ii.necrotic demyelinating lesions that exhibit near complete axon loss typically occur in the optic nerves and cervical spinal cord
b.symptoms: rapid-onset of bilateral painful vision loss, and symptoms of complete spinal cord injury associated with back pain (back pain is rare for spinal multiple sclerosis); may exhibit a relapsing course, but more commonly is fulminant
i.the optic neuritis may be bilaterally simultaneous or sequential
c.diagnostic testing: different than opticospinal multiple sclerosis in that
i.neuroimaging demonstrates a normal brain in 90% of cases, and a spinal cord lesion two spinal segments in length
ii.cerebrospinal fluid analysis demonstrates oligoclonal bands only in 30% of cases and then only transiently; elevated protein and a neutrophil pleocytosis are observed
d.treatment
i.acutely treat with glucocorticoids, plasmapheresis, or IVg
ii.treat relapsing-remitting cases chronically with interferon therapy, prednisone azathioprine, or cyclophosphamide

e.prognosis: 90% 5-year survival in fulminant disease; 70% 5-year survival in relapsing-remitting disease
2.Marburg’s variant of multiple sclerosis
a.pathophysiology: the pathophysiology is generally considered to be the same as that for multiple sclerosis, although there is evidence that Marburg’s variant is related to expression of an immature or modified form of myelin basic protein
i.histology: a large, acute demyelinating lesion of a single hemisphere (60% of cases) that rapidly increases in size and is associated with mass effect due to edema
b.symptoms: predominantly relate to rapidly increased in intracranial pressure
c.diagnostic testing
i.cerebrospinal fluid is usually normal except for a minimal pleocytosis
ii.neuroimaging: lesions are often mistaken for tumor or cerebritis because the lesion contrast enhances and shrinks after steroid treatment
d.treatment: glucocorticoids, IVg, plasmapheresis, mitoxantrone, cyclophosphamide
e.prognosis: mortality is usually due to mass effect and herniation, or to involvement of the brainstem in the demyelinating process
3.Balo’s concentric sclerosis (Box 4.2)
a.histology: demyelinating lesions that are centered around a perivascular collection of inflammatory leukocytes, wherein demyelination and inflammation occur in concentric rings that alternate with areas of preserved myelin that are of decreasing severity with increasing distance from the center
i.areas of preserved myelin exhibit small amounts of demyelination and remyelination, suggesting active suppression of the disease process or else a lesion involving acute and chronic demyelination
b.symptoms: headache, impaired cognitive and behavioral function, seizures
c.diagnostic testing
Box 4.2
Schilder’s Disease
This unclear disease entity typically is thought to have a monophasic course of focal neurological injury and elevated intracranial pressure.
Neuroimaging often demonstrates large enhancing lesions with mass effect.
i.neuroimaging demonstrates multiple concentric ring-enhancing lesions that may have mass effect (Fig. 4–3); typical multiple sclerosis lesions are also observable
ii.cerebrospinal fluid usually exhibits monocyte pleocytosis and occasionally oligoclonal bands
d.treatment: as per multiple sclerosis
e.prognosis: usually has a fatal outcome within a few weeks, although spontaneous remission is possible; survivors do not reliably develop multiple sclerosis
IV. Acute Disseminated Encephalomyelitis (ADEM)
1.Pathophysiology: typically develops during or within 2 weeks of a preceding infection with measles, rubella, smallpox, or chickenpox, or after a vaccination (rabies or smallpox vaccines), although a preceding infection or vaccination is not necessary to establish the diagnosis
a.mumps, influenza, and Mycoplasma pneumoniae upper respiratory tract infection are rare associations
2.Histology: diffuse and extensive demyelination with axonal sparing involving perivascular cuffing with lymphocytes and reactive astrocyte hyperplasia; demyelination often involves the cortical–subcortical junction
3.Epidemiology: common in children
4.Symptoms: acute-onset multifocal neurological abnormalities that increase in severity over a few days and that may involve any part of the brain and spinal cord; ADEM typically has encephalopathy, fever, meningismus, and (possibly) seizures
Figure 4–3 Balo’s concentric sclerosis demonstrating the hallmark alternating layers of demyelination and preserved myelin (straight arrow), as well as a confluent area of demyelination (curved arrow). (From Gharagozloo AM, Poe LB, Collins GH. Antemortem diagnosis of Balo concentric sclerosis. Radiology 1994; 191:818, Fig. 2A. Reprinted by permission.)
Acute Disseminated Encephalomyelitis (ADEM)
111
4 Disorders of Myelination
112
5.Diagnostic testing
a.cerebrospinal fluid exhibits mild lymphocytosis and increased protein but a normal opening pressure
i.differentiate ADEM from Reye syndrome, which has normal cerebrospinal fluid and elevated serum ammonia levels; oligoclonal bands are likely to be present in postinfectious ADEM
ii.serology also for measles, mumps, varicella zoster, and herpes simplex virus (HSV)
b.neuroimaging: unlike the MRI lesions of multiple sclerosis, ADEM may involve the basal ganglia and thalamus, and have large confluent lesions with mass effect (Fig. 4–4)
6.Treatment: High-dose glucocorticoids; IVg, cyclophosphamide, or plasmapheresis in refractory cases
7.Prognosis: 20% mortality and at least 35% eventually will develop multiple sclerosis
Figure 4–4 Acute disseminated encephalomyelitis. (From McKhann GM et al. Q&A Color Review of Clinical Neurology and Neurosurgery. Stuttgart, Germany: Georg Thieme; 2003:145, Fig. 146a. Reprinted by permission.)
a.focal neurological deficits generally improve but 95% survivors with infectious ADEM and 30% with postvaccine ADEM have residual psychiatric disorders, dementia, or epilepsy
V. Acute Necrotizing Hemorrhagic Encephalomyelitis
1.Pathophysiology: essentially a hyperacute form of ADEM that may be associated with Mycoplasma pneumoniae upper respiratory tract infection
a.histology: intense lymphocytic perivascular inflammation with necrosis and hemorrhage from small blood vessels, causing petechiae; progresses to liquefactive destruction of the white matter of both hemispheres, brainstem, and cerebellar peduncles
2.Symptoms: initially meningitis-like symptoms but rapidly develops seizures and focal neurological injury; progresses to coma that rapidly is fatal
3.Diagnostic testing
a.systemic evidence of infection (e.g., increased WBC count)
b.cerebrospinal fluid exhibits increased opening pressure, monocytosis, increased protein, and normal glucose
c.neuroimaging: large areas of increased T2 signal intensity with small areas of reduced T2 signal representing hemorrhage; lesions have significant edema and mass effect
4.Treatment: as per multiple sclerosis
5.Prognosis: usually fatal within several days
VI. Nutritionand Electrolyte-Related
Demyelinating Disorders
1.Central pontine myelinosis
a.pathophysiology: usually occurs as a reaction to the rapid correction of severe, long-standing (i.e., 2-day duration) hyponatremia
i.prolonged systemic hyponatremia causes loss of sodium and potassium from the brain interstitial fluid into the cerebrospinal fluid, and the loss of intracellular organic molecules (e.g., myoinositol, taurine, glutamate) that maintain cellular tonicity; rapid increases in the extracellular tonicity do not permit water equilibration with the intracellular environment, thereby causing cell death
ii.particularly at risk are alcoholics (e.g., 10% occurrence after Wernicke’s encephalopathy), burn victims, patients with liver failure or transplantation, anorexics, malnourished patients, and patients on prolonged diuretic therapy
Figure 4–5 Central pontine myelinosis. (From McKhann GM et al. Q&A Color Review of Clinical Neurology and Neurosurgery. Stuttgart, Germany: Georg Thieme; 2003: 11, Fig. 2. Reprinted by permission.)
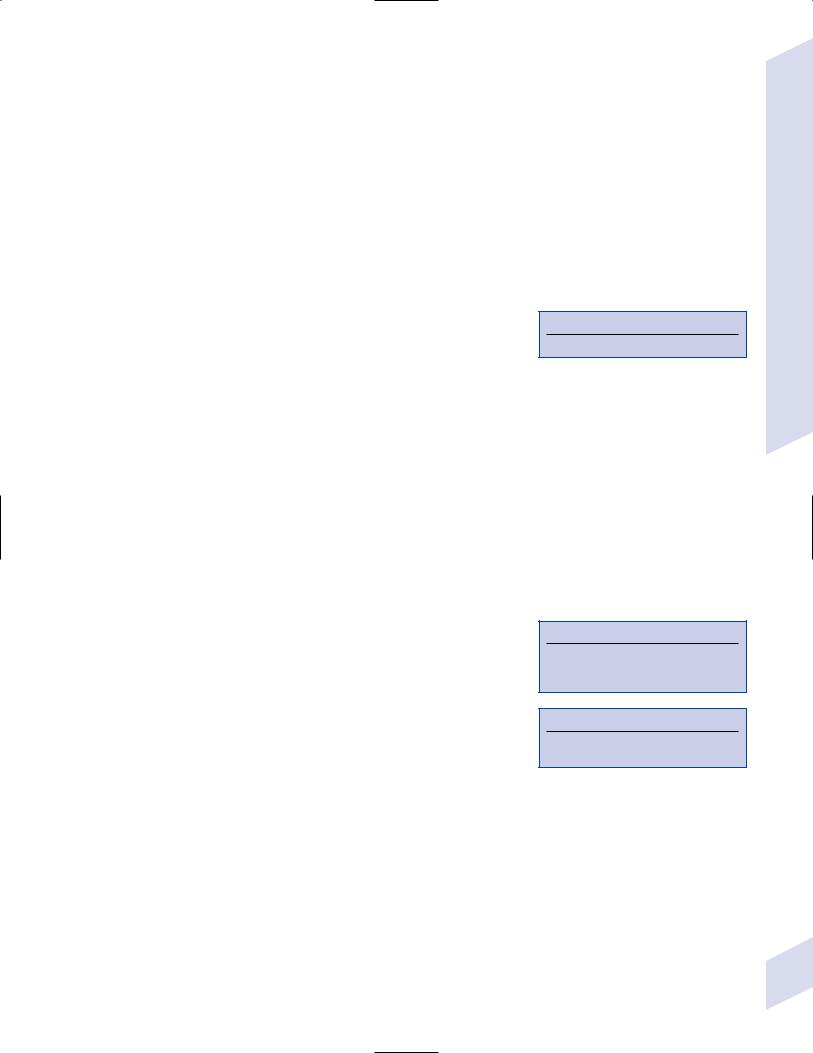
iii.histology: symmetric areas of myelin sheath splitting with some axon swelling that does not lead to axon loss; inflammation develops several days after injury
(1)involves a large demyelinating lesion in the center of the pons that spares a rim of the tegmentum and does not involving the tectum (Fig. 4–5); 10% also have demyelinating lesions elsewhere in the brain
b.symptoms: usually has a biphasic course that progresses over several days
i.encephalopathy and seizures caused by hyponatremia, which remit with correction to normonatremia
ii.dysarthria and dysphagia developing several days after correction to normonatremia, progressing to flaccid quadriparesis {locked-in syndrome}
(1)involvement of the tegmentum may result in pupillary and ocular motor abnormalities
c.treatment: prevention with gradual correction of hyponatremia, for example, no more than 8 mmol/L in 24 hours using 3% saline
required sodium in mmol ([Na]target [Na]actual) • (30 L) (Box 4.3) wherein 1 mL of 3% saline has 0.5 mmol Na
d.prognosis: unpredictable recovery
2.Marchiafava-Bignami disease
a.pathophysiology: classically described in chronic alcoholics who drink red wine but may occur in any alcoholic or malnourished patient; no known relation to nutritional deficiency, although associated with niacin deficiency (i.e., pellagra; see Chapter 13)
i.histology: symmetric areas of demyelination develop in the corpus callosum with histological changes similar to central pontine myelinosis but more frequently involving necrosis; demyelination may occur in any other region of white matter including the optic nerves
b.symptoms: most commonly resembles a rapid-onset frontal lobe dementia but its clinical course is highly variable and dependant upon the location of demyelination
c.treatment: vitamin supplementation
d.prognosis: often remits but may be acutely fatal
VII. Dysmyelinating Disorder (Box 4.4)
1.Pelizaeus-Merzbacher disease
a.pathophysiology: caused by abnormalities of the proteolipid protein (PLP) gene (Box 4.5); alternative splicing of this gene produces PLP (which accounts for 50% of myelin protein) and DM-20 protein (a minor myelin protein), which are necessary for myelin compaction
i.abnormalities in the PLP gene
(1)segmental aberrations: cause PLP overproduction; phenotype is of variable severity, depending upon involvement of surrounding genes
(2)point mutations that prevent PLP translation: causes dysmyelination; phenotype is mild
(3)point mutations that cause incorrect PLP folding: causes oligodendrocyte apoptosis; phenotype is severe
(4)complete gene deletion: prevents PLP production; causes dysmyelination; phenotype is mild
ii.X-linked inheritance; mosaic inactivation of the X chromosome in heterozygous females leads to
(1)no symptoms if the abnormal PLP gene causes oligodendrocyte apoptosis because the few dead oligodendrocytes are replaced by healthy oligodendrocytes
Box 4.3
30 L is estimated body volume
Box 4.4
“Dysmyelinating disease” is a misnomer— these diseases are not always characterized by abnormal myelin.
Box 4.5
PLP is also mutated in a hereditary spastic paraplegia (SPG-2)
Dysmyelinating Disorder
113
4 Disorders of Myelination
(2)minimal, transient symptoms during childhood if abnormal PLP gene only causes abnormal myelin compaction
iii.histology
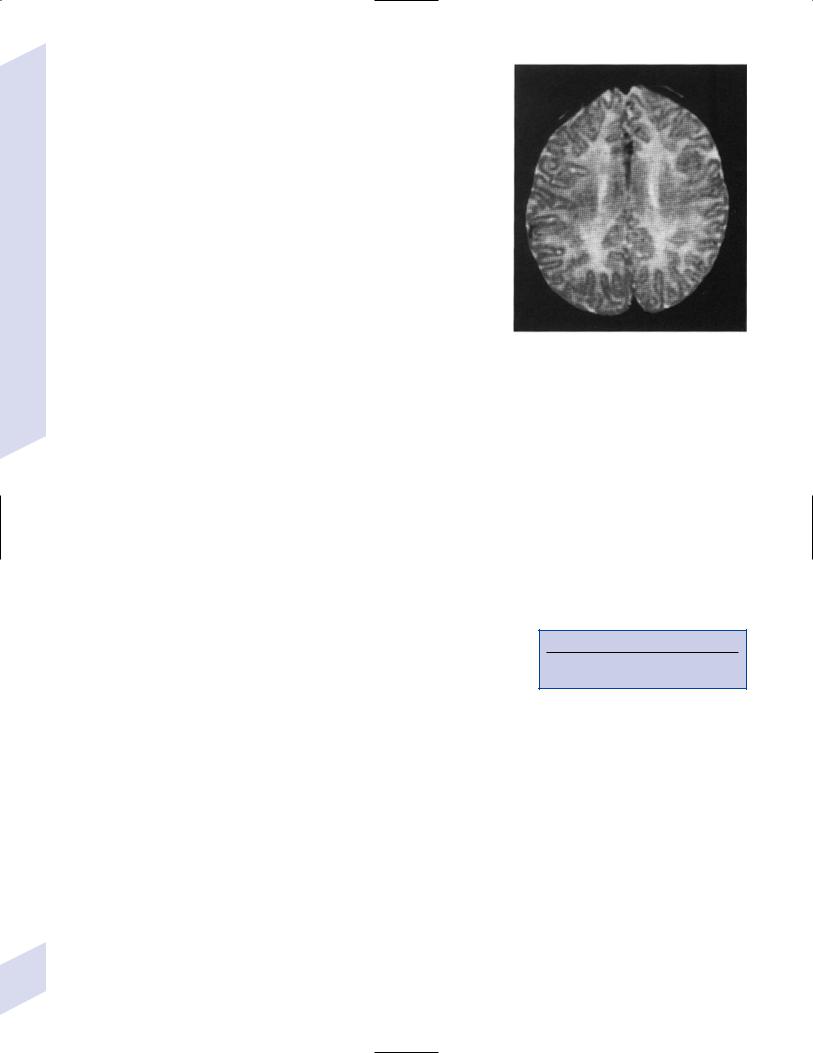
(1)with oligodendrocyte apoptosis: patchy, tiger skin-like (“tigroid”) myelination with a reduced number of oligodendrocytes (Fig. 4–6); severity may be such that no myelin or oligodendrocytes are present in affected areas
(2)with abnormal oligodendrocyte myelination: diffusely reduced myelin staining of the white matter with a normal number of oligodendrocytes
b.symptoms (Table 4–2)
c.diagnostic testing: genetic testing for PLP duplications
i.neuroimaging: large, symmetric areas of increased T2 signal in the white matter with diffuse atrophy
d.treatment: none specific
e.prognosis: severe disease with stridor causes death by 10 years of age
2.Alexander’s disease
a.pathophysiology: caused by mutations in
i.glial fibrillary acidic protein (GFAP) (in 90% cases): mutations cause a toxic gain-of-function that disrupts the normal ability of astrocytes to maintain oligodendrocyte viability
ii.nicotinamide adenine dinucleotide (NADH) ubiquinone oxidoreductase flavoprotein-1: an uncommon association that has yet to be substantiated by autopsy confirmation
b.histology
Figure 4–6 T2-weighted MRI scans through the centrum semiovale demonstrate inhomogeneous signals corresponding to areas of myelination in otherwise widely unmyelinated white matter {tigroid myelination}. (From Plecko B et al. Degree of hypomyelination and magnetic resonance spectroscopy findings in patients with Pelizaeus Merzbacher phenotype. Neuropediat 2003; 34:132, Fig. 4A. Reprinted by permission.)
i.infants: a failure of normal central nervous system myelination followed by demyelination that is associated with eosinophilic inclusion bodies in swollen astrocyte processes {Rosenthal fibers}; areas with these abnormalities develop in a perivascular and subependymal distribution
ii.adult form: central nervous system demyelination is minimal; the key histological feature is a predominance of Rosenthal fibers that contain GFAP, heat-shock proteins, and - -crystallin
(1)difficult to distinguish from multiple sclerosis or conditions with reactive gliosis, which also have Rosenthal fibers
c. symptoms (Box 4.6)
i.infantile form: onset of symptoms 2 years of age
(1)developmental regression; megalencephaly with progressive head enlargement that can be (but is not always) due to underlying hydrocephalus
(2)spasticity
(3)seizures
Table 4–2 Symptoms of Diseases with Proteolipid Protein (PLP) Abnormalities
|
|
|
Spastic paraplegic |
|
Neonatal PMD |
Classic PMD |
type 2 |
|
|
|
|
Intellectual impairment |
Severe |
Mild |
Mild |
Motor impairment |
Severe |
Mild |
Mild |
Speech acquisition |
None |
Normal |
Motor aphasia |
Ataxia |
Severe |
Mild |
Mild |
Nystagmus |
Present |
Present |
Absent |
Other features |
Stridor, seizures |
Dystonias |
Vision loss |
|
|
|
|
Abbreviations: PMD, Pelizaeus-Merzbacher disease.
Box 4.6
Symptomatically, Alexander’s disease is indistinguishable from Canavan’s disease
114

ii.juvenile form: onset at 3–6 years of age; symptoms are a mix between infantile and adult forms, with more signs of brainstem dysfunction but a slower progressive dementia
iii.adult form (rare): exhibits relapsing-remitting course like multiple sclerosis; symptoms include spastic paraparesis, ataxia, and palatal myoclonus
d.diagnostic testing
i.neuroimaging: decreased white matter density initially in the frontal lobes that progresses to all areas of white matter throughout the brain, often contrast enhancing in areas of active demyelination
(1)initially the subcortical white matter, diencephalon, and brainstem are spared
(2)late in the disease, generalized brain atrophy with cystic white matter degeneration develops, often with hydrocephalus
ii.brain biopsy demonstrates demyelination and Rosenthal fibers; electron microscopy demonstrates increased number of cytoplasmic projections on astrocytes
e.treatment: none specific
f.prognosis: usually death within 10 years of symptomatic onset
3.Canavan’s disease/spongy degeneration of infancy
a.pathophysiology: caused by an autosomal recessive deficiency of aspartoacylase causes accumulation of N-acetylaspartate (NAA), which acts as an osmolite producing cellular edema that leads to spongiform degeneration of white matter
b.symptoms: develop by 1 years of age
i.neonatal hypotonia that develops into spasticity
ii.progressive macrocephaly
iii.dysphagia
iv.vision loss from optic nerve atrophy
v.stimulation-induced posturing that involves extension of the lower extremity, flexion of the upper extremity, and head extension
vi.seizures
c.diagnostic testing
i.NAA levels in the urine 50-fold normal levels
ii.reduced aspartoacylase activity in fibroblasts
iii.magnetic resonance spectroscopy demonstrates an increased NAA peak
d.treatment: none specific; reduce intracranial pressure with acetazolamide
4.Vanishing white matter disease/childhood ataxia with central nervous system hypomyelination/myelopathia centralis diffusa
a.pathophysiology: caused by mutations in the gene for the EIF2B translation regulating protein on chromosome 3, which acts to downregulate protein synthesis in response to cell stressors
i.histology: leukoencephalopathy with cavitation but no evidence of myelin products in the form of inclusion bodies
b.symptoms: progressive ataxia and spasticity with relative preservation of cognition; optic atrophy and seizures develop late in the disease
i.fever or other metabolic stress can produce clinical deterioration
c.diagnostic testing
i.cerebrospinal fluid exhibits elevated glycine levels, although this is nonspecific for leukoencephalopathies or other diseases of the white matter
ii.neuroimaging
Dysmyelinating Disorder
115

4 Disorders of Myelination
(1)MRI demonstrates diffuse increases in T2 signal in the subcortical white matter with evidence of cystic degeneration that leaves intervening strands of relatively normal tissue
(2)magnetic resonance spectroscopy demonstrates reduced NAA, choline, and creatinine levels, and an increase in glucose and lactate levels; such changes are consistent with a replacement of tissue with cerebrospinal fluid
d.treatment: none specific
5.Megaloencephalic leukoencephalopathy with subcortical cysts/vacuolating leukoencephalopathy
a.pathophysiology: caused by autosomal recessive-inherited mutations of the MLC1 gene on chromosome 22, which has an unknown function
i.histology: oligodendrocytes exhibit abnormal compaction of the outermost layers of the myelin sheath
b.symptoms: macrocephaly with developmental delay particularly in motor function; mild mental retardation; seizures
c.diagnostic testing
i.neuroimaging: MRI demonstrates diffusely increased T2 signal in the subcortical white matter associated with cystic degeneration that is particularly pronounced in the temporal lobes
(1)cystic degeneration progresses with time; white matter signal changes are static
d.treatment: none specific
6.Aicardi-Goutieres syndrome
a.pathophysiology: caused by autosomal recessive-inheritance of the ASG1 locus, which causes an increased expression of interferonthereby causing demyelination and diffuse atrophy; appears to be associated with corticallybased infarctions as well
i.histology: areas of inflammatory demyelination involve cavitation and infiltration with gemistocytic macrophages that contain neutral fats; dystrophic calcifications in the white matter, basal ganglia, thalamus, cerebellar nuclei, and cerebral arteries also occur
(1)such abnormalities are commonly found after in utero infection with the toxoplasma, other viruses, rubella, cytomegalovirus, herpes simplex (TORCH) organisms
b.symptoms: acquired microcephaly with mental retardation and loss of developmental milestones; skin lesions (erythema, acrocyanosis)
c.diagnostic testing
i.cerebrospinal fluid demonstrates a chronic lymphocytosis and increased levels of interferon-
ii.neuroimaging demonstrates dystrophic calcifications, infarctions, and white matter loss
d.treatment: none specific
7.Adrenoleukodystrophy
a.pathophysiology: X-linked inherited mutation in the ALD gene, which encodes peroxisomal acyl-CoA synthetase that is involved in very long chain fatty acid (VLCFA) oxidization in the peroxisome; causes accumulation of VLCFA in blood and tissues
i.histology: perivascular lymphocyte infiltration and demyelination that spares only arcuate fibers; typically begins in the parietal and occipital lobes
b.symptoms
i.general symptoms: adrenal failure; testicular atrophy
ii.neurological symptoms, according to subtype
(1) |
childhood adrenoleukodystrophy: onset around 4 years of age but |
116 |
may develop in adolescence |
(a)hyperactivity, initially appearing as attention deficit hyperactivity disorder
(b)dementia progressing to vegetative state within 2–3 years
(c)gait ataxia
(d)vision and hearing loss
(e)seizures
(2)adult-onset adrenoleukodystrophy (rare): symptoms include slowly progressive dementia, and psychosis or mood disorders
(3)adrenomyeloneuropathy: onset around 30 years of age
(a)progressive spastic paraparesis with bowel and bladder incontinence, and sexual dysfunction
(b)sensorimotor neuropathy
(c)mild dementia, mood disorders
c.diagnostic testing
i.increased VLCFA levels in blood or amniotic fluid
ii.reduced urine androgens and glucocorticoids
iii.genetic testing to confirm carriers, 10% of whom have normal VLCFA levels
iv.neuroimaging: increased white matter T2 signal, particularly in posterior regions
d.treatment
i.glucocorticoid supplementation for adrenal failure
ii.dietary fatty acid supplements (“Lorenzo’s oil”) does not reduce disease progression, but it may be beneficial if initiated in the presymptomatic state
iii.bone marrow transplantation only when neurological symptoms are mild and there is enhancement of brain lesions on the MRI
8.Metachromatic leukodystrophy
a.pathophysiology: autosomal recessive inherited mutation of arylsulfatase A (Box 4.7), which converts sulfatide to galactocerebroside in the sphingolipid metabolic pathway; alternatively, may be caused by loss of the saposin B cofactor for arylsulfatase A
i.histology: diffuse demyelination of the brain sparing U-fibers and the peripheral nerves; accumulation of sulfated lipids in neurons of the central and peripheral nervous system causes a change from blue to brown or red {metachromasia} when stained with toluidine blue or cresyl violet, respectively
b.symptoms according to subtype
i.infantile metachromatic leukodystrophy: vision loss from optic atrophy; spastic ataxia with minimal extrapyramidal features; dementia; seizures
ii.juvenile metachromatic leukodystrophy: behavioral change and mental retardation; spastic ataxia with extrapyramidal features
iii.adult metachromatic leukodystrophy: behavioral change, psychosis, and dementia
c.diagnostic testing: increased urine sulfatides, confirmed by reduced arylsulfatase activity or saposin B levels
d.treatment: bone marrow transplantation, performed before the onset of significant neurological disease
9.Krabbe’s disease/globoid cell leukodystrophy
a.pathophysiology: autosomal recessive inherited deficiency of galactosylceramidase, which converts galactosylceramide into ceramide in the sphingolipid metabolic pathway; affects both central and peripheral nervous systems
Dysmyelinating Disorder
Box 4.7
Benign deficiency of arylsulfatase A occurs in 1% of the population {aminosalicylic acid (ASA) pseudodeficiency}.
117

4 Disorders of Myelination
i.histology: galactosylceramide attracts and is phagocytized by macrophages that store the indigestible PAS-positive material {globoid cells}; advanced disease exhibits extensive fibrillary astrocytosis in areas of demyelination
b.symptoms according to subtype
i.infantile Krabbe’s disease: begins with an exaggerated startle reflex, fevers, and behavioral regression, but progresses to involve atrophy and spastic weakness
ii.juvenile Krabbe’s disease: symptoms include vision loss, ataxia, behavioral regression, and spastic paraparesis
c.diagnostic testing: enzymatic assay to evaluate for deficiency of galactosylceramidase
d.treatment: bone marrow transplantation, performed before the onset of significant neurological disease
10.Giant cell neuropathy (see p. 221); phenylketonuria (see p. 279)
Appendix 4–1 |
Glucocorticoids* |
|
|
|
|
|
|
|
|
|
|
|
Mineralocorticoid |
|
|
Anti-inflammatory |
Prednisone dose |
potency |
|
Steroid |
potency (vs. prednisone) |
equivalents |
(vs. prednisone) |
Notes |
|
|
|
|
|
Prednisone |
– |
– |
– |
PO form only |
Prednisolone |
1.25 |
1 |
1 |
PO and IV forms |
Methylprednisolone |
1.25 |
0.8 |
0 |
|
Triamcinolone |
1.25 |
0.8 |
0 |
Can be administered by IM |
|
|
|
|
injection; high rate of steroid |
|
|
|
|
myopathy |
Dexamethasone |
7.5 |
0.15 |
0 |
|
Fludrocortisone |
2.5 |
0.4 |
830 |
Useful in orthostatic hy- |
|
|
|
|
potension and dysautonomia |
|
|
|
|
|
*Complications of glucocorticoid use: Psychosis, headache, from intracranial hypertension; peptic ulcers; hypertension from fluid retention (when using steroids with high mineralocorticoid potency); glucose intolerance and iatrogenic diabetes; opportunistic infections and poor wound healing (long-term use); cataracts, osteoporosis (long-term use); adrenal suppression: truncal obesity with muscle atrophy, hypertension, depression, hirsutism, amenorrhea, cutaneous striae.
Complications of glucocorticoid withdrawal: Nausea, weight loss; diffuse myalgias and arthralgias; fatigue; orthostatic hypotension. Abbreviations: IM, intramuscularly.
118