


5
Tumors of the Nervous System
Note: Significant diseases are indicated in bold and syndromes in italics.
I. Central Nervous System Tumors
A. Astrocytic Tumors
1.Fibrillary astrocytomas
a.tumor grades (Table 5–1) (Fig. 5–1): all exhibit nuclear atypia; can be cytologically defined as malignant according to MIB-1 antibody labeling of
the Ki-67 nuclear antigen, which is a protein of unknown function that is expressed only during mitosis (Box 5.1)
b.genetics (Fig. 5–2)
i.mutations of the p53 gene, which encodes a cell-cycle regulator that inhibits progression through G1 phase of mitosis; uncommon in primary glioblastoma
ii.mutations of the MDM2 gene, which encodes a protein that binds p53 protein
(1)mutations may cause overexpression of MDM2 protein, which leads to tumor formation by reducing levels of p53
(2)MDM2 protein also binds mutant p53, thus functional loss of MDM2 may allow abnormally active p53 to cause tumor formation
iii.mutations of the epidermal growth factor (EGF) receptor gene, which encodes the receptor for EGF and tumor growth factor (TGF)-
(1)mutations may cause overexpression of mutant EGF receptor that are constitutively active (i.e., do not require ligand binding)
(2)even without mutations, wild-type EGF receptor and related receptors (i.e., fibroblast growth factor receptor, vascular endothelial growth factor receptor) are commonly overexpressed in low-grade astrocytomas
iv.platelet-derived growth factor- (PDGF), which is involved in cell proliferation
Box 5.1
MIB-1-labeled Ki-67 ratio The number of labeled cells divided by the number of unlabeled cells
Histologically benign tumors with a good
prognosis have a low MIB-1-labeled Ki-67 ratio, if not undetectable MIB-1 labeling
Histologically benign tumors with a poor
prognosis have a high MIB-1-labeled Ki-67 ratio
Central Nervous System Tumors
Table 5–1 Grades of Astrocytomas
|
|
Pleomorphic |
Vascular |
|
|
Grade* |
Mitosis |
cells |
proliferation |
Necrosis |
Other features |
|
|
|
|
|
|
II: Diffuse |
No |
No |
No |
No |
– |
III: Anaplastic |
Yes |
No |
No |
No |
– |
IV: Glioblastoma |
Yes |
Yes |
Yes |
Yes |
Microvascular, |
multiforme |
|
|
|
|
glomeruli, drape-like |
|
|
|
|
|
cellularity {festoons}; |
|
|
|
|
|
“pseudopalisading” |
|
|
|
|
|
|
*WHO grade I tumor (pilocytic astrocytoma) is an uncommon astrocytic tumor (see p. 121).
119
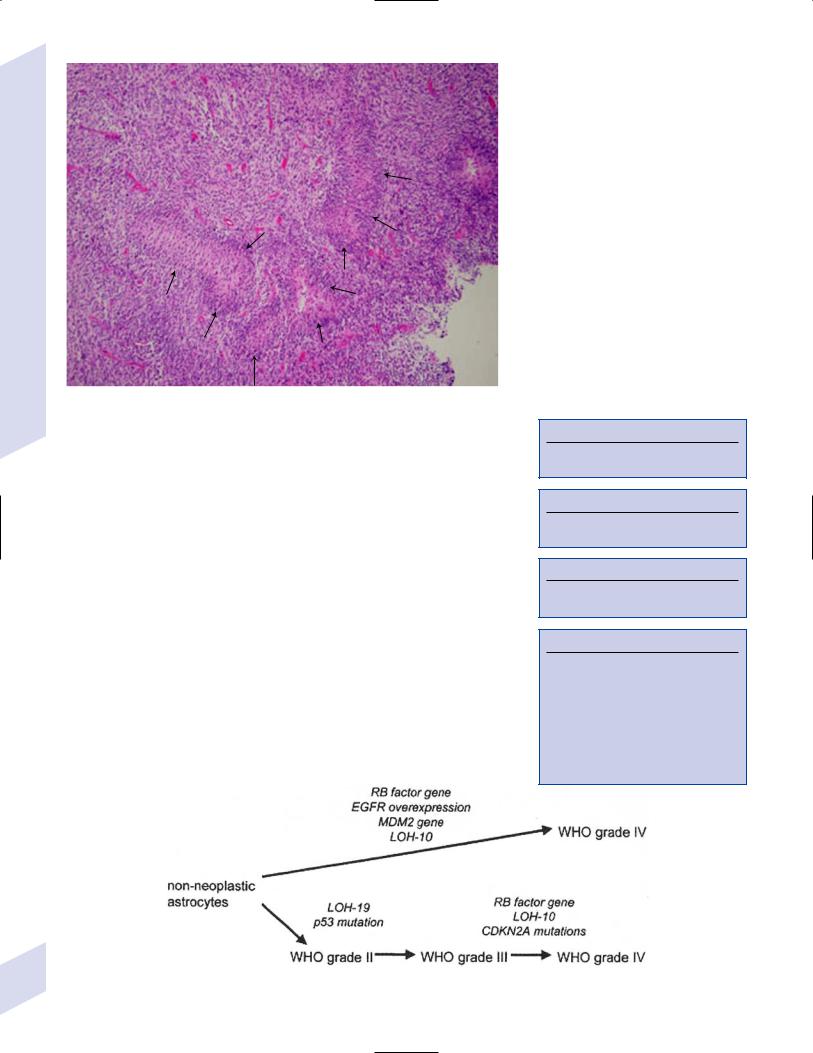
5 Tumors of the Nervous System
Figure 5–1 Glioblastoma multiforme. Arrows identify pseudopalisading and festoons.
v.mutations in the retinoblastoma (RB) factor gene (Box 5.2), which encodes an inhibitory binding protein for transcription factor E2F that promotes proliferation; mutations of RB factor allows inappropriate release of active E2F and progression through G1 phase of mitosis
vi.mutations of the CDKN2A (Box 5.3), which encode protein kinases that inhibit progression through the cell cycle by affecting the function of RB and p53
vii.loss of heterozygosity (LOH) on chromosomes 19 and 10: deletion of an unknown gene or genes on one pair of the chromosomes
(1)LOH 10 region includes
(a)the leucine-rich glioma-inactivated (LGI-1) protein (Box 5.4)
(b)PTEN, a tumor suppressor gene
c.tumor location: supratentorial infratentorial, rarely cerebellum or spinal cord; may be multifocal
i.tumors grow along white matter tracts and vasculature, occasionally disseminating into the CSF {seeding}, particularly with tumors in the cerebellum, corpus callosum, or along the ventricles (Box 5.5)
d.epidemiology: accounts for 60% of primary brain tumors in adults; only known risk factor is brain irradiation
Box 5.2
Retinoblastma factor gene is also mutated in cases of retinoblastoma
Box 5.3
CDKN2A also mutated in oligodendrogliomas.
Box 5.4
LGI-1 is mutated in autosomal dominant partial epilepsy with auditory features
Box 5.5
Tumors with High “Seeding” Potential
Glioblastoma
Medulloblastoma
Primitive neuroepidermal tumors
Ependymoma
Pineoblastoma
Germinoma
Choroid plexus papilloma
120 Figure 5–2 Genetic abnormalities of astrocytomas. (EGFR, epidermal growth factor receptor; LOH, loss of heterozygosity; RB, retinoblastoma.)
e.treatment
i.grade II/diffuse astrocytoma: observation, or treatment with irradiation or chemotherapy
ii.grade III/anaplastic astrocytoma: surgery irradiation chemotherapy
iii.grade IV/glioblastoma multiforme: surgery chemotherapy (temozolomide) irradiation
f.prognosis
i.grade II/diffuse astrocytoma: 5-year survival
ii.grade III/anaplastic astrocytoma: 2.5-year survival
iii.grade IV/glioblastoma multiforme: 1-year survival
2.Uncommon types of astrocytomas
a.juvenile pilocytic astrocytoma—grade I astrocytoma of the WHO classification
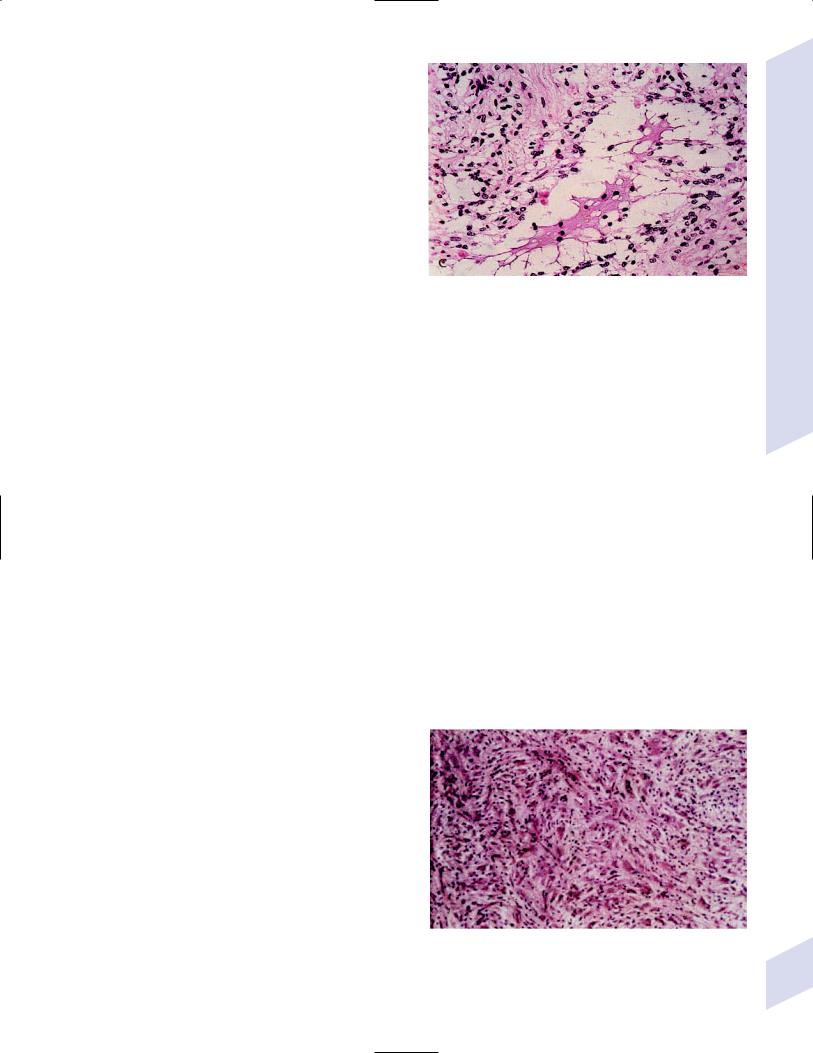
Figure 5–3 Juvenile pilocytic astrocytoma exhibits alternating compact and spongy areas that give a “floating” appearance to some of
i.histology: tumor exhibits a dense cellular areas the cells. Multinucleated giant cells are also seen. (From Keating R
filled with long bipolar astrocytes and numerous |
et al. Tumors of the Pediatric Central Nervous System. Stuttgart, |
Rosenthal fibers, which are interspersed with |
Germany: Georg Thieme; 2001:53, Fig. 5–6c. Reprinted by permission.) |
|
|
relatively cell-free regions (Fig. 5–3); also exhibits |
|
eosinophilic granular bodies |
|
(1)forms a large cystic mass when located in the cerebellum but a solid tumors when located elsewhere
(2)strongly associated with neurofibromatosis I
ii.tumor location: cerebellum hypothalamus, optic nerve
iii.epidemiology: common only in children
iv.treatment: surgery is usually curative; tumors in the optic-hypothalamic area respond to chemotherapy with carboplatin and vincristine
b.subependymal giant cell astrocytoma
i.histology: tumor typically consists of giant astrocytes mixed with differentiated neuron-like cells in a heavily calcified stroma (Fig. 5–4)
(1)no anaplastic potential although exhibits locally invasive growth
(2)typically grow along ependymal surface of lateral ventricles, may block cerebrospinal fluid flow causing hydrocephalus
(3)associated with
(a)soft-tissue sarcomas and breast cancer {Li-Fraumeni syndrome}
(b)multiple intestinal polyps {Turcot syndrome}
(c)phakomatoses: tuberous sclerosis, neurofibromatosis-1 (see Chapter 12)
ii.treatment: observation; surgery if the tumor grows or causes hydrocephalus
c.pleomorphic xanthoastrocytoma
i.histology: tumor exhibits pleomorphic astrocytes (therefore is glial fibrillary acidic protein- [GFAP] positive) filled with lipid vacuoles like adipocytes, but that are rarely mitotic; exhibits a dense reticulin network and a lymphocytic infiltrate, and can be cystic
(1)very benign growth (rarely degenerate into malignant tumors), and only are symptomatic when they cause seizures
ii.epidemiology: common in children
iii.tumor location: cortex (particularly temporal lobe), often extending into the subarachnoid space
iv.treatment: surgery is usually curative
Figure 5–4 Subependymal giant cell astrocytoma. (From Keating R et al. Tumors of the Pediatric Central Nervous System. Stuttgart, Germany: Georg Thieme; 2001:330, Fig. 23–3. Reprinted by permission.)
Central Nervous System Tumors
121
B. Oligodendrogliomas
1. Histology: composed of small, round cells that are GFAP-negative and that develop from oligodendrocytes
|
a. |
cells exhibit “fried egg” artifact and |
|
|
|
|
|
|
“chicken wire” vasculature after forma- |
|
|
|
|
|
|
lin fixation (Fig. 5–5), resembling the |
|
|
|
|
|
|
non-artifactual appearance of clear cell |
|
|
|
|
|
|
ependymomas or central neurocytomas |
|
|
|
|
System |
b. |
50% of oligodendrogliomas have a |
|
|
|
|
|
mixed histology with pronounced as- |
|
|
|
|
|
|
trocytoma features {oligoastrocytoma} |
|
|
|
|
|
Nervous |
|
that may ultimately degenerate into a |
|
|
|
|
|
malignant astrocytoma |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
i. both oligodendrocyte and astro- |
|
|
|
|
the |
|
cyte components show evidence of |
|
|
|
|
|
malignancy in oligoastrocytomas, |
|
|
|
|
|
of |
|
that is, astrocytes are not simply |
|
|
|
|
|
trapped in an oligodendroglioma |
|
|
|
|
|
Tumors5 |
d. |
|
|
|
|
|
tumor location: often developing at |
Figure 5–5 Oligodendroglioma histology. (Courtesy of Dr. C. Yamada) |
|||||
|
c. |
frequently are calcified, or have hem- |
||||
|
|
orrhagic or cystic components |
|
|
|
|
|
|
the gray–white junction and growing toward the cortex, particularly in the |
|
|
|
|
|
|
frontal lobe |
|
|
|
|
2. Tumor grades: histologically defined as low grade or anaplastic by the presence |
|
|
|
|||
|
of mitosis, pleomorphism, vascular proliferation, and necrosis, as per astrocy- |
|
|
|
||
|
tomas; cytologically defined as malignant according to MIB-1 antibody labeling, |
|
|
|
||
|
as per astrocytomas |
|
|
|
|
|
3. |
Genetics |
|
|
|
|
|
|
a. |
p53 gene mutations |
|
|
|
|
|
b. |
epidermal growth factor, platelet-derived growth factor, and vascular en- |
|
|
|
|
|
|
Box 5.6 |
|
|||
|
|
dothelial growth factor overexpression occurs without gene amplification |
|
|||
|
c. |
loss of heterogeneity (LOH): low-grade oligodendrogliomas have LOH on |
|
LOHs and CDKN2A mutations define two |
||
|
|
chromosomes 1 or 19; high-grade oligodendrogliomas also have LOH on |
|
major subtypes of oligodendrogliomas |
||
|
|
chromosomes 9 or 10 (Box 5.6) |
|
|
|
|
d.mutations in the CDKN2A gene, which encode the p16 protein cycle regulator (Box 5.7)
4.Tumor location: frontal temporal, parietal occipital cortex; location is proportionate to the amount of white matter in these areas
5.Epidemiology: bimodal peak incidences in children 8 years of age and adults 40 years of age
6.Diagnostic testing: Exhibits heterogeneous densities on neuroimaging, most of which are calcified (“chunky” or “popcorn-like” calcification) (Fig. 5–6)
7.Treatment
Box 5.7
CDK 4/6 genes are mutated in astrocytomas
a.surgery irradiation (may only observe low-grade oligodendrogliomas after surgery)
b.chemotherapy: highly sensitive to PCV treatment (procarbazine, lomustine [CCNU], vincristine) or temozolomide (Temodar), particularly rapidly enlarging tumors with ring enhancement
i.response to chemotherapy is not related to MIB-1-labeled Ki-67 ratio but it is proportionate to the amount of oligodendroglial-like cells in the tumor
ii.tumors with LOH 1 or 1 19 have greater chemotherapy responsiveness
122
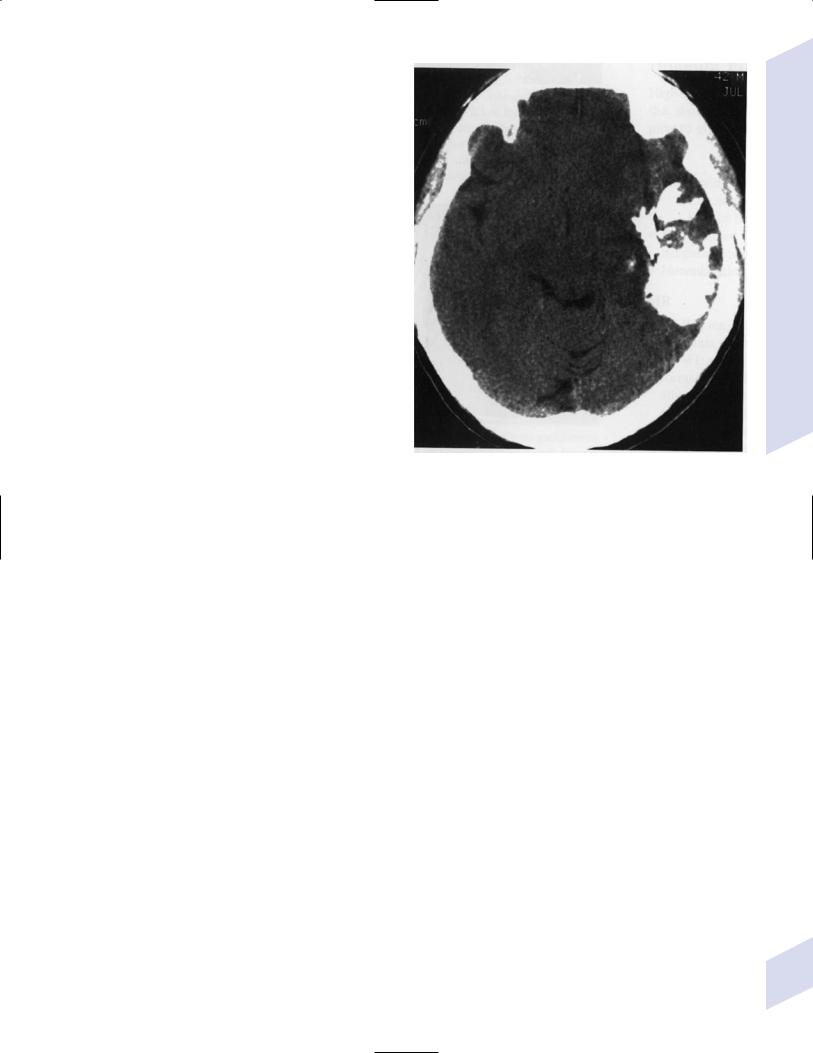
8.Prognosis: Tumors with LOH 1 or 1 19 are associated with longer survival times; tumors with CDKN2A gene deletions are associated with shorter survival times
a.histologically defined benign oligodendroglioma
i.MIB-1-labeled Ki-67 ratio 5% 80% 5-year survival
ii.MIB-1-labeled Ki-67 ratio 5% 25% 5-year survival
b.histologically defined anaplastic oligodendroglioma: 20% 5-year-survival overall
c.oligoastrocytoma: 60% 5-year survival
C. Gliomatosis Cerebri
1.Histology: a diffuse infiltration of brain parenchyma by neoplastic glial cells, generally occurring in multiple cerebral lobes but occasionally infratentorial
a.can develop from glial tumors (20%), but usually develops without preexisting tumor
b.exhibits oligodendroglial, astrocytic, or mixed subtypes
2.Symptoms: generally presents with seizures; may have a focal deficit (30%), dementia (30%), or headache (10%)
3.Diagnostic testing: often is nonenhancing on neuroimaging
4.Treatment: depends upon histology but typically treated with irradiation chemotherapy
Figure 5–6 Oligodendroglioma calcifications. (From Fischbein NJ et al. Teaching Atlas of Brain Imaging. Stuttgart, Germany: Georg Thieme; 2000:10, Fig. A. Reprinted by permission.)
5.Prognosis: 12-month-survival untreated, 2.5-year-survival treated
a. patients with oligodendroglial subtype have a significantly longer survival
D. Meningiomas
1.Histology (Fig. 5–7): the MIB-1 antibody labeling of the Ki-67 antigen can provide a cytological definition of malignancy, as with astrocytomas and oligodendrogliomas
a. WHO grade I subtypes
i.meningothelial meningioma—exhibits groups of tumor cells surrounded by thin collagenous septa that are difficult to visualize, giving the impression of a syncytium
ii.fibrous meningioma—appears similar to dense connective tissue
iii.transitional meningioma—exhibits clumps of cells surrounded by whorls of more cells {onion bulbs}; has elements of meningothelial and fibrous subtypes
iv.psammomatous meningioma—exhibits numerous laminated calcifications at the center of onion bulbs {psammoma bodies} that may become confluent and calcified
v.angiomatous meningioma—appears similar to the meningothelial subtype but has a pronounced vascular component
vi.secretory meningioma—focal epithelial differentiation forms a lumen containing accumulation of glucose polymers (i.e., periodic acid Schiffpositive)
vii.lymphocyte-rich meningioma—exhibits chronic inflammatory infiltrates
Central Nervous System Tumors
123
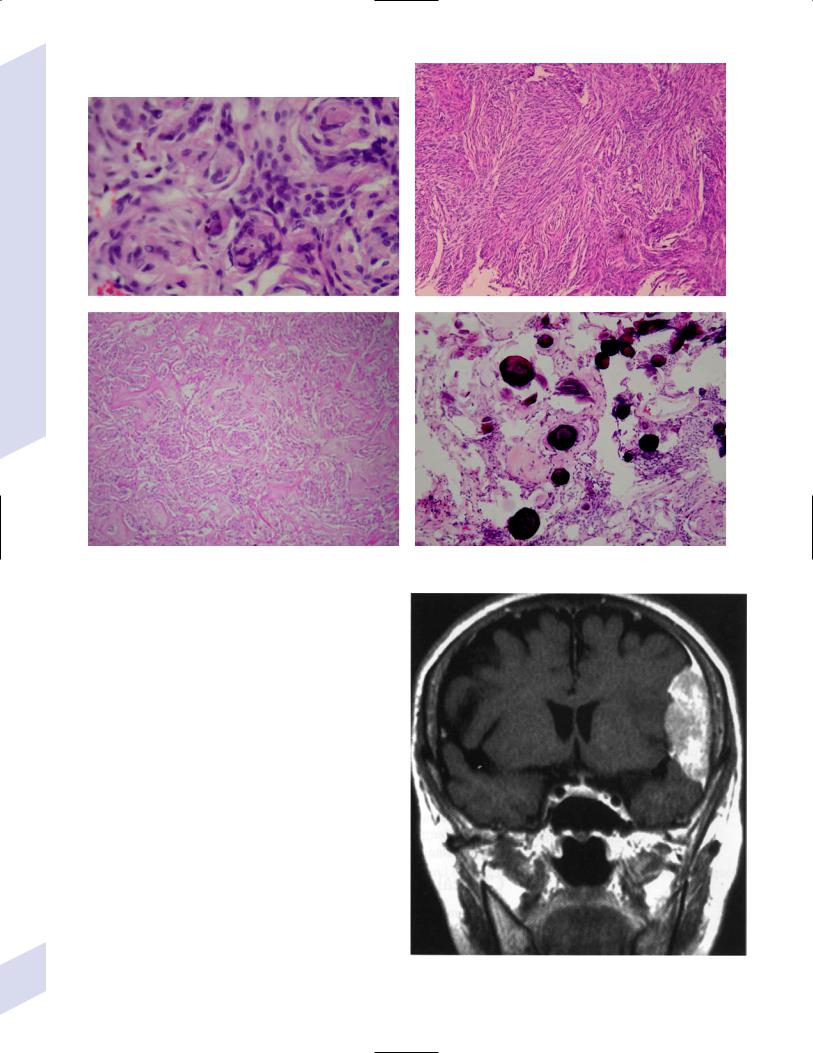
5 Tumors of the Nervous System
A
C
B
D
Figure 5–7 Types of meningiomas: meningothelial (A); fibrous (B); transitional, demonstrating onion bulbs (C); psammomatous, demonstrating psammoma bodies (D). (Courtesy of Dr. C. Yamada)
b.WHO grade II subtypes
i.clear cell meningioma—meningothelial-like but cells have clear, glycogen-rich cytoplasm
ii.chordoid meningioma—exhibits cartilagelike myxoid extracellular matrix
iii.atypical meningioma—exhibits increased mitotic activity, hypercellularity, and necrosis
c.WHO grade III subtype
i.anaplastic/malignant meningioma—exhibits high mitotic index ( 20 mitoses per highpowered field)
2.Tumor location: falx cerebri olfactory groove, sphenoid ridges, parasellar region
3.Genetics: risk of meningioma formation is increased by prior irradiation; exhibits direct or progressive development of aggressive tumors, as with astrocytomas
a.mutations in the neurofibromatosis 2 (NF-2) gene on chromosome 22, which encodes the merlin/ schwannomin protein that acts as a tumor suppressor: promotes development of low-grade meningiomas as part of neurofibromatosis type 2
b.numerous loss of chromosomal heterogeneities promote development of malignant meningiomas
1244. Diagnostic testing: neuroimaging demonstrates duralbased masses with tails (Fig. 5–8)
Figure 5–8 Meningioma with dural tails. (From Fischbein NJ et al. Teaching Atlas of Brain Imaging. Stuttgart, Germany: Georg Thieme; 2000:205, Fig. D. Reprinted by permission.)

a.growth may occur entirely along meningeal surface without forming a tumor mass {en plaque meningioma}
b.edema is prominent only with the secretory, atypical, or anaplastic/ malignant subtypes
5.Treatment: surgery; may add irradiation for atypical and anaplastic/malignant subtypes
6.Prognosis: 20% 20-year-recurrence rate following complete resection, considering all subtypes; however, up to 80% recurrence rate with anaplastic meningiomas
E. Pineal Tumors (Box 5.8)
1.General histology: All subtypes develop from pinealocytes that have neuroendocrine potential
a.pinealocyte-derived cells express the synaptic marker protein synaptophysin, neuron-specific enolase, tau protein, and serotonin
2.Pineocytoma
a.specific histology: tumors show lobular aggregates of well-differentiated pinealocytes arranged as rosettes (Fig. 5–9)
i.under electron microscopy, pinealocytes may exhibit annulate lamellae-like photoreceptor cells of the retina
b.treatment: surgery
c.prognosis: 85% 5-year survival
3.Pineoblastoma—a primitive neuroepidermal tumors (PNET; see below)
Box 5.8
Pediatric Pineal Region Tumors (In
Order of Occurrence)
Germinoma
Astrocytoma
Pineocytoma
Teratoma
Dermoid
Pineoblastoma
F. Primitive Neuroepidermal Tumors
1.General histology: A group of densely cellular tumors similar in appearance to Ewing’s sarcoma, rhabdomyosarcoma, and lymphoma {small blue-cell tumors}
2.Pineoblastoma
a.specific histology: similar to pineocytoma, except rosettes do not have a true lumen
i.associated with familial bilateral retinoblastoma {trilateral retinoblastoma syndrome}
Figure 5–9 Pineocytoma rosettes. (From Keating R et al. Tumors of the Pediatric Central Nervous System. Stuttgart, Germany: Georg Thieme; 2001:315, Fig. 22–8. Reprinted by permission.)
(1)frequently is metastatic via cerebrospinal fluid
b.genetics: mutations of the RB gene are common in cases of trilateral retinoblastoma (Box 5.9)
c.epidemiology: occurs in all ages, but mostly before 20 years of age
d.treatment: surgery irradiation, with prophylactic irradiation of the entire neuraxis for possible metastases
Box 5.9
RB factor gene is also mutated in some astrocytomas
e.prognosis: without metastasis, 60% 5-year survival
i.presence of RB mutations is associated with lower survival
3.Neuroblastoma
a.specific histology: tumor exhibits numerous mitotic figures and focal necrosis; cells form Homer-Wright rosettes and may secrete bioactive substances (e.g., vasoactive intestinal polypeptides [VIP], catecholamines)
i.tumor location: peripheral tumors in mediastinum, paravertebral sympathetic chain ganglia, and adrenal glands; tends to occur around the olfactory nerves when they develop inside the cranium, otherwise it is a metastasis to the brain (Box 5.10)
Box 5.10
Neuroblastoma is the most common brain metastasis in children
Central Nervous System Tumors
125

5 Tumors of the Nervous System
126
ii.diffuse peripheral metastases associated with a small primary tumor in children 1 year of age often spontaneously resolve; rarely neuroblastoma differentiates into a ganglioneuroma
b.genetics
i.segmental rearrangements of chromosomes 17 1 3, 11
ii.amplification of the MYCN gene, which encodes a nuclear transcription factor
(1)particularly common in metastatic neuroblastomas
c.specific symptoms (each occurs in 5% of cases)
i.paraparesis due to in- Figure 5–10 Medulloblastoma demonstrating Homer-Wright rosettes (e.g., arrowhead at
traspinal extension |
center). (From Bernstein M, Berger M. Neurooncology: The Essentials. Stuttgart, Germany: Georg |
|
Thieme; 2000:367, Fig. 364A. Reprinted by permission.) |
ii.diarrhea from VIP secretion
iii.diaphoresis, palpitations, and flushing from sudden catecholamine release
iv.paraneoplastic syndromes: cerebellar degeneration, encephalomyelitis, and/or peripheral neuropathy related to anti-Hu antibodies; opsoclonusmyoclonus syndrome related to anti-Ri antibodies
v.Horner’s syndrome with involvement of lower cervical ganglia
d.prognosis
i.expression of nerve growth factor/trk receptors, polyploidy (versus diploidy), and tumor calcifications are good prognostic signs
(1)nerve growth factor receptors are normally expressed by neural crest cells that are developing into monoamine secreting cells (e.g., adrenal medulla); normal loss of trophic stimulation during development may cause apoptosis and spontaneous tumor regression
ii.presence of MYCN amplification is a poor prognostic sign in all tumor grades
4.Medulloblastoma—The most common PNET
a.specific histology: cells are GFAP-negative, hyperchromatic, and highly
mitotic; tumors exhibit Homer-Wright rosettes with no lumen (Fig. 5–10), and necrosis with pseudopalisading similar to glioblastoma multiforme
i.develops from embryologic cells of the external germinal layer of the cerebellum
ii.adult tumors associated with Turcot’s syndrome and Li-Fraumeni syndrome (see p. 121)
b.tumor location: roof of 4th ventricle; 75% are midline, and 25% extend into the cerebellar parenchyma; frequently metastasize by direct extension through the subarachnoid space
c.epidemiology: mostly occur in children and adolescents; smaller adult peak incidence in adults 30 to 40 years of age
d.diagnostic testing: requires neuroimaging of the spine and cerebrospinal fluid analysis
e.treatment: surgery chemotherapy (CCNU vincristine) irradiation

Figure 5–11 Ependymoma pseudorosettes. (From Hirano A. Color Atlas of Pathology of the Nervous System, 2nd Ed. Tokyo/New York: Igaku-Shoin Press; 1988:110, Fig. 265. Reprinted by permission.)
G. Ependymoma
1.General histology: all subtypes of ependymomas develop from the ependyma and have thin layers of well-differentiated cuboidal or columnar GFAPpositive epithelium (Box 5.11) and spherical or rod-shaped intracytoplasmic inclusions {blepharoplasts}; epithelium forms perivascular pseudorosettes (Fig. 5–11)
a.blepharoplasts are analogous to the basal bodies of cilia and are located at apical portion of cells in ependymal rosettes
b.may be low grade or anaplastic
2.Subtypes
a.classic ependymoma—exhibits perivascular pseudorosettes ependymal rosettes
b.clear cell ependymoma—cells exhibit prominent perinuclear halos resembling “fried egg” artifact of the oligodendroglioma and the central neurocytoma
c.papillary ependymoma—resembles choroid plexus papilloma
d.myxopapillary ependymoma—exhibits microcysts, mucin cuffs around blood vessels, and a highly spindled pilocytic glial background
i. always located in the filum terminale; growth is benign
3.Tumor location: 60% are infratentorial, 90% of which are located in the 4th ventricle growing from the dorsal brainstem surface
a.infratentorial ependymoma is largely intraventricular, but in the 4th ventricle (Box 5.12) they often grow through the foramen of Luschka and Magendie into the subarachnoid space; supratentorial ependymomas can grow into the brain parenchyma
4.Epidemiology: bimodal distribution in children (1 to 5 years of age) and adults (20 to 30 years of age)
5.Diagnostic testing: requires spinal imaging and cerebrospinal fluid analysis to rule out drop metastases
a.CT: hypo-/isodense mass with heterogenous enhancement; 50% have “chunky” or “popcorn” calcifications like oligodendrogliomas
Central Nervous System Tumors
Box 5.11
GFAP-Positive Tumors
Astrocytomas
Oligodendrogliomas
Ependymoma
Primitive neuroepidermal tumors (PNETs)
Gangliocytomas
Hemangioblastomas
Box 5.12
The growth pattern for the infratentorial ependymoma is the same as in choroid plexus papilloma.
127
5 Tumors of the Nervous System
128
Table 5–2 Choroid Plexus Papilloma versus Papillary Ependymoma
Choroid plexus papilloma |
Papillary ependymoma |
|
|
Organized cells |
Scattered cells |
Transthyretin-positive |
Transthyretin-negative |
GFAP-negative |
GFAP-positive |
Homogeneous enhancement |
Heterogenous enhancement |
Little stroma |
Abundant stroma |
|
|
Abbreviations: GFAP, glial fibrillary acidic protein. |
|
b.MRI: hypo-/isointense on T1 with heterogenous enhancement; hyperintense on T2 (Box 5.13)
6.Treatment: surgery irradiation
7.Prognosis: 45% 5-year survival
H. Choroid Plexus Papilloma
1.Histology: Tumors arise from the epithelium of the choroid plexus and exhibit a modified ependymal covering with a heavily vascularized stromal core that contains nests of transthyretin/prealbumin-positive cells
a.15% undergo malignant degeneration, but even then they do not invade brain parenchyma
b.disorganization and crowding of the columnar cells in the fibrovascular stroma distinguishes choroid plexus papillomas from papillary ependymomas (Table 5–2)
2.Tumor location: in children, the atria of lateral ventricle; in adults, the 4th ventricle
a.metastasize via cerebrospinal fluid; may grow through the foramen of Luschka and Magendie into the subarachnoid space like ependymomas
3.Epidemiology: most common in children 2 years old
4.Diagnostic testing
a.CT: hyperdense, homogenous enhancement, frond-like morphology i. 25% are calcified; internal hemorrhage is common
b.MRI: homogenous enhancement with flow voids; often associated with hydrocephalus
5.Treatment: surgical, possibly with irradiation and chemotherapy if it proves carcinomatous
I. Colloid Cyst
1.Histology: a cystic cavity lined by cuboidal or columnar epithelial cells that rests on a thin capsule composed of collagen and fibroblasts; epithelial cells are uniformly benign
a.tumor enlarges by accumulation of epithelial secretions that form a gelatinous material ( “crank case oil” of craniopharyngioma or Rathke’s pouch cyst)
2.Tumor location: anterior portion of the 3rd ventricle between the columns of the fornix, and attached to the ventricular roof and choroid plexus (Box 5.14)
3.Symptoms: often causes transient neurological symptoms; may even be fatal from acute hydrocephalus
4.Diagnostic testing
a.CT: cyst usually is hyperdense, not hypodense
b.MRI: cyst usually is low on T2 signal, not high; may exhibit hydrocephalus
Box 5.13
In comparison with the ependymoma, choroid plexus papilloma homogeneously enhances and medulloblastoma is denser and rarely calcified.
Box 5.14
Tumors in the ventricles
Lateral ventricle—Subependymoma (frontal); choroid plexus papilloma (atrium)
Third ventricle—Colloid cyst, central neurocytoma
Fourth ventricle—Ependymoma; ependymoblastoma (brainstem side); medulloblastoma (cerebellum side)

5.Treatment: surgery; stereotactic aspiration or cerebrospinal fluid shunting is also sufficient
J. Central Neurocytoma
1.Histology: tumors exhibit densely packed clear cells, similar to oligodendroglioma and clear cell ependymoma; has immunohistochemical and ultrastructural features suggesting neuronal differentiation (e.g., cells are synaptophysin-positive, GFAP-negative)
2.Tumor location: arise from the septum pellucidum and 3rd ventricle, often filling lateral ventricles and obstructing cerebrospinal fluid outflow through foramen of Monroe; rarely involves periventricular brain parenchyma, and symptoms are usually due to hydrocephalus
a. despite its name, tumor is not always located along midline
3.Diagnostic testing: neuroimaging demonstrates solid intraventricular tumor; cysts and calcification are common
4.Treatment: surgery
K. Mixed Neuronal-Glial Tumors
1.General symptoms: a common cause of medically refractory complex partial seizures in children and young adults
2.Dysembryoplastic neuroepithelial tumor
a.histology: tumors are composed of neuronal elements with bundled axons that project into the subcortical white matter, and glial elements that resemble low-grade oligodendrogliomas; may actually be a type of hamartoma
b.tumor location: mesial temporal lobe other cortical areas
c.treatment: surgery is curative
3.Gangliocytoma/ganglioglioma
a.histology: tumors contain both neural and glial elements; commonly has cyst formation and calcifications
i.neuronal component is large, mature multipolar neurons arranged in groups
(1)neuronal component always has dysplastic features but is nonproliferative
(2)neurons are synaptophysin-positive
ii.glial component is astrocytic in nature
(1)if glial components are neoplastic, the tumor is a ganglioglioma; otherwise it is a gangliocytoma
b.tumor location: temporal, occipital, and frontal lobes
c.epidemiology: common in children and young adults
d.treatment: surgery; irradiation is necessary for nonresectable tumors or for malignant tumors
e.prognosis: determined by neoplastic activity of the glial component
L. Germinoma
1.Pathophysiology: a type of germ cell tumor that is similar to testicular seminomas and ovarian dysgerminomas; likely develops from germ cells that abnormally migrate from the yolk sack of the embryo and survive in certain regions of the brain
a.genetics: 90% of cases are associated with abnormalities on chromosome 12; also associated with polyploidy of chromosome X or 21
i.increased risk of germ cell tumors occurs in Down’s and Klinefelter’s syndromes
Central Nervous System Tumors
129

5 Tumors of the Nervous System
2.Histology: composed of large regularly shaped cells often with interspersed lymphocytes; exhibits reactivity for alkaline phosphatase
3.Tumor location: midline structures (e.g., pineal gland or suprasellar region); metastatic in 15%
4.Diagnostic testing: cerebrospinal fluid often exhibits increased alkaline phosphatase levels; diagnosis must be established with tissue biopsy
5.Treatment: irradiation surgery
6.Prognosis: 95% 5-year survival
M. Hemangioblastoma
1.Histology: Cystic, noninfiltrative masses with sharp demarcation, formed from capillary overgrowth
a.occasionally express erythropoietin-like hormone from mast cells, causing a pure erythrocytosis (i.e., a secondary polycythemia)
b.tumor location: cerebellar vermis hemisphere
i. occurrence in spinal cord is associated with syringomyelia
c.20% of cases have autosomal dominant-inherited hemangioblastomas of the brain and retina, and visceral mesoderm tumors {von Hippel-Lindau disease}
2.Treatment: surgery
N. Chordoma
1.Histology: composed of groups of epithelial cells embedded in a mucinous or cartilaginous matrix that develops from notochord remnants that did not appropriately form bone
a.becomes symptomatic in adulthood, usually with pain from local bone destruction
2.Tumor location: sacrum sphenoid bone, clivus vertebrae
3.Treatment: surgery irradiation, particularly focused/stereotactic irradiation
4.Prognosis: high recurrence rate
O. Metastasis
1.Epidemiology: 25% risk of developing brain metastasis given all patients with a peripheral primary tumor
a.20% are asymptomatic at time of diagnosis; 75% involve multiple metastases
b.primary tumors below the diaphragm often present as posterior fossa metastases
2.Pathophysiology: metastasis most commonly occurs by hematogenous spread (conversely, brain abscesses can develop by either hematogenous spread or direct extension)
a.location of metastases is proportionate to blood flow, so 80% locate in the cortex in watershed areas that have multiple vascular supplies and relatively slow blood flow, or at the gray–white matter junction
b.development of dedicated tumor vasculature is necessary for growth 1 mm
3.Subtypes
a.brain: lung (50%); breast (15%); skin/melanoma (10%); unknown (10%)
b.epidural spine: breast (20%); lung (15%); prostate (10%); lymphoma (10%)
130
4.Treatment a. medical
i.glucocorticoids for relief of edema; antiepileptics only in patients with seizures
ii.chemotherapy: effective if the primary tumor is sensitive
iii.whole-brain irradiation: 30 Gray irradiation divided over 10 sessions improves survival, regardless of the tumor type
(1)no evidence of benefit of high-dose or highly fractionated irradiation schedules
iv.stereotactic radiosurgery: fewer radiation-related complications than whole-brain irradiation but effective only for metastases 3 cm; can be used for three metastases
b.surgical: excision of single metastases reduces the likelihood of death from a neurological cause but does not improve overall survival
i.anecdotal evidence suggests that resection of two to three metastases may be beneficial
P.Primary Spinal Cord Tumors
1.Tumor location: distinguishing intraparenchymal, extraparenchymal–intradural, and extradural tumors
a. imaging characteristics by location
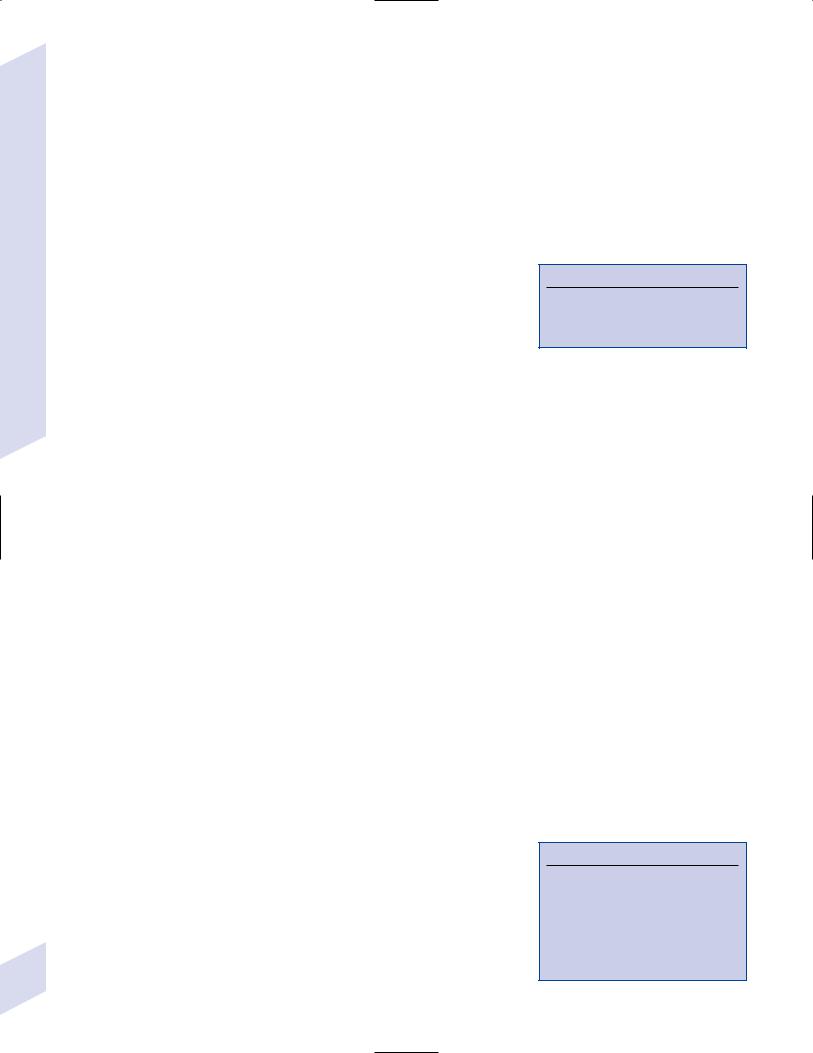
Figure 5–12 Intraparenchymal spinal cord tumor. Note ballooning of the spinal cord parenchyma and the lapering of the CSF space around it. (From McKhann GM et al. Q&A Color Review of Clinical Neurology and Neurosurgery. Stuttgart, Germany: Georg Thieme; 2003:139, Fig. 2. Reprinted by permission.)
i.intraparenchymal tumor causes widening of the cord (must be demonstrated in multiple views) with T2 signal abnormalities in the cord (Fig. 5–12); may or may not contrast enhance, depending upon the tumor type
ii.extraparenchymal–intradural tumor causes widening of the ipsilateral subarachnoid gutter with cord displacement (“capping defect” appearance); contrast enhances
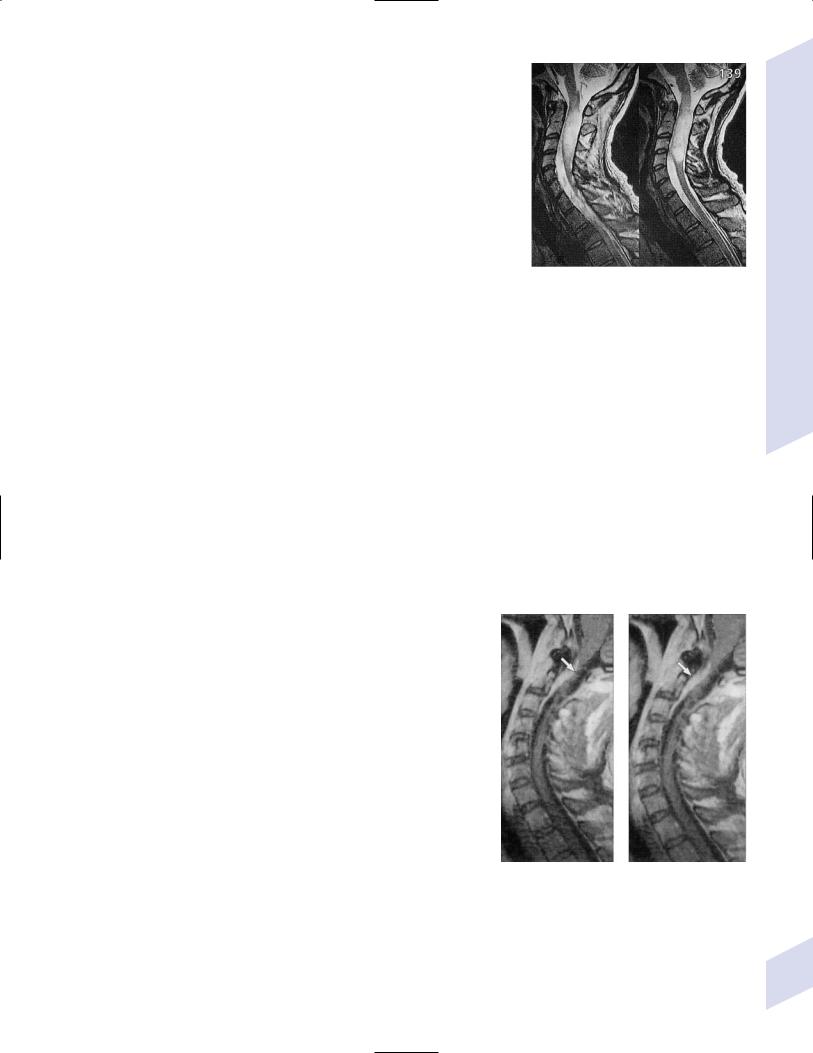
iii.extradural tumor causes thinning of the dural sac (“paint brush” appearance) (Fig. 5–13); tumors also cause cord edema and can occur at multiple levels
2.Types of tumors
a.intraparenchymal (5%): low-grade astrocytoma, ependymoma
b.extraparenchymal–intradural (40%): meningioma, neurofibroma, lipoma
c.extradural (55%): metastasis (breast, prostate, lymphoma), chordoma, bone tumors (multiple myeloma)
Q. Carcinomatous Meningitis
1.Pathophysiology: occurs in 8% of all cancer patients on autopsy, although often it is microscopic and was asymptomatic during life
a.causative metastatic tumors: breast (35%), lung (20%), melanoma (20%), gastrointestinal (10%), lymphoma/leukemia (10%), unknown (5%)
b.causative primary brain tumors: PNETs (i.e., medulloblastoma, ependymoblastoma), choroid plexus papilloma, primary central nervous system (CNS) lymphoma
2.Symptoms
a.polyradiculopathy (80%)
b.multiple cranial neuropathies (65%)
Figure 5–13 Extradural spinal tumor. Note compression of the spinal cord parenchyma. (From McKhann GM et al. Q&A Color Review of Clinical Neurology and Neurosurgery. Stuttgart, Germany: Georg Thieme; 2003:160, Fig. 139. Reprinted by permission.)
Central Nervous System Tumors
131
|
|
c. |
multifocal neurological injury (50%) |
|
|
|
d. |
endocrine abnormalities from hypothalamic involvement (5%) |
|
|
|
e. |
headache, nausea, and poorly defined visual changes |
|
|
|
f. |
stroke, from invasion of Virchow-Robins spaces by tumor |
|
|
3. |
Diagnostic testing |
||
|
|
a. |
lumbar puncture shows mildly increased pressure with Lundberg waves |
|
|
|
|
(see p. 35), mild pleocytosis (occasionally eosinophilic with leukemias and |
|
|
|
|
lymphomas), elevated protein, and decreased glucose ( 10 g/dL is nearly |
|
System |
|
|
pathognomonic); rarely xanthochromia or frank hemorrhage with carci- |
|
|
|
nomatous meningitis from melanoma |
||
|
|
i. repeat lumbar puncture for large-volume cytological analysis at least |
||
|
|
twice before accepting a negative result ( 90% accuracy) |
||
Nervous |
|
b. |
MRI: nonspecific findings of dural thickening and enhancement on contrast |
|
|
|
|||
|
|
|
T1 images; may involve nerve root enhancement, filling of the subarach- |
|
|
|
|
noid space, or small tumor masses or clumping of nerve roots in the cauda |
|
the |
|
|
equina because of tumor-induced fusion |
|
4. |
Treatment: irradiation to symptomatic areas intrathecal chemotherapy |
|||
of |
||||
|
(methotrexate, cytarabine, thiotepa) |
|||
Tumors |
|
|||
|
a. |
may require ventricular access device (e.g., Ommaya reservoir) for in- |
||
|
|
|||
|
|
|
trathecal chemotherapy administration or ventriculoperitoneal shunt for |
|
5 |
|
|
hydrocephalus |
|
|
b. |
most intraventricular drugs are rapidly eliminated by bulk flow and poorly |
||
|
|
|||
penetrate tissue
5. Prognosis: treatment nonresponders 2-month-mean survival; treatment responders 6-month-mean survival
R. General Medical Therapies of Brain Tumors
1.Glucocorticoids: reduces tumor edema within hours of administration, which can improve focal neurological symptoms as well as reduce intracranial pressure
a.should be used prophylactically for 2 weeks after any irradiation
b.cancer patients should simultaneously receive prophylaxis for Pneumocystis carinii (i.e., with Bactrim or dapsone) when on prolonged glucocorticoid treatment for 3 weeks
2.Anticonvulsants: no evidence for prophylactic use in patients who have not had a seizure
3.Anticoagulation: not contraindicated irrespective of type of tumor and its hemorrhage risk
a.no established benefit of using inferior vena cava filter instead of anticoagulants
II. Pituitary Tumors (Box 5.15)
A. Adenomas
1.Pathophysiology: develop in anterior pituitary may be hormone-secreting or nonsecreting
a.poorly differentiated tumors rarely secrete multiple hormones
b.most adenomas are a hypertrophic response to prolonged stimulation by hypothalamic-releasing hormones
c.nonsecreting macroadenomas are incidental findings in 20% of the population
d.posterior pituitary tumors tend to be astrocytomas
2.Histology: often exhibit evidence of small hemorrhages or amyloid deposits from collection of excess hormone protein (an endocrine amyloidosis)
a.rarely exhibits cellular features of malignancy but adenomas reliably invade
132 |
bone and dura |
Box 5.15
Empty sella syndrome:
Chronic compression of the pituitary by a protrusion of the subarachnoid space forces the pituitary into posterior sella
Occurs mostly in obese women; incidental finding in 5%
Causes headache, visual field cuts, pituitary failure only in children

3.Genetics
a.mutations of the GSPT1 gene, which encodes for stimulatory G-protein that limits growth hormone-releasing hormone (GHRH) stimulation of pituicytes; occurs in 40% of growth hormone- (GH) secreting adenomas
b.many aggressive adenomas exhibit loss of chromosomal heterogeneity at several sites
4.Epidemiology: common in adults 30 to 50 years of age
5.Symptoms
a.bitemporal hemianopsia
b.microadenoma ( 1 cm) or pituitary hypertrophy: symptoms caused by hormone secretion (in order of occurrence)
i.prolactin: causes amenorrhea and galactorrhea in women; impotence in men
ii.GH: causes
(1)gigantism in children; acromegaly in adults
(2)nerve entrapment
(3)hyperthyroidism secondary to goiter formation
(4)heart failure
iii.adrenocorticotropic hormone (ACTH): causes Cushing’s syndrome often with hyperpigmentation due to coincident secretion of - melanocyte stimulating hormone
c.macroadenomas ( 1 cm): symptoms are from hormone secretion and mass effect
i.mass effect causes headache, bitemporal hemianopia, and/or multiple cranial nerve palsies from involvement of the cavernous sinuses
ii.hormone effects (75% of macroadenomas)
(1)follicle stimulating hormone (FSH)/luteinizing hormone (LH): causes decreased libido, usually identified in men
(2)thyroid stimulating hormone (TSH): causes hyperthyroidism
d.pituitary apoplexy—caused by hemorrhage or infarction into a pituitary adenoma; symptoms include acute-onset headache, ophthalmoplegia, bilateral vision loss, and somnolence progressing to coma
6.Treatment
a.bromocriptine for prolactin-secreting tumors
b.surgery irradiation for all other types
c.propylthiouracil or methimazole can be used temporarily to control hyperthyroidism
Pituitary Tumors
B. Craniopharyngioma
1.Pathophysiology: In adults, likely develops after dedifferentiation of pituicytes into squamous epithelial-like cells that then form a tumor; in children, may be derived directly from Rathke’s pouch (Box 5.16)
a.usually have a cystic component filled with cholesterol-rich fluid {crank case oil}
2.Tumor location: develop from the top of the pituitary near the optic chiasm, and often extend into the 3rd ventricle
3.Symptoms: obstructive hydrocephalus with 3rd ventricle extension (rare in adult cases); variable visual field cuts; endocrine dysfunction; chemical meningitis with cyst rupture
4.Treatment: complete surgical removal
5.Prognosis: high operative morbidity and mortality with hypothalamic damage; significant recurrence tendency, which requires irradiation as well as repeat surgery
Box 5.16
Rathke’s pouch also gives rise to the nonneoplastic Rathke’s pouch cyst, which can be treated just by drainage.
133

5 Tumors of the Nervous System
Figure 5–14 Schwannoma. Transition between the cellular Antoni A area and the looselyarranged Antoni B area. Palisading Schwann cells with elongated nuclei characteristic of a Verocay body (arrow). (From Bernstein M, Berger M. Neurooncology: The Essentials. Stuttgart, Germany: Georg Thieme; 2000:28, Fig. 2–10. Reprinted by permission.)
III. Peripheral Nervous System Tumors
A. Schwann Cell Tumors
1.Schwannoma
a.histology: neoplastic Schwann cells are surrounded by small amounts of connective tissue that is encapsulated and largely external to the nerve
i.tumor exhibits different areas that are highly cellular (Antoni type A) or loose myxoid connective tissue (Antoni type B) (Fig. 5–14)
(1)Verocay body: cellular palisades that occur in Antoni type A areas
ii.minimal risk of malignant transformation
iii.exhibits intense staining for the astrocyte marker protein S-100 throughout the tumor
b.tumor location: most occur on peripheral nerves but 8% are intracranial on cranial nerves at the cerebellopontine angle (i.e., the acoustic neuroma, which actually develops from the vestibular portion of CN VIII)
i.schwannomatosis: condition of multiple schwannomas without achieving the diagnostic criteria for neurofibromatosis
c.symptoms: usually are asymptomatic swellings, although pressure on the tumor causes pain; rarely cause motor or sensory dysfunction in the affected nerve despite stretching of the nerve fibers around the outside of the tumor
d.treatment: surgical resection of symptomatic tumors
2.Neurofibroma
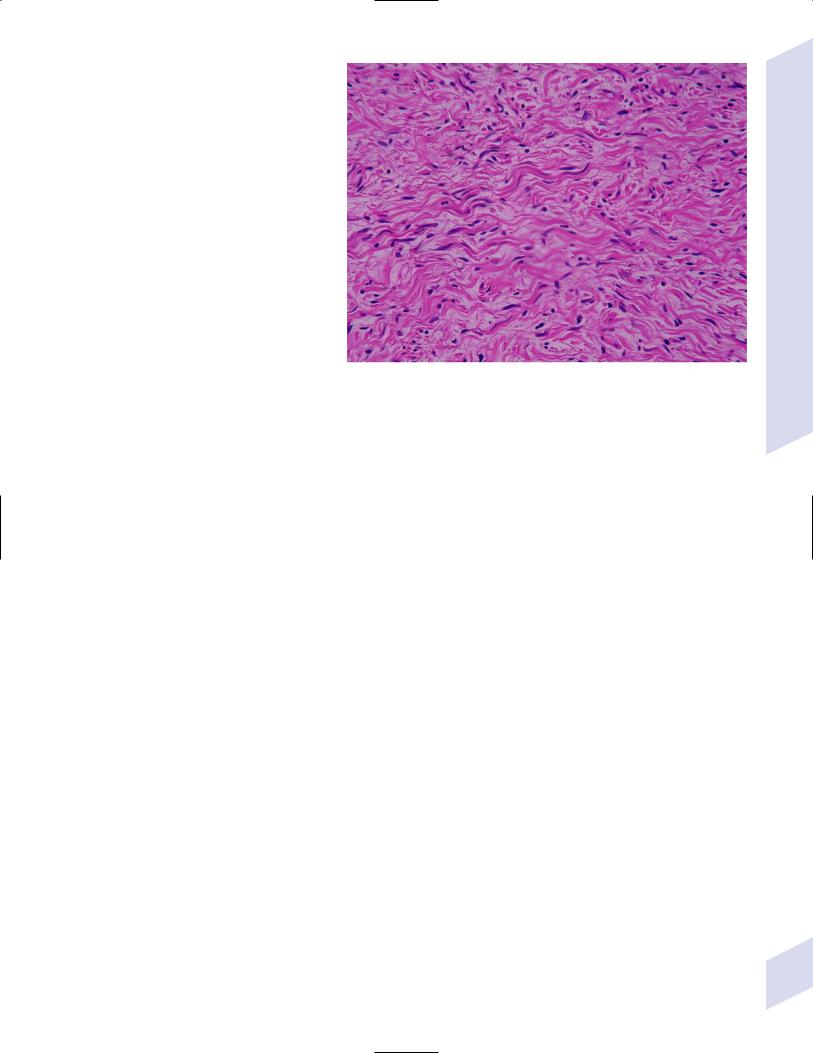
a.histology: tumors are composed neoplastic Schwann cells encased in a large amount of connective tissue similar to Antoni type B area of schwannoma (Fig. 5–15); tumors grow into the nerve
i.rarely undergoes anaplastic transformation, and then usually only after irradiation
ii.exhibits patchy staining for the astrocyte marker protein S-100
b.tumor location: cutaneous branches of peripheral nerves
c.subtypes
134 |
i. solitary (90%) |
ii.multiple: tumors usually are large and involve deep nerves
iii.plexiform: solitary neurofibromas that grow along the length of the nerves causing fusiform enlargements; associated mostly with neurofibromatosis
iv.diffuse: associated with neurofibromatosis
d.symptoms: painful swelling, but pressure on tumor does not exacerbate pain; motor and/or sensory dysfunction in distribution of the nerve
e.treatment: difficult to excise surgically
3.Malignant peripheral nerve sheath tumor
a.pathophysiology: generally develop de novo or rarely from degeneration of a neurofibroma
i.4% prevalence in neurofibromatosis
ii.target large nerves (e.g., sciatic)
and the plexuses; metastasizes to
lung, liver, and bone Figure 5–15 Neurofibroma demonstrating intrinsic nerve fascicles. (Courtesy of
Dr. Yamada)
b. histology: hypercellular tumors with mitotic figures and areas of necrosis;
tumors form fusiform masses that invade surrounding tissues
i. may express S-100 protein, which is commonly considered an astrocyte marker
c. symptoms: pain, swelling, motor and sensory dysfunction of involved nerve
d. treatment: irradiation surgical removal involving limb amputation or en-bloc resection
e. prognosis: 40% 5-year survival
Paraneoplastic Syndromes
IV. Paraneoplastic Syndromes
1.Epidemiology: develops in 0.01% of cancer patients, except
a.Lambert-Eaton myasthenia 3% of small-cell lung cancer (SCLC)
b.myasthenia gravis 15% of thymomas
2.General pathophysiology
a.the paraneoplastic syndrome is often the presenting feature of the tumor by months or even years because the autoantibodies that cause it are also effective against tumor antigens and thereby limiting tumor growth
i.tumors in patients with paraneoplastic syndromes are better recognized by the immune system (e.g., infiltrated with leukocytes), in comparison with similar tumors in patients without paraneoplastic syndromes
b.development of a paraneoplastic syndromes may indicate tumor recurrence after an effective treatment
3.General diagnostic testing: evaluation for primary tumor is best done with PET scanning
a.serum antibodies (see below)
b.lumbar puncture demonstrates mild T lymphocyte pleocytosis early in course
4.General treatment: eliminations of causative tumor may improve paraneoplastic syndrome in 30%, but treatment of paraneoplastic syndrome should be initiated immediately as they may limit further neurological injury
a.immunosuppression: glucocorticoids, cyclophosphamide, tacrolimus, mycophenolate
b. IVIg plasmapheresis: particularly effective in myasthenia gravis, Lambert- |
135 |
Eaton myasthenia, and stiff-man syndrome |
|

5 Tumors of the Nervous System
136
Table 5–3 Antibody-Mediated Paraneoplastic Syndromes*
Syndrome |
Antibodies |
Associated tumor |
|
|
|
Myasthenia gravis (see Chapter 10) |
Nicotinic ACh receptor |
Thymoma |
|
Striated muscle |
|
|
Titan |
|
|
Ryanodine calcium channel |
|
|
|
|
Isaac syndrome/neuromyotonia (see Chapter 10) |
Nicotinic ACh receptor |
SCLC, thymoma, lymphoma |
|
Voltage-gated potassium channels |
|
|
|
|
Lambert-Eaton myasthenia (see Chapter 10) |
P/Q-type V-gated calcium channel |
SCLC |
|
PCA-2 |
|
|
|
|
Cerebellar degeneration |
Hu |
SCLC, neuroblastoma, prostate |
|
Yo/PCA-1 |
SCLC, ovary, breast |
|
PCA-2 |
SCLC |
|
Ma-1 |
SCLC |
|
CV-2 |
SCLC, thymoma |
|
Tr |
Hodgkin’s disease |
|
Metabotropic Glu receptor type 1 |
Hodgkin’s disease |
|
|
|
Brainstem encephalitis |
Ma-1 |
SCLC |
|
Ta/Ma-2 |
Testicle |
|
|
|
Limbic encephalitis |
Voltage-gated potassium channels |
SCLC |
|
|
|
Encephalomyelitis |
Amphiphysin |
SCLC, breast |
|
Hu |
SCLC, neuroblastoma, prostate |
|
PCA-2 |
SCLC |
|
CV-2 |
SCLC |
|
|
|
Neuropathy |
MAG |
Waldenstrom’s macroglobulinemia |
|
Hu |
SCLC, neuroblastoma, prostate |
|
CV-2 |
SCLC |
|
|
|
Retinopathy |
Antiretinal |
Melanoma |
|
|
|
Stiff-man syndrome (see Chapter 8) |
Amphiphysin |
SCLC, breast |
|
|
|
Opsoclonus–myoclonus |
Ri |
SCLC, breast, bladder, neuroblastoma |
|
|
|
*Best bets for testing Hu/ANNA1, Yo/PCA-1, and CV-2.
Abbreviations: ACh, acetylcholine; SCLC, small-cell lung cancer.
5.General prognosis: patients with paraneoplastic syndrome generally have a better oncologic outcome than do tumor patients without paraneoplastic syndromes
6.Subtypes (Table 5–3): symptoms evolve over a period of days to weeks
Appendix 5–1 Imaging Characteristics of Tumors
Calcified tumors |
Contrast-enhancing tumors |
Hemorrhagic tumors |
|
|
|
Meningioma |
Metastases |
Metastases, particularly melanoma and choriocarcinoma |
Oligodendroglioma |
Meningioma |
Astrocytoma (high-grade) |
Astrocytoma (low-grade) |
Astrocytoma (high-grade) |
Lymphoma |
Ependymoma |
Schwannoma/acoustic neuroma |
|
Craniopharyngioma |
Pituitary tumors |
Pituitary tumors |
Vascular tumors |
Vascular tumors |
Vascular tumors |
|
|
|