

7 Behavioral Neurology and Psychiatry
7
Behavioral Neurology
and Psychiatry
Note: Significant diseases are indicated in bold and syndromes in italics.
I. Dementias
A. Mild Cognitive Impairment (MCI) (Box 7.1)
1.Definition: cognitive impairment that does not achieve the criteria for dementia because the degree of impairment does not significantly affect the patient’s activities of daily living
2.Subtypes
a.amnestic MCI/age-related memory loss: an objective impairment of memory function in comparison with the normal range for the patient’s age, which does not involve significant impairment of other cognitive functions or impair the patient’s activities of daily living
i.15% of cases/year develop into Alzheimer’s disease, which is 10 times the incidence of non-MCI patients; the incidence of progression to Alzheimer’s disease is increased in MCI patients who express the apolipoprotein E (APOE) 4 allele
(1)small hippocampal and/or entorhinal cortex volumes on a MRI scan may also predict progression of MCI to Alzheimer’s disease, although the rate of volume loss is not helpful
b.MCI with impairment in a nonmemory domain (Box 7.2)
c.MCI with impairment in multiple domains
3.Treatment: none yet established
B. Definition of Dementia
1.In general, impairment of at least two of five functional domains (memory, language, visuospatial, emotion, executive) that affects the patient’s activities of daily living
a.dementia is usually suspected in patients who have a mini-mental status exam (MMSE) score 24, but performance is dependent upon the patient’s age and education background; therefore, use the MMSE only for screening, not for establishing the diagnosis
i. MMSE 24 often does not identify non-Alzheimer’s dementias
2.Subtypes of dementia: cortical versus subcortical dementias (Table 7–1)
C. Reversible Causes of Dementia
1.Reversible causes exist in 10% of dementia cases, and include
a.metabolic abnormalities: hyponatremia, hypocalcemia, hypoglycemia
b.organ-failure: respiratory failure (hypoxia or hypercarbia); liver, renal, or cardiac failure
Box 7.1
Types of Memory by Subject
Declarative/episodic—knowledge of events (hippocampus)
Procedural—riding a bike (basal ganglia, cerebellum)
Conditioning—associative learning (cerebellum)
Semantic—factual or definitional knowledge (hippocampus-independent)
Types of Memory by Duration
Immediate/working (seconds) Short-term/anterograde ( 10 minutes) Long-term/remote ( 10 minutes)
Box 7.2
Non-amnestic MCI may progress to a nonAlzheimer’s type dementia.
154
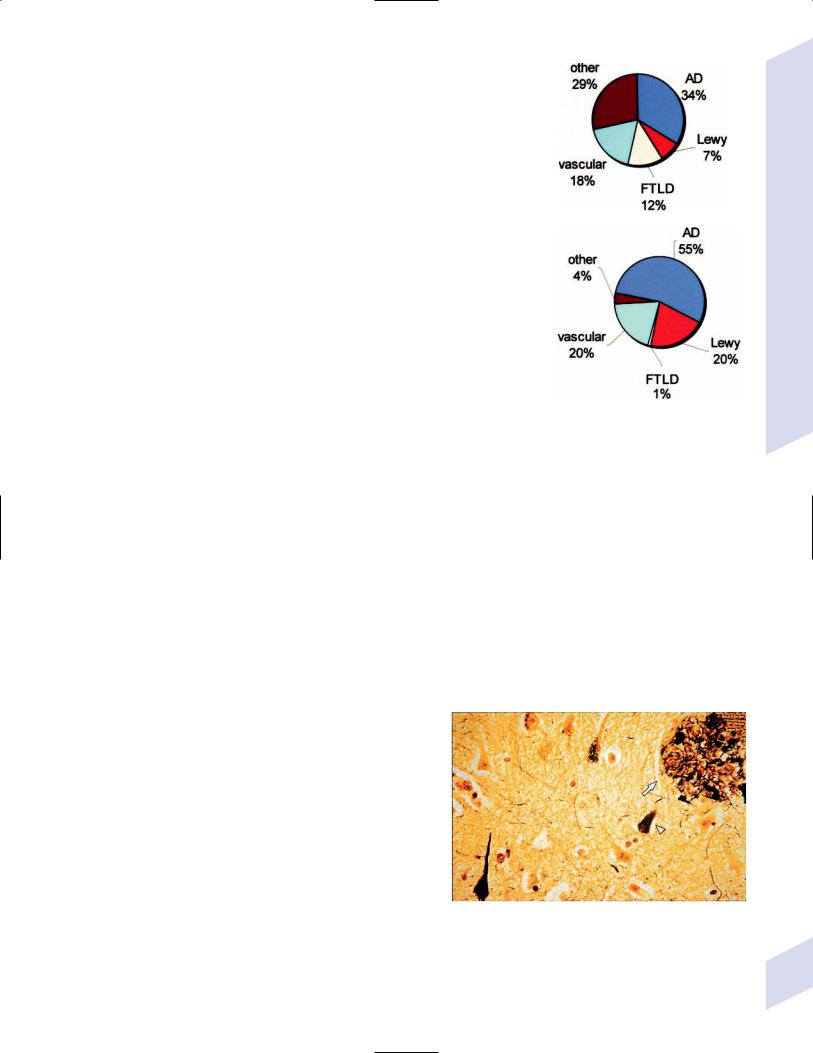
Table 7–1 Cortical versus Subcortical Dementias
Functional domain |
Cortical dementia |
Subcortical dementia |
|
|
|
|
|
Memory |
Learning retrieval deficit |
Retrieval learning deficit |
|
Language |
Aphasic |
Dysarthric |
|
Emotion |
Disinhibited |
Depressed |
|
Visuospatial |
(Indistinguishable) |
|
|
Executive |
Impaired and does not |
Impaired, but improves |
|
|
improve with cues |
with cues |
A |
|
|
|
c.endocrine disorders: hypoand hyperthyroidism, hyperparathyroidism, adrenal failure or Cushing’s syndrome
d.toxin-induced: heavy metal exposure, drugs, alcohol
e.vitamin deficiency, particularly B1/thiamine or B12
f.medication use, particularly benzodiazepines, opioids, and anticholinergic agents
g.chronic brain infections (e.g., HIV, syphilis) or inflammatory conditions
h.intracranial mass lesions or hydrocephalus
i.normal pressure hydrocephalus
D.Epidemiology of Major Dementing Disorders (Fig. 7–1)
E.Alzheimer’s Dementia (AD)
B
Figure 7–1 Epidemiology of major dementing disorders. (A) 65 years old; (B) 65 years old. (AD, Alzheimer’s dementia; Lewy, Lewy body dementia)
1. Histology: primary cortical atrophy involving the loss of synapses, and neurons, |
|
|
|
||
and the shrinkage of surviving neurons; atrophy initiates in the mesial temporal |
|
|
|
||
|
Box 7.3 |
||||
and posterior cingulate cortex (cortex layers V-VI) (Box 7.3), but eventually |
|
||||
|
|
|
|||
involves the hippocampus and all of the cortex sparing only the primary motor, |
|
Cortical histopathology in AD is in compar- |
|||
sensory, and visual cortices |
|
|
ison to the loss of layers I–III in frontotem- |
||
a. neurons in the nucleus basalis of Meynert, locus coeruleus, and raphe are |
|
poral lobar degeneration disorders |
|||
|
|
|
|||
also reduced in number early in the disease |
|
|
|
|
|
b. hippocampal atrophy is predominant in the CA1 area (unlike Lewy body |
|
|
|
||
dementia, which affects CA2–3) |
|
|
|
|
|
c. the cortex and hippocampus lose choline acetyltransferase nicotinic and |
|
|
|
||
muscarinic acetylcholine receptors well into the course of the disease, there- |
|
|
|
||
fore these are not the primary defects; also exhibit reduced acetylcholine |
|
|
|
||
release and decreased choline reuptake (necessary for acetylcholine |
|
|
|
||
synthesis), which may relate to a decreased trophic |
|
|
|
|
|
responsiveness to nerve growth factor |
|
|
|
|
|
d. plaques and tangles (Fig. 7–2) |
|
|
|
|
|
i. neurofibrillary tangle: intraneuronal inclusions of |
|
|
|
|
|
hyperphosphorylated tau protein (a microtubule- |
|
|
|
|
|
associated protein) and ubiquitin (which binds and |
|
|
|
|
|
marks other proteins for degradation) |
|
|
|
|
|
(1) neurofibrillary tangles are associated with an |
|
|
|
|
|
early age of disease onset and a more severe |
|
|
|
|
|
disease course |
|
|
|
|
|
(2) neurofibrillary tangles may not occupy the whole |
|
|
|
|
|
neuron, and may only fill a dendrite {neuropil |
|
|
|
|
|
thread} |
|
|
|
|
|
ii. plaques: all are extracellular |
Figure 7–2 Neurofibrillary tangles (arrowhead) and neuritic |
||||
(1) neuritic/senile plaque: composed of abnormal |
plaque (arrow) in Alzheimer’s disease. (From McKhann GM et al. |
||||
Q&A Color Review of Clinical Neurology and Neurosurgery. |
|||||
neuronal processes (dystrophic neurites), and |
|||||
Stuttgart, Germany: Georg Thieme; 2003:123, Fig. 121. Reprinted |
|||||
glial processes surrounding an amyloid core; also |
by permission.) |
|
|
|
|
Dementias
155
7 Behavioral Neurology and Psychiatry
156
includes tau and other molecules (acetylcholinesterase, ubiquitin, chymotrypsin, complement proteins, glycosaminoglycans)
(2)ghost plaque: develop only after cell death when extracellular proteases degrade the neurofibrillary tangle, leaving only the dense amyloid core
(3)diffuse plaque: similar histological appearance to senile plaques; composed of amorphous nonfibrillar-amyloid or finely fibrillar -amyloid deposits
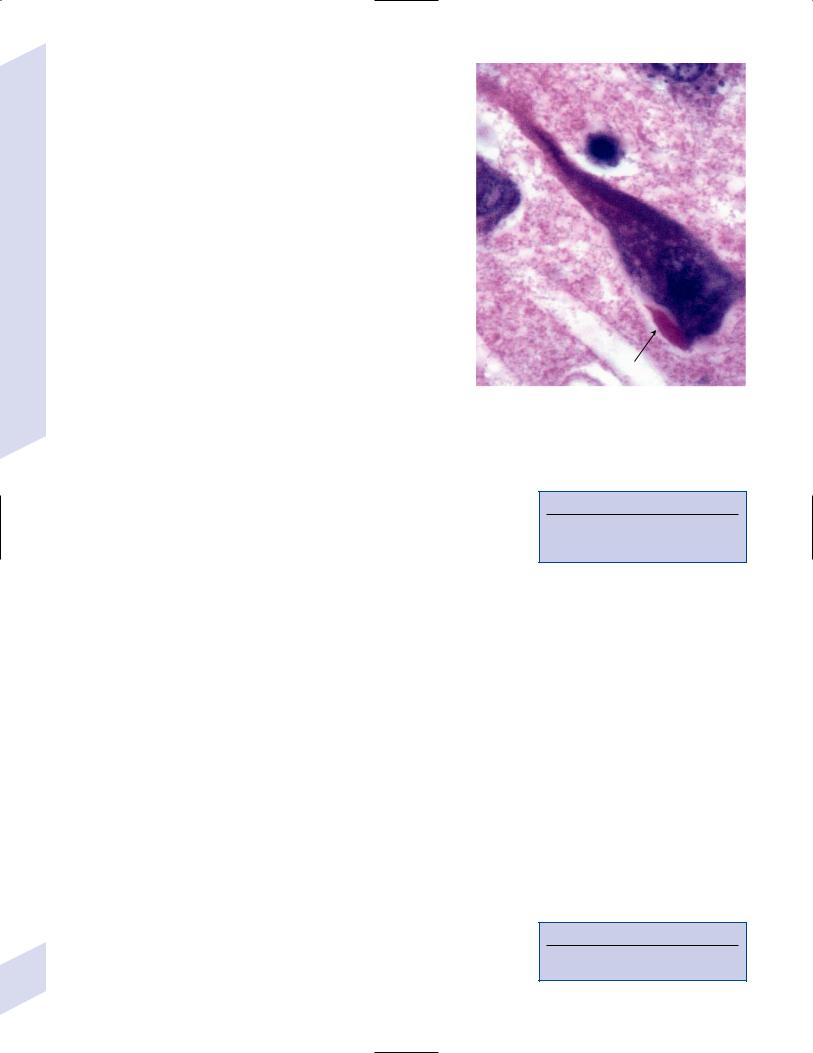
iii.Hirano bodies (Fig. 7–3): eosinophilic intracellular inclusions in hippocampal pyramidal cells that have a “herringbone” appearance on electron microscopy
iv.Lewy bodies (see p. 162): more commonly found in the synucleinopathies (Parkinson’s disease, Lewy body dementia, multisystem atrophy), but are present in the substantia nigra in 20% of AD
e.amyloid angiopathy caused by the -amyloid protein coexists in 90% of AD patients (see p. 65)
2.Subtypes
a.sporadic AD (95% of cases): symptomatic onset 65 years of age, typically with a slowly progressive course at time of diagnosis (i.e., at the time of diagnosis, the patient is 2–3 years into the course of the disease)
i.risk factors
Figure 7–3 The Hirano body (arrow). (From Hirano A. Color Atlas of Pathology of the Nervous System, 2nd Ed. Tokyo/New York: Igaku-Shoin Press; 1988:94, Fig. 222. Reprinted by permission.)
(1)increasing age
(2)APOE alleles: APOE is component of very low-density lipoprotein (VLDL) and chylomicron particles, which facilitates their uptake by muscle and fat cells (Box 7.4)
(a)the APOE 4 allele is present in 50% of sporadic AD cases; no clear association with the 4 allele and the severity of the disease
(b)APOE 4 binds -amyloid, creating an insoluble complex that may form plaques
(c)APOE 3 allele may stabilize tau protein thereby preventing neurofibrillary tangle formation, but only the rare APOE 2 allele is known to be protective against AD
(3)family history of AD without clear inheritance pattern
(4)low level of education
(5)female gender
(6)history of severe head trauma
(7)stroke and vascular risk factors
(8)small head size
(9)Down’s syndrome
b.familial/early-onset AD (5% of cases): patients are generally 65 years old at onset, and exhibit a more rapid progression of symptoms than do sporadic AD patients; although the disease course is still insidious
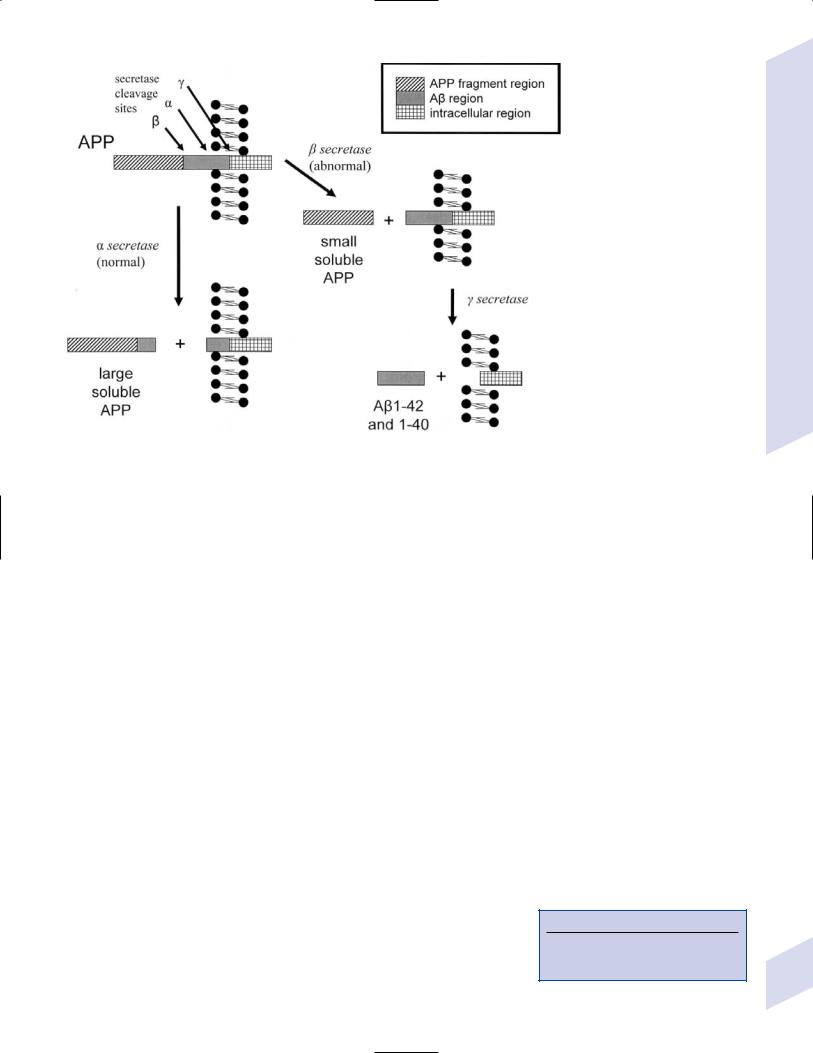
i.causative genetic mutations (Fig. 7–4): all have autosomal dominant inheritance
(1)the AD1 mutation in the amyloid precursor protein (APP) gene (chromosome 21), which occurs in a region that codes for the-amyloid protein, accounts for 2% of familial AD
(a)overexpression of the APP in trisomy 21/Down’s syndrome may account for the early-onset AD in these patients
(2)presenilin 1 (PS1, chromosome 14): accounts for 75% of familial AD (Box 7.5)
Box 7.4
Other apolipoprotein disorders–APOA1 deposits in some types of amyloidosis; APOB deficiency in abetalipoproteinemia
Box 7.5
Presenilin 1 forms the catalytic subunits of the secretase complex that cleaves APP
(3)presenilin 2 (PS2, chromosome 1)
(4)2 macroglobulin (chromosome 12)
3.Epidemiology
a.age distribution: prevalence at 60 years of age 3%; prevalence at 90 years of age 50%
b.prevalence as a cause of dementia is likely underestimated because
i.Lewy body dementia has a high coincidence with AD
ii.50% of vascular dementia cases have elements of AD histology on postmortem examination
4.Symptoms: course of impairment is typically memory loss before involvement of any other cognitive functions or behavioral impairment
a.memory loss: early and severe impairment of short-term memory longterm or immediate memory
i.episodic memory is most severely affected
ii.procedural memory is well-preserved, although mild apraxia can be detected early in the disease
b.language impairment: initial language abnormality is anomia (due to lateral temporal dysfunction, not memory impairment) that progresses to a transcortical sensory aphasia
c.behavioral impairment: more common late in the disease
i.agitation and aggressive behaviors (70%), but rarely sexual disinhibition (10%)
ii.depression (50%), which is proportionate to cell loss in locus coeruleus
iii.simple delusions (30%) that are often paranoid in nature
iv.hallucinations (20%)
d.visuospatial impairment: manifests primarily as difficulty with navigation
e.executive impairment: impaired judgment and reasoning, and poor abstraction and decision-making abilities occur late in the disease as frontal lobes become involved (Box 7.6)
Figure 7–4 Key enzymes of Alzheimer’s disease. (APP, amyloid precursor protein)
Box 7.6
Agnosias can occur in AD, but they are distributed topographically (e.g., visual agnosias to the occipital lobe).
Dementias
157
7 Behavioral Neurology and Psychiatry
158
f.generally does not involve focal neurological signs, seizures, motor disturbances (i.e., parkinsonism), or impairment in the level of arousal until late in the disease; the physical exam generally is nonfocal, although multiple frontal lobe release reflexes (e.g., glabellar reflex, grasp, snout reflex) are often present (Box 7.7)
i.myoclonus occurs in 10% of AD cases, which can be confused with prion diseases
ii.pyramidal signs (increased reflexes, Babinski reflexes, hypertonus) are unlikely to be due to AD, as the primary motor and sensory cortices are not involved in the disease process until very late
5.Diagnostic testing: none are well-established or necessary for diagnosis
a.genetic markers: the low incidence of familial AD makes them impractical as screening tools
b.cerebrospinal fluid protein markers: protein markers correlated with pathologically determined AD include
i.increased tau protein and hyperphosphorylated tau protein levels
ii.combined increases in -amyloid protein and tau protein levels
c.EEG: loss of alpha activity and increased theta and delta frequencies in 80% of AD cases with 4-year disease duration; useful for differentiating against the frontotemporal lobar degeneration disorders in which EEG is relatively normal
d.neuroimaging
i.SPECT scans demonstrate hypoperfusion in the temporal and parietal lobes
ii.PET scans demonstrate hypometabolism in the temporal and parietal lobes
iii.MRI and CT demonstrates reduced hippocampal, entorhinal cortex, and posterior cingulate cortex volumes (Fig. 7–5)
6.Treatment
a.preventative
i.chronic use of NSAIDs is associated with a decreased risk of developing AD, but it is not an established prophylactic treatment
ii.hormone replacement therapy appears to increase, not reduce, the risk of dementia
b.symptomatic treatment
i.acetylcholinesterase inhibitors: donepezil (Aricept), rivastigmine (Exelon), galantamine (Reminyl)
(1)improve functional abilities and delay institutionalization in patients with mild or moderate dementia; also appear to be beneficial in severe dementia
(2)tacrine also improves cognitive performance but it has a high rate of side effects (particularly hepatotoxicity) and requires q.i.d. dosing, therefore it is rarely used
ii.memantine: acts as an N-methyl-D-aspartate (NMDA) glutamate receptor antagonist; improves patient’s functional abilities and reduces caregiver burden in patients with moderate or severe dementia
iii.no evidence for use of vitamin E, NSAIDs, or estrogens
c.treatment of behavioral impairment
i.depression: SSRIs or trazodone, which have minimal anticholinergic side effects
ii.agitation: behavioral modification; buspirone; atypical antipsychotics (olanzapine, quetiapine, risperidone, clozapine)
(1)avoid benzodiazepines because they worsen cognitive function
iii.insomnia: improve sleep hygiene, avoid daytime naps; trazodone
Box 7.7
20% of normal young persons have one frontal lobe release sign, but the palmomental reflex is always pathological
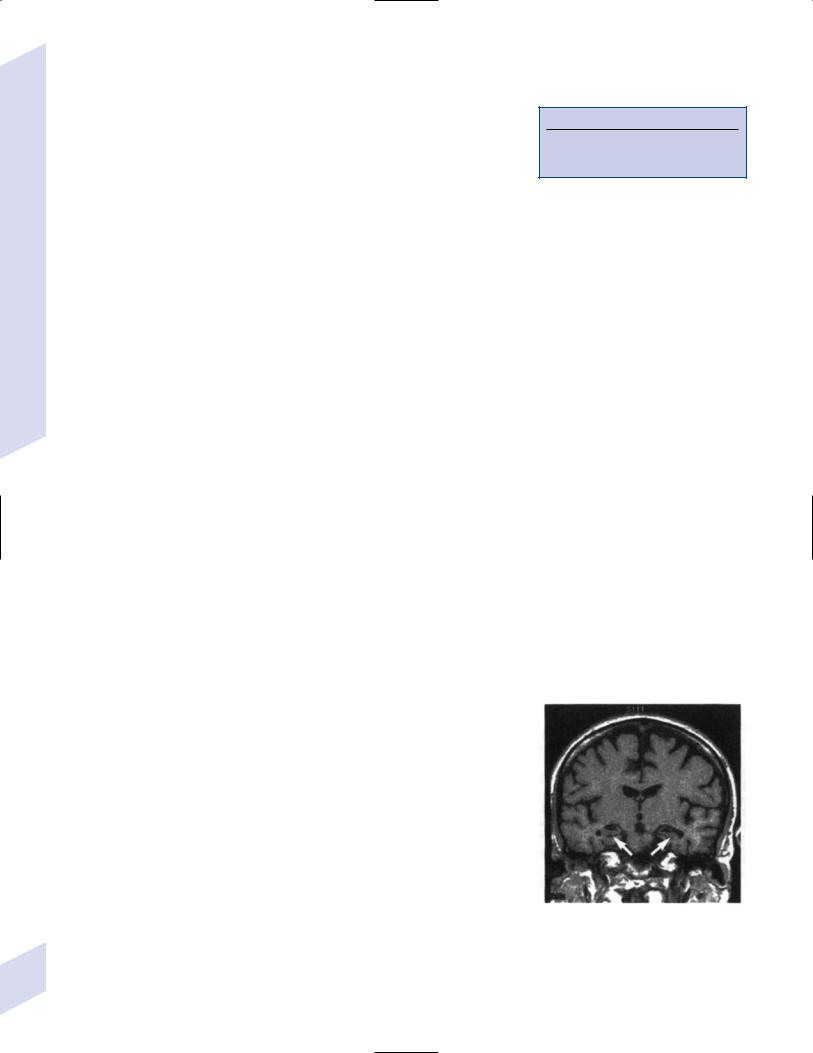
Figure 7–5 Coronal T1 MRI from a patient with Alzheimer’s disease. Note the pronounced hippocampal atrophy (arrows). (From Nestor P, Hodges J. Non-Alzheimer’s dementias. Semin Neurol 2000, 20:443, Fig. 1b. Reprinted by permission.)

Table 7–2 Differentiating Alzheimer’s Dementia (AD) from Other Dementias
Type |
Symptoms different from AD |
Signs different from AD |
|
|
|
Pseudodementia/ |
Rapid onset |
Good performance on cognitive |
depression with |
|
tests; self-referral to physician |
cognitive distur- |
|
|
bances |
|
|
Vascular dementia |
Classically (but not commonly) |
Focal deficits; bulbar signs |
|
has an abrupt onset and step- |
|
|
wise deterioration |
|
Lewy body |
Fluctuating cognitive abilities & |
Parkinsonism; high sensitivity to |
dementia |
attention; visual hallucinations; |
the side effects of antipsychotics |
|
sleep disturbance; frequent falls |
|
|
from syncope |
|
Frontotemporal |
Impulsivity; compulsivity; |
Lack of EEG slowing; lack of visu- |
lobar degeneration/ |
emotional lability; disinhibition; |
ospatial impairment or other |
Pick’s disease |
aphasia |
parietal lobe findings |
Prion dementias |
Abrupt onset, rapid progression; |
Cerebellar symptoms; visual loss; |
(e.g., Creutzfeldt- |
early aphasia; extrapyramidal |
myoclonus; CSF positive for 14–3-3 |
Jakob disease) |
symptoms |
protein; diffuse slow-wave EEG with |
|
|
periodic complexes; neuroimaging |
|
|
abnormalities (see p. 265) |
Parkinson’s disease |
Subcortical dementia features; |
Parkinsonism |
|
subcortical dementia symptoms |
|
Huntington’s |
Impulsivity; hypersexuality; |
Chorea |
disease |
psychosis; subcortical dementia |
|
|
symptoms |
|
|
|
|
Abbreviations: CSF, cerebrospinal fluid; EEG, electroencephalogram.
7.Prognosis
a.nursing home placement occurs on average 3 years after diagnosis
b.70% 5-year mortality, with death usually being attributed to complications of immobility or malnutrition (Table 7–2)
F. Vascular/Multi-Infarct Dementia
1.Pathophysiology: caused by any combination of cortical infarction, lacunar infarction, and/or leukoaraiosis; also involves microscopic areas of neuron loss and gliosis {microinfarcts}
a.requires 50 cc of tissue loss to achieve symptomatic dementia
b.risk factors are as per stroke, but also include the cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) gene mutation and the APOE 4 allele (as in AD)
c.epidemiology: incidence increases with age; 25% of patients will become demented within 3 months of a stroke
2.Subtypes
a.large infarct dementia: caused by a few cortical and/or subcortical infarcts that are larger than lacunes
b.small infarct dementia: typically caused by multiple lacunes {l’etat lacunaire}, leukoaraiosis, CADASIL, or amyloid angiopathy
c.strategically placed infarct dementia: caused by single lesions in the hippocampus/medial temporal lobe, medial thalamus, caudate nucleus, or nondominant parietal lobe; such infarcts tend to have a preference for the dominant hemisphere
d.hypoxic-ischemic encephalopathy
e.hemorrhagic dementia secondary to subdural hemorrhage, subarachnoid hemorrhage, or intracerebral hemorrhage
Dementias
159

7 Behavioral Neurology and Psychiatry
3.Symptoms: dementia must be preceded within 3 months by a stroke, and focal neurological deficits should be present; exhibits distinguishable corticaland subcortical-type dementias
4.Diagnostic testing
a.the size and exact number of infarcts are not as predictive of dementia as is cortical atrophy
b.isolated lacunar infarcts occur in 20% of the general population, but are associated with worse cognitive performance and an increased risk of dementia
c.periventricular white matter T2 hyperintensities on an MRI scan {leukoaraiosis} is not specific for vascular dementia or any dementia, but is associated with worse performance on cognitive tests
5.Treatment: as per stroke prevention, with particular attention to the reduction of hypertension (reduces incidence of dementia by 50%) and reduction of elevated homocysteine levels; acetylcholinesterase inhibitors; memantine
6.Prognosis: 30% 5-year survival, which is worse than other types of dementia or nondemented stroke; however, cognitive deterioration progresses more slowly than in AD
G. Frontotemporal Lobar Degeneration/Pick’s Disease (Box 7.8)
1.Histology: Selective atrophy of the frontal (particularly orbitofrontal) and/or anterior temporal lobes often involving the hippocampus, amygdala, and/or underlying basal ganglia (particularly the caudate); atrophy tends to spare the sensory cortices (as with AD) but also the parietal lobe (unlike AD)
a.pronounced astrocyte gliosis in cortex and subcortical white matter with loss of cortical layers I–III (versus AD that affects layers V–VI predominantly)
b.loss of acetylcholine receptors choline acetyltransferase (in comparison with AD)
c.inclusion bodies: argentophilic cellular inclusions {Pick bodies} and swollen neurons {Pick cells} are fairly specific for the frontotemporal lobar degeneration disorders, although they are found in 50% of cases (Fig. 7–6); cases with Pick bodies and cells tend to be more rapidly progressive
Box 7.8
Frontotemporal lobar degeneration is not the same as frontotemporal dementia, which is a subtype of it
Figure 7–6 The Pick body (arrow). (From Hirano A. Color Atlas of Pathology of the Nervous 160 System, 2nd Ed. Tokyo/New York: Igaku-Shoin Press; 1988:98, Fig. 233. Reprinted by permission.)
i.Pick bodies contain ubiquitin and tau protein tau
ii.in cases without Pick bodies or cells, intranuclear ubiquitin inclusions can often be identified
2.Pathophysiology
a.genetics: 20% of cases have a familial pattern that is usually autonomic dominant
i.tau gene mutations: mutations in the tau gene on chromosome 17 are responsible for 10% of familial frontotemporal lobar degeneration disorders, particularly those that involve parkinsonian features (i.e., the FTDP-17 variant)
ii.tau gene haplotypes: of the two tau haplotypes (H1 and H2) (Box 7.9), the H2 haplotype is particularly common in the semantic subtype of frontotemporal lobar degeneration disorders
b.associated with Parkinson’s disease and amyotrophic lateral sclerosis (ALS)
3.Subtypes: all have an insidious progression and symptomatic onset before 65 years of age (Box 7.10); lack of parietal lobe dysfunction differentiates against AD
a.frontotemporal dementia—common symptoms include impersistence of attention, apathy or disinhibition, and irritability; occasionally may also involve
i.Kluver-Bucy syndrome (usually incomplete): placidity, hypersexuality, hyperorality, and/or increased manual exploration
ii.perseverative behaviors with stereotypies
iii.echolalia and perseveration of speech progressing to mutism
b.primary progressive aphasia—caused by disease predominantly in the dominant frontal or temporal lobes
i.symptoms: aphasia of either a nonfluent or fluent type; progresses to dyslexia, agraphia, and apraxias, suggesting parietal lobe involvement
(1)isolated aphasia is usually present 2 years before the development of other symptoms
c.semantic dementia—symptoms include reduced verbal fluency and pronounced naming difficulties; usually seen in patients with anterior temporal lobe atrophy
d.progressive prosopagnosia—usually seen in patients with nondominant temporal lobe (i.e., fusiform gyrus) atrophy
4.Diagnostic testing
a.neuroimaging may reveal an asymmetric atrophy pattern (Fig. 7–7)
b.PET demonstrates hypometabolism and SPECT demonstrates hypoperfusion in the frontal and temporal lobes and often the basal ganglia
5.Treatment: None with proven efficacy; SSRIs for management of behavioral abnormalities irrespective of depression
H. Lewy Body Dementia
1.Histology: degeneration of the temporal and parietal cortices (like AD), but also of the occipital cortex; hippocampal atrophy predominantly affects the CA2–3 regions, unlike AD that predominantly affects CA1
a.exhibit significant loss of acetylcholine but limited loss of neurons in the cholinergic basal forebrain nuclei (i.e., basal nucleus of Meynert), unlike AD
b.exhibits loss of basal ganglia dopamine as in Parkinson’s disease, but also loss of basal ganglia D2 receptors
c.80% of cases exhibit some Alzheimer’s-like histological abnormalities, particularly neuritic-like plaques that do not contain tau protein
d.inclusion bodies
Box 7.9
H1 haplotype is common in progressive supranuclear palsy and corticobasal ganglionic degeneration
Box 7.10
Eventually all frontotemporal lobar degeneration subtypes will progress to frontotemporal dementia.
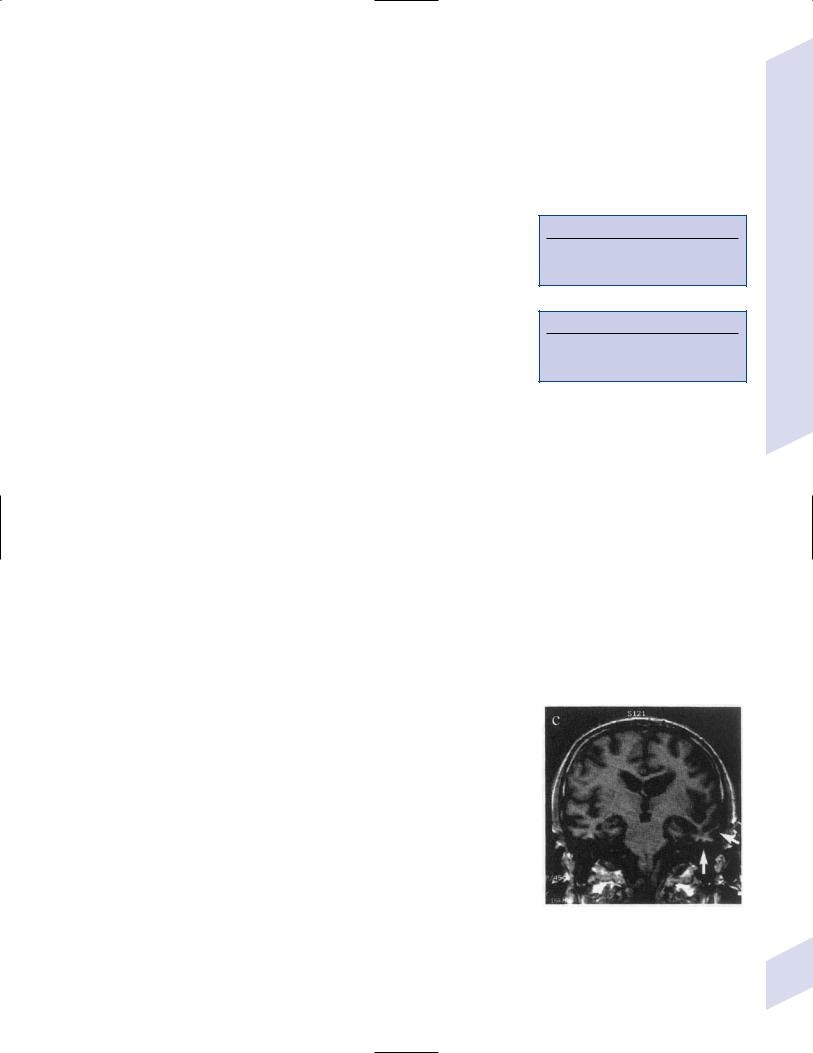
Figure 7–7 Coronal T1 MRI from a patient with semantic dementia. Note the pronounced atrophy of the left temporal lobe (arrows). (From Nestor P, Hodges J. NonAlzheimer’s dementias. Semin Neurol 2000, 20:443, Fig. 1c. Reprinted by permission.)
Dementias
161
7 Behavioral Neurology and Psychiatry
Figure 7–8 The Lewy body (arrow). Courtesy of Dr. C. Yamada.
i.Lewy body (Fig. 7–8): eosinophilic inclusions surrounded by a halo that contain -synuclein and ubiquitin; found predominantly in the hippocampus and parahippocampal gyrus, but are widely distributed throughout the cortex, brainstem, and spinal cord (Box 7.11)
(1) Lewy bodies are identical to those found in Parkinson’s disease
ii.Lewy neurites: intracellular inclusions of ubiquitin and -synuclein, which are limited to dendrites
2.Pathophysiology: Rare familial disease patterns are weakly associated with cytochrome P450 mutations or -synuclein mutations
3.Symptoms
a.fluctuating cognitive abilities and level of arousal that may relate to cyclic sleep disturbance
b.autonomic dysfunction: orthostatic hypotension; incontinence; sexual dysfunction (Box 7.12)
c.visual hallucinations (80%); also illusions and paranoid delusions (Box 7. 13)
i.hallucinations may be caused by intrusion of REM sleep while the patient is awake (i.e., as in peduncular hallucinosis)
d.sleep disturbance, particularly REM (rapid eye movements) sleep behavioral disorder (RSBD) that may precede the dementia by decades
i.conversely, an unspecified dementia occurring in conjunction with RSBD is likely to be Lewy body disease
e.impairment of visuospatial abilities without ophthalmologic abnormality (Box 7.14)
f.Parkinsonism: symptoms are symmetric and poorly responsive to dopaminergic agonists; tremor is mild and often involves a positional as well as resting component
i.the parkinsonism must develop no sooner than 1 year before the dementia to be diagnosed as Lewy body disease and not as Parkinson’s disease dementia
4.Diagnostic testing
a.polysomnography may demonstrate RSBD (i.e., the lack of atonia during REM sleep)
Box 7.11
Lewy body-like inclusions occur in Parkinson’s disease, Alzheimer’s disease, multiple system atrophy, corticobasal ganglionic degeneration, Hallervorden-Spatz disease, ataxia telangiectasia, and frontotemporal lobar degeneration amyotrophic lateral sclerosis.
Box 7.12
Fluctuating cognitive abilities and autonomic dysfunction together account for episodes of loss of consciousness.
Box 7.13
Capgras syndrome—The delusion that imposters have replaced family members
Box 7.14
Impairment of visuospatial abilities and parkinsonism together may account for frequent falls.
162

b.SPECT demonstrates parietal and occipital hypoperfusion, and PET demonstrates hypometabolism in these regions
c.neuroimaging and EEG provide no reliable or specific information
5.Treatment
a.acetylcholinesterase inhibitors, which do not seem to worsen the disease’s parkinsonian features
b.atypical antipsychotics (olanzapine, quetiapine, risperidone, clozapine) for treatment of psychosis
c.low-dose dopamine agonists for parkinsonism, although they are poorly effective and readily exacerbate the hallucinosis and orthostatic hypotension
I. Normal Pressure Hydrocephalus/Hakim-Adams Syndrome
1.Pathophysiology: a communicating hydrocephalus that does not involve constantly increased intracranial pressure; potential mechanisms include dilation caused by transiently increased intracranial pressure and/or altered elastic properties of the ventricle walls
2.Epidemiology: more common in males 60 years old
3.Symptoms (in order of appearance)
a.gait disturbance: Bruns’ frontal lobe ataxia (which is really an apraxia; see p. 5)
b.“dementia” (60%), which is really an attentional deficit (i.e., a confusional state)
c.urinary incontinence (spastic bladder); urgency may precede incontinence
4.Diagnostic testing: No test exhibits high diagnostic accuracy
a.lumbar puncture
i.demonstrates normal pressure but continuous cerebrospinal fluid pressure monitoring may exhibit prolonged or transient pressures20 mm Hg (Lundberg waves; see p. 35)
ii.measurement of the resistance to fluid injection into the subarachnoid space is correlated with the severity of symptoms but is not predictive of the response to shunting
b.radionuclide cisternography: retention of contrast in the ventricles 24 hours after administration is not useful for establishing the diagnosis or predicting the clinical response to shunting
c.MRI cerebrospinal fluid flow study: absence of flow near the cerebral aqueduct does not predict the clinical response to shunting
5.Treatment: low-pressure ( 3-mm opening pressure) ventriculoperitoneal shunt
a.patient should be sat upright slowly over several days after shunt placement
b.lumboperitoneal shunts often results in over-shunting and have high complication rates
6.Prognosis: incontinence and gait generally improve more than the “dementia”; responsiveness to shunting is predicted by
a.the presence of the classic triad of symptoms with gait disturbance as the first symptom
b.clinical improvement after lumbar puncture
c.a near-normal opening pressure
d.high transient pressures on continuous cerebrospinal fluid pressure monitoring
e.the absence of periventricular increased T2 signal on MRI
f.symptoms 1 year in duration
Dementias
163

II. Sleep Disorders
A. Normal Sleep
7 Behavioral Neurology and Psychiatry
1.Physiology
a.arousing systems
i.mesencephalon
(1)ascending reticular activating system (ARAS): the major component is the cholinergic nuclei of the mesencephalic and pontine reticular formation (pedunculopontine tegmental nucleus), but it also includes the noradrenergic locus coeruleus and the serotonergic raphe nuclei
(2)ventral tegmental area (dopaminergic)
(3)mesencephalic raphe nuclei
ii.hypothalamus: includes the posterior nucleus (histaminergic) and posterolateral nucleus (orexinergic/hypocretinergic) of the mamillary region
iii.basal forebrain: includes the septal nuclei and the nucleus basalis of Meynert (cholinergic)
iv.thalamus, specifically the intralaminar nuclei (glutamatergic)
b.somnogenic systems
i.hypothalamus: the lateral preoptic nucleus sends inhibitory projections (GABA, galanine) to the arousing centers, thereby inhibiting wakefulness
ii.medulla
(1)dorsolateral region of medullary reticular formation and anterior solitary tract nucleus
(a)synchronize the EEG and may induce non-REM sleep
(b)also trigger atonia prior to the onset of REM sleep via projections to nucleus gigantocellularis of the medullary reticular formation that forms the lateral reticulospinal tract
(2)nucleus reticularis pontis: may trigger REM sleep via projections to mesencephalic ARAS nuclei; responsible for ponto-geniculo- occipital (PGO) spikes detected by parenchymal electrodes in animals during REM sleep
iii.hypothalamus: lateral preoptic nucleus sends inhibitory projections (GABA, galanin) to the arousing centers of the brainstem, basal forebrain, and hypothalamus, thereby inhibiting wakefulness
iv.thalamus: the reticular nucleus sends GABAergic projections to the other thalamic nuclei, thereby inhibiting their excitation of the cortex
v.basal forebrain: exhibits changes in adenosine levels (a somnogenic compound) that increase with wakefulness and decrease with sleep; adenosine receptors in this region may mediate caffeine’s alerting effects as an antagonist on these receptors
vi.pineal: produces melatonin (Fig. 7–9) from serotonin and begins releasing it during the evening with a peak at 3–5 AM; may act to fix sleep–wake behaviors with the light–dark cycle
(1)the melatonin cycle is regulated by projections from the suprachiasmatic nucleus and sympathetic afferents from the superior cervical ganglia
c.circadian systems: the suprachiasmatic nucleus of the hypothalamus acts as the major circadian pacemaker and regulates other hypothalamic nuclei including the histaminergic and orexinergic portions of the posterior hypothalamus; affects melatonin release by sympathetic nervous system
164 |
projections from the superior cervical ganglion |
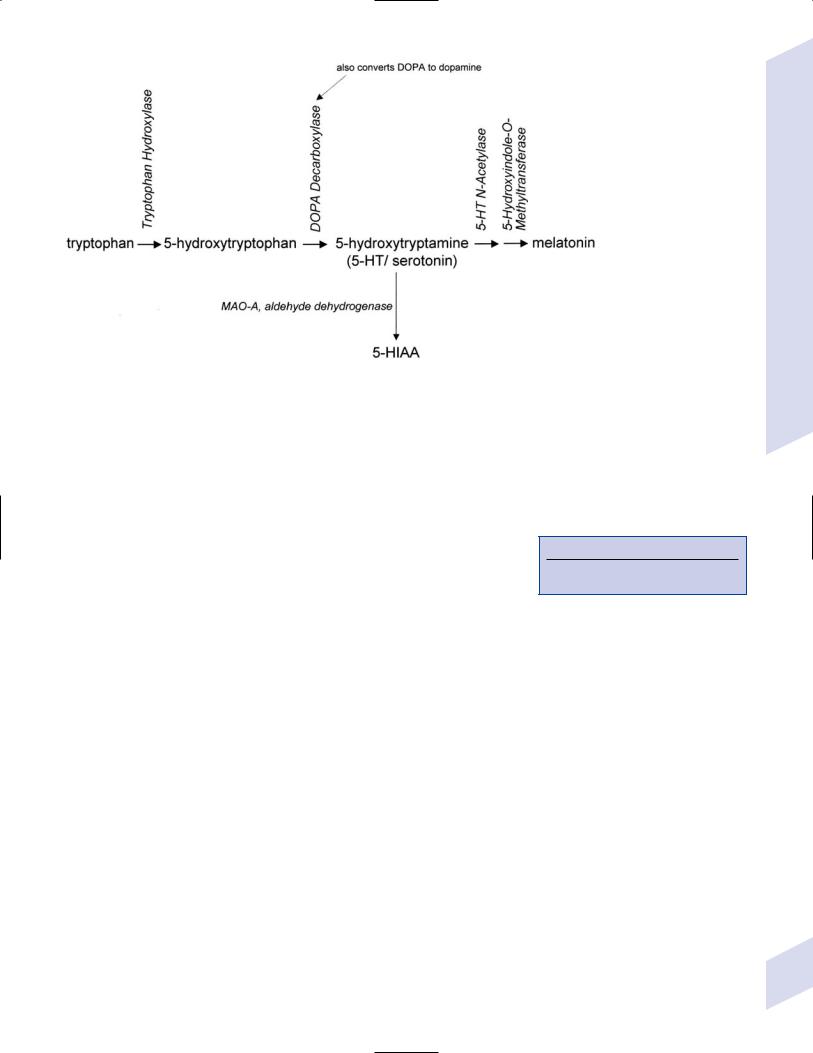
Figure 7–9 Biosynthesis of melatonin and serotonin.
2.Sleep stages
a.wakefulness: EEG demonstrates desynchronized activity of predominant frequency; frequency is normally reduced by mental activity or by drowsiness
i.closing the eyes results in a synchronization and increased amplitude of the occipital alpha frequency
ii.all three neurotransmitter systems of the ARAS (acetylcholine, catecholamines, and serotonin) are active
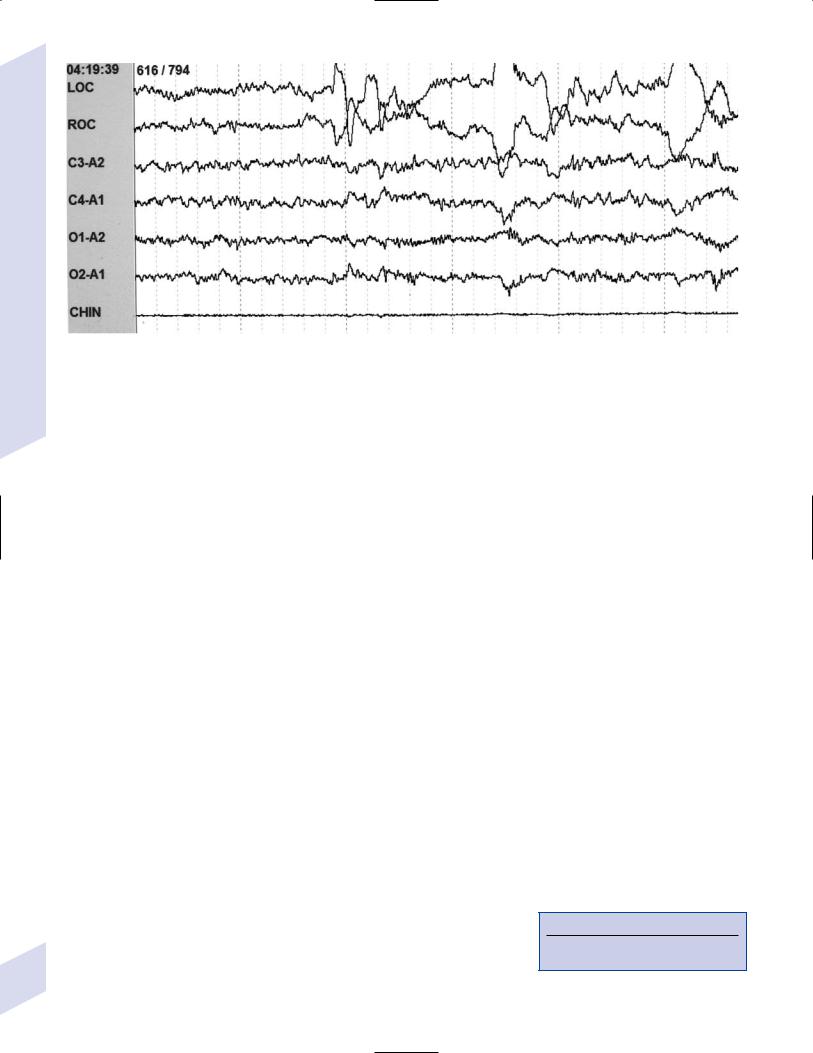
b.non-REM/slow-wave sleep (Fig. 7–10) (Box 7.15): all exhibit synchronized, high-voltage low-frequency background and are associated with a low level of motor activity and decreased muscle tone; all neurotransmitter systems of the ARAS are reduced
Box 7.15
non-REM sleep can have dreams, but they are relatively unformed.
Sleep Disorders
Figure 7–10 The EEG in wakefulness and the various |
|
slow-wave sleep stages. Transition from wakefulness to |
|
stage I sleep (arrow, top line). K-complexes (arrow, third |
|
line) and sleep spindles (underline) are characteristic of |
|
stage II sleep. (From Sinton CM, McCarley RW. Neuro- |
|
physiological mechanisms of sleep and wakefulness. |
|
Semin Neurol 2004, 24: 213, Fig. 1A. Reprinted by per- |
165 |
mission.) |
7 Behavioral Neurology and Psychiatry
166
Figure 7–11 REM sleep. Courtesy of Dr. P. Manthena.
i.stage 1 sleep exhibits 50% alpha frequency (versus being awake with eyes closed) and vertex waves
ii.stage 2 sleep exhibits sleep spindles and K complexes, as well as vertex waves
iii.stage 3 sleep exhibits 20–50% delta frequency of at least 75 mV amplitude
iv.stage 4 sleep exhibits 50% delta frequency of at least 75 mV amplitude
c.REM sleep: exhibits desynchronized, low-voltage mixed frequency background involving theta alpha and beta frequencies (Fig. 7–11); acetylcholine levels from the ARAS are relatively high, but catecholamines and serotonin levels are reduced
i.PGO spikes are seen during REM sleep in animals prior to the appearance of rapid eye movements, but it is not known if they exist in humans
ii.bursts of hippocampal theta waves {sawtooth waves} are characteristic
iii.somatic activity: atonia and absence of movements except in the ocular muscles and diaphragm, but including oropharynx; rarely twitching can be observed in the facial muscles
(1)paralysis of nondiaphragmatic respiratory muscles may lead to collapse of the rib cage in infants or to airway collapse in adults (as in obstructive sleep apnea)
(2)erections occur in REM sleep; absence of erections during REM sleep supports the diagnosis of biogenic (versus psychogenic) impotence
iv.dreams: are relatively complex and involve emotions
d.sleep stage distribution: in normal sleep, there is an orderly progression through slow-wave sleep stages, which is followed by REM sleep; this cycle repeats itself 4–5 times/night, although the majority of slow-wave sleep occurs during the first half of the night and the majority of REM sleep occurs during the second half
B. Dyssomnias (Box 7.16)
1.Disorders of initiating and maintaining sleep {insomnias} a. subtypes
Box 7.16
Dyssomnias require the complaint of daytime sleepiness.

i.transient insomnias ( 1-month course): usually caused by psychological stress, anxiety disorders, depression, changes in the patient’s sleep schedule, or medications/drug use
ii.chronic insomnias ( 1-month course)
(1)primary insomnia
(2)insomnia secondary to chronic medical condition, psychiatric disorders, or medications/drugs
b.symptoms: difficulty falling asleep and/or difficulty staying asleep with early morning awakenings; nonrestorative sleep
c.treatment
i.improve sleep hygiene: daytime exercise; avoid daytime naps; avoid light exposure at night; avoid caffeinated beverages; avoid heavy meals or drinking 3 hours before bedtime; strictly limiting the use of the bed to sleeping; regular bed and waking times
ii.relaxation therapy; biofeedback; cognitive–behavioral therapy
iii.sleep restriction therapy: limiting the time spent in bed until 85% of the time is in sleep; after this is achieved, the patient can increase the total time spent in bed
iv.benzodiazepines (Box 7.17), which cause significant morning sleepiness
(1)act by increasing the duration of sleep stage 2 and increasing the number of sleep cycles at the expense of REM sleep, which produces a refreshing sleep but which may lead to REM rebound when the medication is discontinued
v.hypnotics: zolpidem (Ambien) and zaleplon (Sonata), which are GABA receptor agonists
(1)exhibit minimal respiratory or cognitive side effects, and significantly less tolerance or rebound than benzodiazepines
(2)hypnotics do not decrease REM sleep as much as benzodiazepines, therefore there may be no REM rebound phenomenon upon discontinuation
vi.sedating antidepressants (amitriptyline, trazodone, amoxapine); other antidepressants (e.g., SSRIs) are less likely to be effective unless the patient has a psychiatric disorder
2.Disorders of excessive daytime sleepiness: The most common cause is selfimposed sleep restriction, which must be excluded by a careful history
a.narcolepsy
i.pathophysiology: destruction (possibly autoimmune) of the hypocretin/ orexin-containing neurons in the posterolateral nucleus of the mamillary region of the hypothalamus, which normally act to stimulate the cholinergic, histaminergic, and monoaminergic components of the reticular activating system; this causes instability between waking and REM sleep, and allows one to intrude into the other
(1)associated with HLA-DQ and -DR types
ii.symptoms: develops at 20–30 years of age
(1)excessive daytime sleepiness with the sudden onset of sleep {sleep attacks}, which are refreshing for the patient; sleep attacks are not specific for narcolepsy
(2)sudden-onset diffuse weakness that is precipitated by intense emotional responses, particularly laughter {cataplexy}; pathognomonic for narcolepsy
(a)cataplexy may develop years after the other symptoms of narcolepsy
(b)cataplexy likely represents development of REM atonia while awake
(c)may only last a few seconds and involve the face and/or arms
Box 7.17
Benzodiazepines use only sparingly, never scheduled, and always in conjunction with behavioral modification.
Sleep Disorders
167
7 Behavioral Neurology and Psychiatry
168
(3)paralysis at the onset of sleep or upon awakening, with intact consciousness {sleep paralysis}
(4)hallucinations at sleep onset {hypnagogic} or upon awakening {hypnopompic}
(a)hallucinations may or may not be accompanied by sleep paralysis
(b)hallucinations may be of any type but are usually visual
(c)some awareness of surroundings is preserved
(5)fragmented sleep patterns with frequent arousals
iii.diagnostic testing
(1)multiple sleep latency test: in narcoleptics, evaluation of 4–5 daytime naps (one every 2 hours) will demonstrate sleep-onset latencies of 5 minutes and two or more naps with REM sleep beginning immediately after sleep onset {sleep-onset REM}; nonnarcoleptics do not exhibit sleep-onset REM and have longer sleep-onset latencies
(a)should be performed with an overnight polysomnogram the previous evening to rule-out other sleep disorders
(2)maintenance of wakefulness test: determines the patient’s ability to stay awake for 40 minutes in a dark, relaxed environment
(3)overnight polysomnography to rule-out other causes of excessive daytime sleepiness
iv.treatment
(1)behavioral: scheduled naps; avoid heavy meals and alcohol consumption
(2)for daytime sleepiness: modafinil (Provigil) or amphetamines
(3)for cataplexy: tricyclic antidepressants, fluoxetine, venlafaxine, sodium oxybate
b. sleep breathing disorders (Box 7.18) (Box 7.19)
i.subtypes
(1)obstructive sleep apnea—may be caused by pharyngeal collapse, dysfunction of the airway dilator muscles, or anatomical narrowing of the airways
(a)risk factors include increased body mass index, neck circumference 17 inches, hypothyroidism, witnessed sleep apneas, gasping during waking, and hypertension
(b)exhibits continued respiratory activity during apneic episodes
(2)central sleep apnea: caused by bilateral medullary lesions, usually in the context of postanoxic brain injury, encephalitis, Leigh’s disease (see p. 48), or disorders of the autonomic nervous system (e.g., Hirschsprung’s disease)
(a)occurs normally several times during each sleep cycle
(b)does not exhibit respiratory activity during apneic episodes
ii.symptoms: transient arrest of breathing during sleep that can be complete {apnea} or partial {hypopnea} that must be 10 seconds in duration and occur 5 times per hour to be considered pathological; other symptoms include
(1)excessive daytime sleepiness
(2)snoring, gasping, or snorting during sleep; dry mouth or sore throat upon awakening
(3)grinding of teeth during sleep
(4)mood changes: irritability, apathy
(5)sleep terrors
Box 7.18
Ondine’s curse—Repeated central sleep apneas during early postnatal life, caused by abnormal function of chemoreceptors in the central nervous system
Box 7.19
The number of apneic episodes does not correlate with daytime sleepiness.

iii.treatment
(1)nonsurgical treatment: weight loss, continuous positive airway pressure (CPAP), and oral appliances to promote airway patency
(2)surgical treatment: uvulopalatopharyngoplasty; tonsillectomy; adenoidectomy; tracheostomy in severe refractory cases
c.restless legs syndrome
i.pathophysiology: unknown, but may be related to abnormal transport and storage of iron in the brain
ii.subtypes
(1)idiopathic: most common subtype
(2)secondary: associated with iron deficiency, chronic renal failure, pregnancy, peripheral vascular disease, and Parkinson’s disease
(a)iron is a necessary cofactor for tyrosine hydroxylase, which is involved in the synthesis of dopamine
(3)iatrogenic: caused by antidepressants (particularly the SSRIs), caffeine, amphetamines, lithium
iii.symptoms
(1)paresthesias and dysesthesias of the limbs that create the urge to move the affected limb {akisthesia}; paresthesias and dysesthesias are temporarily relieved by movement of the akisthetic limb
(a)pronounced at rest and while inactive, but may be present during the day
(2)periodic limb movements (80%)
iv.diagnostic testing: serum iron and ferritin levels to identify ironresponsive patients
v.treatment
(1)dopaminergic agonists: pramipexole (Mirapex), ropinirole (Requip)
(a)initiation or increasing the dosage often exacerbates symptoms
(2)gabapentin, particularly for patients with painful symptoms
(3)iron supplementation
(4)opiates (hydrocodone, methadone) for refractory cases
d.periodic limb movement disorder
i.pathophysiology: unknown, but likely involves decreased dopaminergic activity that causes disinhibition of the spinal cord
(1)may not even be a distinct disease, but may rather be a feature of other diseases (e.g., restless legs syndrome, sleep apnea, narcolepsy)
ii.symptoms: involuntary movements of the lower upper extremities lasting 5 seconds, typically occurring during sleep stages 1–2; must be repetitive and disruptive of sleep to be considered pathological
iii.treatment: dopaminergic agonists
3.Circadian rhythm disorders
a.pathophysiology: caused by a loss of synchronization between the internal circadian rhythm and the light–dark cycle of the environment, usually from jet lag or shift work but may be due to irregularity of the internal circadian rhythm in patients with hypothalamic lesions or severe vision loss
b.subtypes
i.advanced sleep phase syndrome—symptoms include early sleep onset (before 9 PM) and early awakening (before 5 AM)
ii.delayed sleep phase syndrome—symptoms include a late sleep onset (after 2 AM) and a late awakening
Sleep Disorders
169

7 Behavioral Neurology and Psychiatry
c.treatment
i.bright light exposure
(1)in the evening for an advanced sleep phase
(2)in the morning for a delayed sleep phase
ii.melatonin, given 4–5 hours before the onset of bedtime
C.Parasomnias
1.Non-REM parasomnias
a.pathophysiology: caused by partial arousal from sleep stages 3–4
i.triggers for parasomnia attacks include
(1)an increased amount of slow-wave sleep, e.g., during recovery from sleep deprivation, at the onset of CPAP therapy, or during febrile illnesses
(2)sleep fragmentation as caused by stress, pain, sleep-related breathing disorder (snoring, sleep apnea), pregnancy, alcohol use, or new sleep site (e.g., hotel)
b.epidemiology: male predominance except for nocturnal eating/drinking behavior; more common in the young and in patients with a family history of non-REM parasomnias
c.subtypes
i.sleep terrors
ii.sleep walking {somnambulism}
iii.nocturnal eating/drinking behavior
d.symptoms: episodes last 15 minutes and develop early during sleep when slow-wave sleep is more abundant; patients are poorly responsive to external stimuli during an episode and have only a vague or no recollection of the event
e.treatment: avoid triggers; prevent injuries during attack (door alarms, motion sensors); clonazepam
2.REM parasomnias: REM sleep behavior disorder (RSBD)
a.pathophysiology: may relate to dysfunction of the pedunculopontine tegmental nucleus of the mesencephalic reticular formation that disrupts the atonia generated by the nucleus gigantocellularis and the lateral reticulospinal tract
b.subtypes
i.acute-onset RSBD: typically caused by withdrawal of sedative-hypnotic drugs or alcohol, caffeine use, or a new medication (particularly SSRIs or cholinergic medications)
ii.insidious-onset RSBD: likely a neurodegenerative disorder involving reduced striatal dopamine transporters and dopamine innervation; associated with the synucleinopathies (e.g., Parkinson’s disease, multiple system atrophies, Lewy body dementia), which may develop decades after the onset of RSBD
c.epidemiology: male predominance; increasing incidence with advanced age
d.symptoms: violent behavior directed against an apparent enemy following a brief period of autonomic and behavioral arousal; the patient will have excellent recall of a vivid dream that underlies the event
e.diagnostic testing: polysomnography demonstrates a lack of atonia during REM sleep with intrusion of complex motor behaviors
f.treatment: clonazepam
3.Transitional parasomnias: rhythmic movement disorder/jactatio capitis nocturna
a.pathophysiology: unknown; occurs during sleep onset and during transitions to and from sleep stage 2
i.common in young children but spontaneously resolves by 4 years of age
170b. symptoms: violent movements (head banging, thrashing, kicking) often associated with vocalizations; movements can cause injury
i.persistence of movements into adulthood is common only in patients with autism and mental retardation, in whom the movements may resemble the patient’s waking stereotypies
III.Psychosis
A. Schizophrenia
1.Pathophysiology: unknown cause, but predominant hypotheses include
a.neuroanatomical abnormalities
i.a migrational defect of cortical layers I–III
ii.diffuse brain atrophy with both neuronal and glial cell loss
b.prefrontal cortex dysfunction {hypofrontality}
c.increased mesolimbic dopamine activity
d.genetic factors: strong familial patterns of schizophrenia have been associated with numerous chromosomal abnormalities; increased incidence of schizophrenia occurs in monozygotic twins dizygotic twins nontwin siblings
e.psychosocial factors
i.stress-diathesis model: an environmental stress induces schizophrenia in biologically vulnerable patients (e.g., viral infection, obstetrical complication, hypoxia)
ii.sociocultural and economic factors likely influence the course of schizophrenia rather than directly causing the disorder
2.Symptoms: average age of onset at 21 years of age for men and 27 years of age for women (Box 7.20) (Box 7.21)
a.diagnostic symptoms
i.over a 6-month period, at least two of the following
(1)delusions
(2)hallucinations
(3)disorganized speech with reduced content (similar to mild form of Wernicke’s aphasia)
(4)disorganized behavior, which must impair social activities, work, or self-care
(5)flat affect, apathy, anhedonia, social withdrawal
or
ii.bizarre delusions, auditory hallucinations involving two or more voices, or an auditory hallucination making a running commentary on the patient’s activities/thought
b. other symptoms
i.preoccupation with religious, philosophical, or abstract thought
ii.ideomotor apraxia, sensory aprosody, right–left confusion, denial of illness (all are indicative of parietal lobe dysfunction)
iii.abnormal movements: grimacing, stereotypies, abnormal smooth pursuit eye movements
iv.polysubstance abuse: cigarette smoking (75%), alcohol (50%), cannabis, cocaine
v.depression, particularly after psychotic episodes (25%)
3.Diagnostic testing no laboratory values are diagnostic for schizophrenia and a diagnosis is made solely on clinical features
a.EEG: demonstrates reduced alpha and increased delta and theta frequencies during waking, frequent spikes after sleep deprivation, and increased desynchronization induced by mild auditory stimulation
b.somatosensory evoked potentials exhibit increased early waveforms (N100) and reduced delayed waveforms (P300) that may represent subcortical and cortical processing, respectively
Psychosis
Box 7.20
Subtypes of Schizophrenia
Paranoid—Mostly hallucinations and delusions
Disorganized—Mostly disorganized speech and behavior
Catatonic—Negativism, motiveless resistance to orders, posturing, echolalia, and echopraxia; may be hypoactive or agitated
Box 7.21
Good prognostic signs in schizophrenia:
Late onset; acute onset; married; positive symptoms
Bad prognostic signs in schizophrenia:
Early onset; gradual onset; single; negative symptoms
171
7 Behavioral Neurology and Psychiatry
172
Table 7–3 Antipsychotic Medications
Class |
Receptor effect |
Specific benefits |
Side effects |
|
|
|
|
|
|
Typical antipsychotics |
D2 D1 |
|
Parkinsonism, tardive |
|
|
|
antagonism |
|
disorders, neuroleptic |
|
|
|
|
malignant syndrome |
|
|
|
|
|
|
haloperidol (Haldol) |
High potency |
Sundowning |
Gynecomastia, |
|
|
|
|
impotence |
|
|
|
|
|
|
chlorpromazine |
Low potency |
Antipruritic, |
Anticholinergic effects |
|
(Thorazine) |
|
hiccups |
|
|
|
|
|
|
Atypical antipsychotics |
D2 and 5HT2 |
Minimal Parkin- |
|
|
|
|
antagonism |
sonism; tics |
|
|
|
|
|
|
|
Clozapine (Clozaril) |
|
Effective for |
Agranulocytosis; ↓ |
|
|
|
treatment |
seizure threshold |
|
|
|
refractory cases; |
|
|
|
|
↓ suicide risk |
|
|
|
|
|
|
|
Risperidone (Risperdal) |
5HT2 D2 affinity |
|
Gynecomastia, raises |
|
|
|
|
prolactin levels |
|
|
|
|
|
|
Olanzapine (Zyprexa) |
|
|
Hyperglycemia, ↑ |
|
|
|
|
weight |
|
|
|
|
|
|
Quetiapine (Seroquel) |
|
|
Sedation |
|
|
|
|
|
|
Ziprasidone (Geodon) |
D2 partial agonists |
Minimal weight |
QT prolongation |
|
Aripiprazole (Abilify) |
|
gain and |
|
|
|
|
hyperglycemia |
|
|
|
|
|
|
4.Treatment
a.antipsychotics (Table 7–3): sudden withdrawal may provoke a transient chorea ( tardive dyskinesia)
i.typical antipsychotics good for positive symptoms while relatively ineffective for negative symptoms
ii.atypical antipsychotics: efficacy for positive and negative symptoms with minimal or no potential for extra pyramidal symptoms (EPS)
b.adjuvants: anticonvulsants (carbamazepine, valproate), lithium, benzodiazepines
c.psychosocial treatment including supportive psychotherapy, social skills training, and assertive community treatment
5.Prognosis: symptoms may be continuous, episodic with or without symptoms between episodes
a.15% mortality from suicide, most commonly in the well-educated and young
B. Schizophreniform Disorder
1.Pathophysiology: exhibits abnormalities similar to schizophrenia (poor activation of the prefrontal cortex, diffuse atrophy)
2.Symptoms: at least 1 month, but 6 months, of
a.hallucinations and delusions
b.flat affect
c.absence of a concurrent mood disorder (which indicates schizoaffective disorder)
3.Treatment: antipsychotics, typically for 3–6 months
4.Prognosis: by definition, symptoms must resolve within 6 months; the postpsychosis period is typified by depression and an increased suicide risk

IV. Mood Disorders
A. Major Depressive Disorder
1.Biological pathophysiology
a.monoamine hypermetabolism and depletion
b.neuroendocrine abnormalities
i.hypothalamic-pituitary-adrenal axis: 50% of patients with major depression do not suppress adrenal cortisol secretion in response to administration of dexamethasone (i.e., in the dexamethasone suppression test), reflecting persistent hypothalamic CRH secretion
(1) failure of dexamethasone suppression is not specific for major depression
ii.hypothalamic-pituitary-thyroid axis hypoactivity, which is a nonspecific finding that occurs in many psychiatric disorders
c.circadian rhythm abnormalities: major depression patients exhibit delayed sleep onset, rapid onset of REM sleep, and increased REM density; sleep deprivation may actually improve depressive symptoms
d.genetics: exhibits a multifactorial inheritance pattern, although no candidate genes have been identified
2.Psychosocial pathophysiology
a.stressful life events often precede the initial major depressive episode, but they do not necessarily precede subsequent episodes of depression
i.development of depression is particularly associated with certain stressful events (i.e., death of a parent or spouse)
3.Symptoms: onset commonly between 20–50 years of age; onset 20 years of age is often associated with drug abuse
a.at least 2 weeks of five of the following symptoms (with at least one being depressed mood or anhedonia inflated self-esteem, decreased need for sleep, distractibility, increased talkativeness, racing thoughts, increased physical and mental activity, over-involvement in pleasurable activities (typically sex and/or shopping)
b.no mania or hypomania
4.Treatment
a.psychotherapy: cognitive–behavioral, psychodynamic (insight-oriented), interpersonal, and family therapies
b.medical: antidepressants (Table 7–4)
i.may supplement with lithium, triiodothyronine (T3), mood stabilizers, or atypical antipsychotics if the patient does not respond to antidepressant treatment
5.Prognosis: 30% may remit after successful treatment of the first episode; in nonremitters, subsequent episodes of depression become more severe and prolonged
B. Bipolar Disorders (Box 7.22)
1.Symptoms of manic and hypomanic episodes
a.both types of episodes require at least three of the following
i.mood must be either euphoric, expansive, or irritable
b.manic episode: 1 week of symptoms that impair social or occupational functioning
c.hypomanic episode: at least 4 days of symptoms that do not impair social or occupational functioning
2.Subtypes
a.bipolar I disorder—multiple manic episodes with or without major depressive episodes
i.pathophysiology
Mood Disorders
Box 7.22
Dysthymia: 2 years of depression-like symptoms that do not meet criteria for major depression; treat as per depression
Cyclothymia: 2 years of hypomanic episodes and depressive symptoms that do not meet criteria for a major depressive episode; treat as per bipolar disorder
173
(1) biological: linked to polymorphisms on chromosomes 11 and X

7 Behavioral Neurology and Psychiatry
Table 7–4 Antidepressant Medications
Class/drug |
Specific benefit |
Major side effects |
|
|
|
Selective serotonin reuptake |
Low potential for lethality in |
Insomnia, tremor, sexual |
inhibitors: |
overdose; few anticholinergic |
dysfunction; increased |
Citalopram (Celexa) |
effects, orthostatic |
suicide risk in children |
hypotension, and cardiac |
|
|
Escitalopram (Lexapro) |
|
|
arrhythmias; also useful for |
|
|
Fluoxetine (Prozac) |
obsessive-compulsive, panic, |
|
and eating disorders |
|
|
Paroxetine (Paxil) |
|
|
|
|
|
Sertraline (Zoloft) |
|
|
|
|
|
Serotonin-norepinephrine |
Anxiety disorders |
Hypertension |
reuptake inhibitors: |
|
|
Venlafaxine (Effexor) |
|
|
Duloxetine (Cymbalta) |
|
|
|
|
|
Tricyclics: |
Phobias, hypochondriasis, |
Overdose → prolonged QT, |
|
chronic pain, |
anticholinergic effects |
|
anxiety, & panic disorders |
|
Amitriptyline (Elavil) |
Headache prophylaxis, sleep |
Orthostatic hypotension, |
|
disorders, chronic pain |
sedation |
|
conditions |
|
Clomipramine |
Obsessive–compulsive disorder |
Orthostatic hypotension, ↑ |
|
|
seizure risk* |
Desipramine |
Attention-deficit disorders |
|
Nortriptyline |
Lowest incidence of orthostatic |
Arrhythmia |
|
hypotension among TCAs |
|
|
|
|
Second generation: |
|
|
Bupropion (Wellbutrin) |
Attention deficit, low incidence |
Lowers seizure threshold |
|
of sexual dysfunction |
|
Mirtazapine (Remeron) |
|
Sedation, weight gain, |
|
|
increased appetite, sedation |
|
|
|
Monoamine oxidase |
“Atypical” depression (e.g., with |
Hypertensive reaction to |
(MAO)-A inhibitors: |
hypersomnia and hyperphagia), |
tyramine ingestion; overdose |
|
phobias |
toxicity due to serotonin |
|
|
syndrome (see p. 202) |
Phenelzine |
hypochondriasis |
|
Tranylcypromine |
|
|
|
|
|
*“increased” seizure risk is still 1%. Abbreviations: TCAs, tricyclic antidepressants.
(2)psychosocial: stressful life events often precede the initial manic episode
ii.increased risk in patients with family members who have major depression or bipolar I disorder
b.bipolar II: episodes of major depression mixed with hypomanic episodes but not manic episodes
3.Treatment
a.lithium, anticonvulsants (carbamazepine, valproate, lamotrigine), antipsychotics
i.bupropion may be less likely to induce mania/hypomania in bipolar depressed patients
ii.side effects of lithium include hypothyroidism, tremor, and nephrogenic diabetes insipidus
b.cognitive–behavioral therapy
4.Prognosis: bipolar II disorder patients are at higher risk for suicide than bipolar I patients
a.rapid cycling (four episodes per year) occurs in bipolar I or II patients; may be
174 |
triggered by use of antidepressants (most often by tricyclic antidepressants) |

V. Anxiety Disorders
1. |
Pathophysiology |
|
||
|
a. |
biological: the sensation of anxiety may be related to dysfunction of the |
|
|
|
|
septohippocampal pathway that is caused by hyperactive noradrenergic |
|
|
|
|
input from the locus coeruleus and/or serotonergic input from the raphe |
|
|
|
|
nucleus |
|
|
|
|
i. anxiety disorders are associated with |
|
|
|
|
(1) |
an abnormal hypothalamic-pituitary-adrenal hormone response |
|
|
|
(2) |
an increased baseline sympathetic nervous system activity that |
|
|
|
|
over-responds to stimuli and that poorly adapts to repeated |
|
|
|
|
stimuli |
|
|
|
ii. genetics: associated with polymorphisms of serotonin transporter |
|
|
|
|
genes |
|
|
|
b. |
behavioral: the fear response to specific stimuli become generalized |
Disorders |
|
|
d. |
psychodynamic: anxiety is related to unresolved unconscious conflict or |
||
|
c. |
cognitive: learned response from repeated exposure to anxiety inducing |
|
|
|
|
situations (similar to classic conditioning) |
|
|
|
|
an unsuccessful attempt to diffuse anxiety laden impulses |
Anxiety |
|
2. |
Subtypes |
|
||
|
|
|||
|
a. |
panic attacks—symptoms include recurrent, discrete periods of intense |
|
|
|
|
fear with four or more of the following |
|
|
175
7 Behavioral Neurology and Psychiatry
(1)reexperiencing the trauma (nightmares, flashbacks)
(2)avoiding recollections of the traumatic event
(3)persistent symptoms of hyperarousal including an exaggerated startle response or hypervigilance
ii.short-lived symptoms are suggestive of an acute stress disorder ( 4 weeks duration)
iii.highly associated with substance abuse
iv.treatment: SSRIs, serotonin and norepinephrine reuptake inhibitors (SNRIs; i.e., venlafaxine), beta blockers, MAOIs; psychotherapy
VI. Somatoform Disorders and Factitious Disorders
A. Somatization Disorder (Box 7.23)
1.Pathophysiology
a.psychological: may relate to behavioral patterns of emotional expression that are learned in unstable social settings; 50% coincidence with personality disorders, major depression, bipolar I disorder, anxiety disorders, and/or substance abuse
b.biological: reports of patients exhibiting decreased metabolic activity in several brain regions
i.genetics: exhibits familial patterns
2.Symptoms: almost always begins 30 years of age
a.diagnostic symptoms: at least a 2-year period of multiple medically unexplainable symptoms including
i.at least four different pain complaints (e.g., pain with menstruation, during urination, or during sexual intercourse)
ii.at least two gastrointestinal symptoms (e.g., bloated sensation, flatulence, food intolerance)
iii.at least one symptom related to sexual function (including abnormal menstrual bleeding)
iv.at least one “pseudoneurological” symptom (e.g., dysphagia, glomus sensation), usually milder than those of conversion disorder and nonspecific
3.Treatment: after a thorough medical evaluation (particularly for patients30 years of age)
a.evaluation and treatment of other underlying psychiatric comorbidities, with careful monitoring of medication use due to poor compliance
b.regularly scheduled clinic visits with a single physician
c.psychotherapy
4.Prognosis: Chronic, undulating, and relapsing disorder; symptoms are exacerbated by stress, and the patient is chronically debilitated by the symptoms
B. Conversion Disorder
1.Pathophysiology
a.psychological: may be caused by repression of an unconscious intrapsychic conflict and its representation as a physical symptom
i. associated with major depression, anxiety disorders, and schizophrenia
b.biological: SPECT scanning can occasionally demonstrate hypoperfusion of the basal ganglia (particularly the caudate) and thalamus contralateral to the side of sensorimotor conversion symptoms
2.Symptoms: excludes pain-related symptoms such as those of somatization disorder
Box 7.23
Other Symptoms of Somatization
Disorder
Multiple positives during review-of- systems
History of multiple surgeries
Receiving medical care from multiple physicians
Self-perception of sickly nature
176

a.motor dysfunction
i.weakness may involve muscle atrophy in long-standing conversion disorder
ii.extrapyramidal-like motor dysfunction (e.g., dystonia, tremor)
iii.astasia-abasia—exceptionally ataxic gait with minimal falling that often involves thrashing of the trunk and arm movements that would only seem to worsen an unsteady gait
b.sensory loss: classically in a neuroanatomically inconsistent pattern (e.g., stocking or glove distribution) or precisely splitting the midline
c.vision loss: commonly involves tunnel vision or blindness without significant functional impairment
d.pseudoseizures (see p. 99)
e.inappropriate lack of concern for symptoms {la belle indifference} although it is not considered an accurate indicator of conversion disorder
3.Treatment: Psychomotor and sensory rehabilitation; psychotherapy; may require anxiolytics or antidepressants
4.Prognosis: The majority of symptoms spontaneously resolve but 25% recur
a.upwards of 50% of patients diagnosed with conversion disorder may subsequently be found to have focal neurological disease or a causative medical condition
C. Hypochondriasis
1.Pathophysiology: may represent misinterpretation of, and elaboration upon, common sensations, or a learned behavioral pattern of emotional expression
a.associated with major depression or anxiety disorders in 80%
2.Epidemiology: equally common in men and women; more common in Blacks; not related to socioeconomic status
3.Symptoms: at least 6 months of the false belief that the patient suffers from a disease based upon the misinterpretation of benign physical sensations or signs, wherein the patient cannot be convinced that there is no underlying disease despite normal diagnostic evaluations
a.typically involves only a few physical sensations or signs, unlike somatization disorder
b.hypochondriac beliefs must cause emotional distress and impair the patient’s activities of daily living
4.Treatment: evaluation and treatment of other underlying psychiatric comorbidities; avoid medications except for treatment of psychiatric comorbidities
5.Prognosis: often exhibits episodic course that is exacerbated by stress; 50% spontaneously resolve
VII. Substance Dependence and Abuse
1.Definitions
a.substance abuse: a maladaptive pattern of drug use that interferes with a person’s activities of daily living
b.substance dependence: severe pattern of drug use leading to clinically significant impairment or distress; involves tolerance, withdrawal, unsuccessful attempts to cut down, and continued drug use despite physical or psychological problems that it causes
2.General epidemiology: psychiatric comorbidities occur in 70% of substance abuse patients, particularly polysubstance abuse, antisocial personality disorder, and major depression
Substance Dependence and Abuse
177

7 Behavioral Neurology and Psychiatry
178
3.Specific drugs-of-abuse a. alcohol
i.biochemical effects: acute use increases cell membrane fluidity that indi-
rectly may activate nicotinic acetylcholine-, 5-HT3-, and GABAA-receptors, and inhibit glutamate receptor and voltage-gated calcium channels
ii.acute alcohol intoxication
(1)symptoms include
(a)behavioral disinhibition (Box 7.24), mood lability, and impaired judgment
(b)dysarthria, ataxia, and nystagmus from cerebellar dysfunction
(c)autonomic dysfunction: hypoglycemia, hypothermia
(d)respiratory depression
(e)somnolence progressing to coma
(f)episodes of pure anterograde amnesia {blackouts}
(2)treatment: respiratory support; correction of electrolyte and acidbase abnormalities
iii.chronic alcohol use
(1)neurological complications
(a)malnutrition due to dietary changes and inhibition of vitamin uptake in the intestines, particularly vitamins B1/thiamine and B12
(b)alcoholic neuropathy: a length-dependent axonal sensorimotor neuropathy that frequently involves pain and dysesthesias, trophic changes of the skin (hyperpigmentation, edema, ulceration, cellulitis), and joint injuries
(i)involvement of the vagus nerve (25%) causes hoarseness and orthostatic hypotension
(ii)treatment: abstinence and multivitamin supplementation provides slow, partial recovery
(c)midline cerebellar degeneration—pronounced atrophy of the anterior and superior cerebellar vermis that includes all cell types; mild atrophy of the cerebellar hemispheres, deep cerebellar nuclei, and inferior olive nucleus
(i)symptoms include subacute development of gait and truncal ataxia with minimal limb ataxia (present in the lower extremities, if at all); no nystagmus or ocular dysmetria, dysarthria, hypotonia, or tremor
1.symptoms worsen during periods of active drinking or during intercurrent illness
(ii)treatment: improves significantly with abstinence and multivitamin supplementation
(d)tobacco-alcohol/nutritional amblyopia—likely a multivitamin deficiency state and not a direct toxic effect of alcohol because it occurs in nonalcoholic malnourished patients
(i)symptoms include bilateral painless scotoma and loss of acuity
(ii)treatment: multivitamin supplementation
(e)demyelinating disorders: Marchiafava-Bignami disease involves progressive degeneration of the corpus callosum; rarely central pontine myelinosis (see p. 112)
(2)general treatment
(a)support groups (i.e., Alcoholics Anonymous); psychotherapy focused on situations leading to drinking (most effective if family is involved); behavioral therapy involving operant conditioning
Box 7.24
Alcoholic behavioral disinhibition can result in violent outbursts, which may be an idiosyncratic reaction to the alcohol; rarely occurs with barbiturate intoxication.
(b)disulfiram (Antabuse) prophylaxis: inhibits the second metabolic step in the breakdown of alcohol by alcohol dehydrogenase, which allows accumulation of acetaldehyde that causes flushing and nausea
(c)acamprosate, naltrexone, lithium, topiramate, and SSRIs may reduce alcohol craving and help maintain abstinence
iv.alcohol withdrawal
(1)tremor (develops 8 hours after abstinence)
(2)psychosis (develops 8–12 hours after abstinence)
(3)seizures (Box 7.25) (develops 12–24 hours after abstinence): typically are multiple but 3% develop status epilepticus; may be related to concurrent hypoglycemia, hyponatremia, or hypomagnesaemia
(a)treatment: benzodiazepines
(4)delirium tremens (3–4 days after abstinence): involves delirium, tremor, autonomic instability, wildly fluctuant psychomotor activity, and hallucinations (visual and tactile); typically develops in binge drinkers with 5-year history of alcohol use and some degree of liver failure
(a)treatment: scheduled benzodiazepines (preferably chlordiazepoxide) for prophylaxis; beta blockers for autonomic instability; antipsychotics for persistent psychotic symptoms
b.sedative-hypnotic drugs
i.includes
(1)benzodiazepines
(2)barbiturates: pentobarbital (Nembutal), secobarbital, pentobarbi-
tal |
secobarbital (Amytal) |
(3)barbiturate-like drugs: methaqualone (Quaalude)
ii.biochemical effects: barbiturates are direct agonists of GABAA receptors; differ from benzodiazepines by potentiating GABA-induced chloride currents by prolonging periods of activity rather than increasing frequency of channel opening
(1)long-term use causes downregulation and desensitization of GABAA receptors
(2)all sedative-hypnotics exhibit cross-tolerance with alcohol
iii.acute sedative-hypnotic intoxication: symptoms include
(1)behavioral disinhibition, mood lability, impaired judgment
(2)cerebellar dysfunction: dysarthria, ataxia, nystagmus
(3)memory impairment
(4)somnolence, coma (Box 7.26)
(5)respiratory depression
(6)hypotension
iv.sedative-hypnotic withdrawal: symptoms include
(1)anxiety, dysphoria
(2)insomnia
(3)photoand phonophobia
(4)nausea/vomiting
(5)seizures
(6)visual, tactile, or auditory hallucinations or illusions
v.treatment
(1)acute intoxication treatment with
(a)benzodiazepines: flumazenil; gastric lavage with charcoal
(b)barbiturates: gastric lavage with charcoal
Box 7.25
Patients suspected of alcoholic seizures still should be evaluated for an underlying disease of the CNS; 3% will have a surgically treatable brain lesion.
Box 7.26
Death by overdose-respiratory suppression with benzodiazepines generally occur only when they are used together with other sedative-hypnotic drugs or alcohol.
Substance Dependence and Abuse
179

7 Behavioral Neurology and Psychiatry
(2)detoxification
(a)gradually taper benzodiazepines
(b)convert barbiturates to equivalent doses of long-acting phenobarbital, then gradually taper
(c)psychotherapy or behavioral therapy
c.cocaine
i.biochemical effects: blocks the reuptake transporters primarily for dopamine, but also for norepinephrine, and serotonin, thereby increasing neurotransmitter levels in the synaptic cleft; dopamine activity is increased particularly in the ventral tegmental projections to the limbic system (mesolimbic dopaminergic “reward pathway”) where it acts on D1 and D2 receptors (similar to amphetamines)
(1)use of high-potency forms (i.e., crack) may induce craving after a single use
ii.acute cocaine intoxication: symptoms are the same as for amphetamine intoxication, but overdoses can easily be fatal due to arrhythmia
(1)euphoria, impaired judgment
(2)autonomic dysfunction (e.g., hypertension, hyperthermia)
(3)agitation, insomnia
(4)stereotypies, automatisms
(5)dystonias, dyskinesias
(6)paranoia, psychosis
(7)delirium, particularly in patients with preexisting brain injuries
(8)seizures
(9)stroke (hemorrhagic or ischemic), myocardial infarction
iii.chronic cocaine use: complications include hypertension, vasculitis, and cardiomyopathy
iv.cocaine withdrawal: symptoms include dysphoria, anxiety, fatigue, hypersomnolence, nightmares, and depression
v.treatment
(1)acute intoxication: antipsychotic medications, except in cases with hyperthermia, which they can exacerbate; benzodiazepines
(2)amantadine, bromocriptine may be used to ease withdrawal symptoms
(a)evaluation for underlying psychiatric comorbidities after detoxification, particularly depression and attention-deficit disorder
d.amphetamines
i.include dextroamphetamine, methamphetamine, methylphenidate, and numerous synthetic compounds (e.g., ecstasy, crank, methylenedoxymethamphetamine [MDMA]); ephedrine and propanolamine have similar effects but are not considered true amphetamines
ii.biochemical effects: all amphetamines reverse the dopamine reuptake transporter thereby increasing extracellular dopamine, particularly that of the ventral tegmental projections to the limbic system (mesolimbic dopaminergic “reward pathway”)
(1)the synthetic amphetamines also increase extracellular serotonin levels, which may account for their hallucinogenic properties
iii.acute amphetamine intoxication: symptoms are as for acute cocaine intoxication; it rarely is fatal (unlike cocaine overdose)
(1)MDMA is frequently associated with hyperthermia
iv.chronic amphetamine use: complications include anorexia and cachexia, hypertension, and vasculitis
180

v.amphetamine withdrawal: as per cocaine withdrawal
vi.treatment
(1)acute intoxication: antipsychotics; benzodiazepines
(2)chronic use: inpatient supportive care
e.marijuana/cannabis
i.biochemical effects: tetrahydrocannabinol (THC), which is the active compound in marijuana, binds two specific G-protein-linked cannabinoid receptors that are located predominantly in the basal ganglia, hippocampus, and cerebellum; the endogenous ligands are anandamide and 2-arachidonylglycerol, which are breakdown products of phosphatidylcholine and phosphatidylinositol, respectively
(1)THC does not appear to induce craving, but does induce tolerance
ii.symptoms
(1)acute intoxication
(a)euphoria, disorientation to time, impaired judgment
(b)increased sensitivity to sensory inputs but slowed reaction times
(c)conjunctival injection, hunger, dry mouth
(2)withdrawal: mild irritability, insomnia, and nausea
iii.treatment: none specific
f.opioids
i.include
(1)natural products or their derivatives: morphine, heroin, codeine, hydromorphone (Dilaudid)
(2)synthetic drugs: meperidine (Demerol), methadone, propoxyphene (Darvon)
ii.biochemical effects: appear to involve the ventral tegmental dopamine projections to the limbic system (i.e., the mesolimbic dopaminergic “reward pathway”), but unlike cocaine and amphetamines, opioids may also directly act in the nucleus accumbens through dopamine-independent mechanisms
(1)withdrawal produces rebound noradrenergic activity that is responsible for most of the unpleasant symptoms, which can be reduced by presynaptic 2 receptor agonists (i.e., clonidine) that likely act in the locus coeruleus
iii.symptoms
(1)acute intoxication: euphoria progressing to apathy; psychomotor agitation or retardation; analgesia; impaired memory; dysarthria; somnolence, coma; seizures (with meperidine use)
(2)withdrawal: dysphoria, insomnia; nausea, rhinorrhea, increased lacrimation, diarrhea; sympathetic hyperactivity (pupillary dilation, piloerection, diaphoresis, hyperthermia); diffuse myalgias and arthralgias
iv.treatment
(1)acute intoxication: reverse with naloxone (Narcan) or naltrexone carefully dosed so as not to precipitate a withdrawal reaction
(2)detoxification: conversion to methadone followed by gradual tapering; clonidine; buprenorphine (a partial opioid agonist associated with limited physical dependence and minimal withdrawal symptoms)
g.phencyclidine (PCP) and ketamine
i.biochemical effects: both are NMDA glutamate receptor antagonists; PCP also increases release of dopamine, particularly the ventral tegmental projections to the limbic system (“reward pathway”)
Substance Dependence and Abuse
181
7 Behavioral Neurology and Psychiatry
ii.acute intoxication: aggression and impulsiveness; impaired judgment; psychosis with auditory and visual hallucinations, delirium; cerebellar dysfunction (ataxia, dysarthria, horizontal and vertical nystagmus); autonomic dysfunction (hypertension, hyperthermia); rigidity with rhabdomyolysis; reduced pain sensitivity; hyperacusis; amnesia; seizures
iii.treatment
(1)urine acidification to increase excretion
(2)benzodiazepines, antipsychotics; supportive care is generally insufficient
182