


3 Seizures and Epilepsy
3
Seizures and Epilepsy
Note: Significant diseases are indicated in bold and syndromes in italics.
I. Diagnostic Testing
1.Scalp electroencephalogram (EEG): scalp recordings cannot detect electrical synchronization of 6 cm2 of cortex, and have poor resolution of mesial and orbital frontal cortex, mesial parietal cortex, and mesial temporal cortex (Box 3.1)
a.special electrodes
i.sphenoid electrodes: inserted through temporal and masseter muscles underneath the zygomatic arch and positioned next to the foramen ovale; assesses the mesial temporal cortex
ii.nasopharyngeal electrodes: inserted into the posterior nasopharynx; assesses the mesial temporal cortex
(1)can also be advanced into the ethmoid sinuses to record the orbitofrontal cortex
b.effects of hyperventilation: induce a clinical seizure in 0.1% of generalized epilepsy and 0.5% of partial epilepsy, but increase interictal epileptiform
discharges in 12% of generalized epilepsy and 3% of partial epilepsies; may induce 50% of absence seizures
c.effects of sleep, waking, and sleep deprivation: all can provoke epileptiform activity
i.particularly good at inducing epileptiform activity in cases of infantile spasms and juvenile myoclonic epilepsy
d.effects of photic stimulation: strobe light stimulation may produce
i.photic driving responses: composed of rhythmic activity elicited over the posterior regions; occurs in 80% of epilepsies, but it also can be a normal physiological response
ii.photoparoxysmal response: spike-and-waves or polyspike-and-waves triggered by photic stimulation in 50% of generalized seizures but only 3% of focal seizures
iii.photomyoclonic response: brief repetitive spikes generated by contraction of muscles of the face or scalp that are driven by photic stimulation; seen in normal and epileptic patients
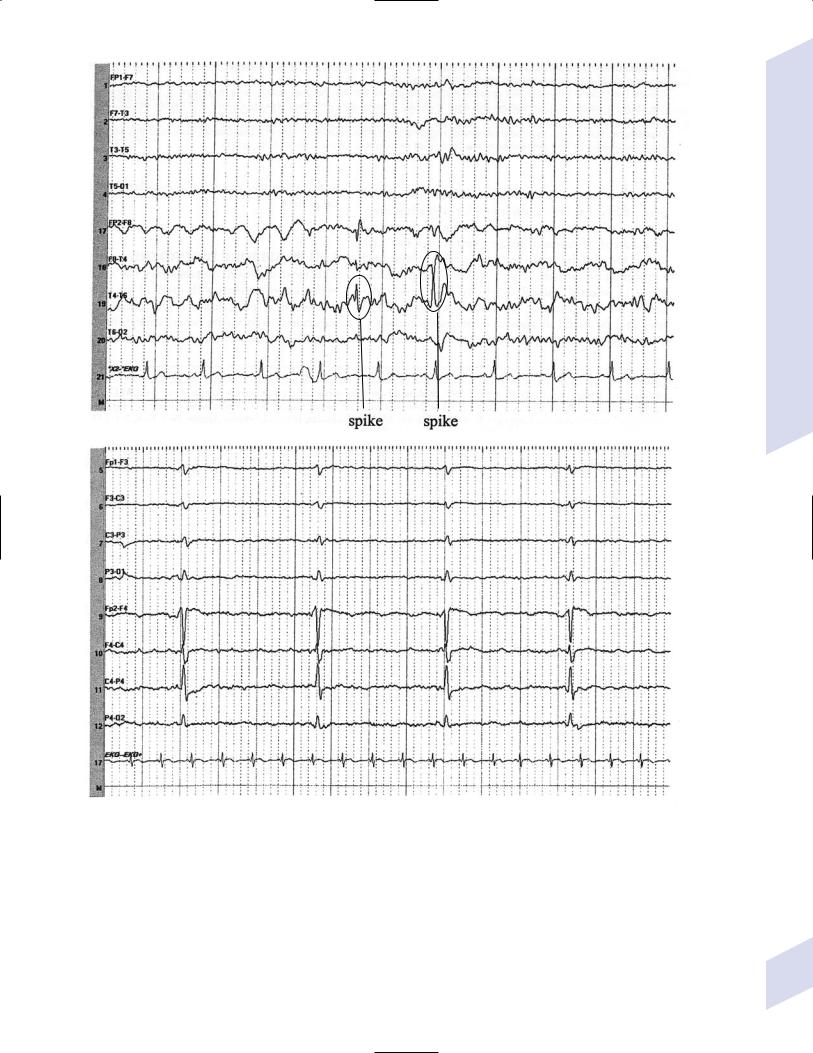
e.abnormalities consistent with seizure foci (Fig. 3–1)
i.sharp waves (also known as sharp-wave complexes, spikes, and spikewave complexes)
(1)occipital sharp waves are commonly seen in children and are not predictive of seizure foci
(2)absence of sharp waves does not rule out seizure foci
ii.amplitude asymmetries: alpha rhythm asymmetries occur normally, but generally one side is augmented rather than decreased; increased amplitudes also occur in the context of removal of portions of the skull {breech rhythm}
Box 3.1
EEG frequencies – delta ( 4 Hz); theta (4–7 Hz); alpha (8–13 Hz); beta ( 13 Hz)
80
Diagnostic Testing
A
B
Figure 3–1 Abnormalities consistent with a seizure focus. (A) Spike-and-wave discharge; (B) periodic lateralizing epileptiform discharges (PLEDS).
(1)reduced amplitudes may occur because of cortex injury or tissue collection between the cortex surface and the electrodes (e.g., subdural hemorrhage)
iii.frequency asymmetries: hemispheric background frequencies should be within 1 Hz of each other; asymmetry is a very nonspecific finding
iv.polymorphic delta activity: delta activity that is variable in morphology and frequency; when present focally, it usually indicates a lesion of the white matter, but it localizes the lesion quite poorly
81
3 Seizures and Epilepsy
C
Figure 3–1 (Continued) (C) frontal intermittent rhythmic delta activity (FIRDA). (From Drazkowski J. Use of EEG in a consultative role. Semin Neurol 2000, 23: 296, Fig. 1; 300, Fig. 6; 302, Fig. 8. Reprinted by permission.)
v.rhythmic delta activities: typically are associated with subcortical or even brainstem lesions when the background rhythm is otherwise normal
(1)frontal intermittent rhythmic delta activity (FIRDA): occurs in adults, particularly with renal or hepatic encephalopathies or hydrocephalus
(2)occipital intermittent rhythmic delta activity (OIRDA): occurs in children, particularly in the context of absence epilepsies
vi.periodic lateralizing epileptiform discharges (PLEDS): focal bursts epileptiform discharges that occur repetitively every 1–5 seconds; indicates acute or subacute focal cortical injury, or occasionally a metabolic disorder (particularly nonketotic hyperglycemia) superimposed on a chronic, focal cortical injury
(1)bilateral PLEDS is suggestive of diffuse cortical injury or encephalopathy
2.Prolactin level: Useful only if collected within 15–20 minutes of the seizure
a.prolactin levels are increased over baseline level in 80% of generalized seizures and 70% of complex-partial seizures
b.prolactin levels are rarely increased after pseudoseizures, after prefrontal seizures that symptomatically resemble the bizarre behaviors of pseudoseizures {pseudo-pseudoseizures}, or after status epilepticus
c.seizures also increase CRH and LH secretion (Box 3.2)
II. Partial Seizures
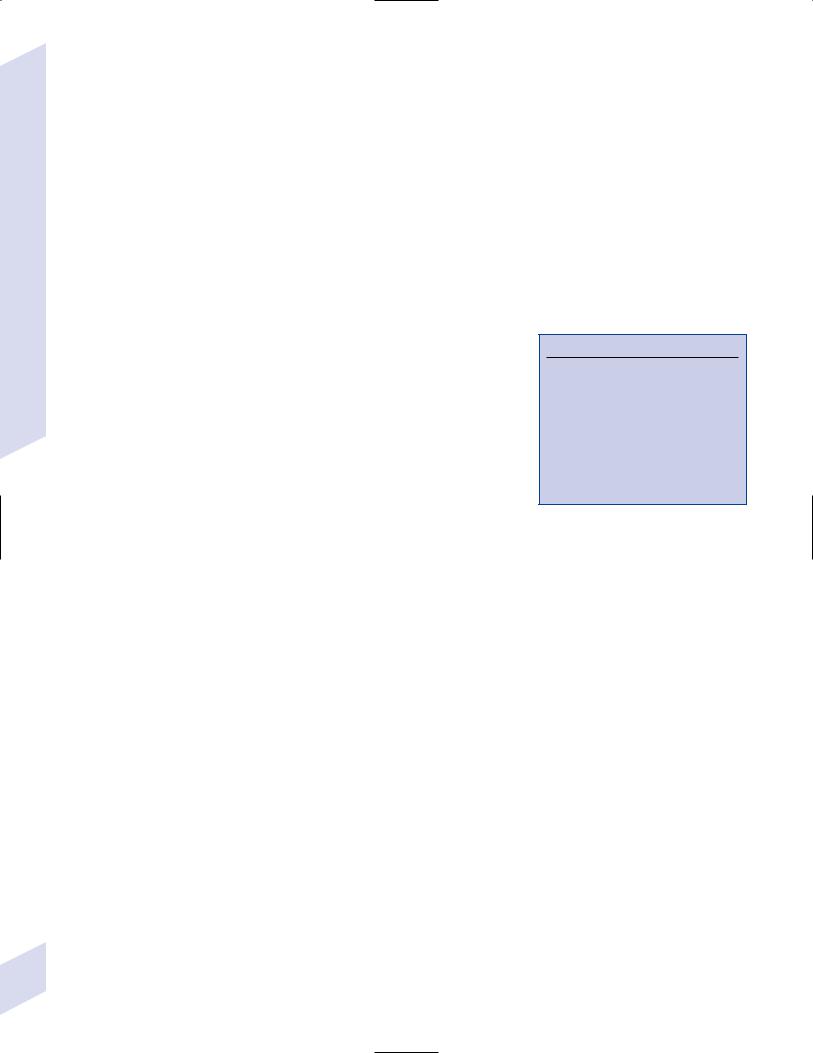
1.Simple partial seizures (Fig. 3–2): do not involve impairment or loss of consciousness irrespective of the complexity or distribution of convulsive activity; form the aura of complex partial seizures
82 |
a. simple partial motor seizures |
Box 3.2
Recurrence of Seizures: Overall risk of seizure recurrence is 40% after two years.
Low-recurrence risk with idiopathic or cryptogenic seizures and a normal EEG (25%)
Medium-recurrence risk with an abnormal neurological exam, neuroimaging, or EEG (50%)
High-recurrence risk with an abnormal neurological exam, neuroimaging, and EEG (65%)
The likelihood of developing epilepsy is increased by having antecedent conditions associated with an increased risk of epilepsy, seizures occurring during sleep, a family history of seizures, an abnormal EEG, particularly one with epileptiform abnormalities, or partial seizures

Partial Seizures
Figure 3–2 Symptomatic manifestations of focal seizures according to the seizure focus. (From Mumenthaler M. Neurological Differential Diagnosis. 2nd ed. Stuttgart, Germany: Georg Thieme; 1992:56, Fig. 20. Reprinted by permission.)
83
3 Seizures and Epilepsy
84
i.primary motor cortex focus (Brodmann area 4): causes clonic or tonic-clonic movements according to the motor homunculus; rarely causes weakness without movements
(1)Jacksonian march—progression of convulsive movements as the epileptiform activity migrates along the motor homunculus
ii.supplementary motor area focus (Brodmann area 6): frequent, brief seizures occurring in clusters with a rapid onset and offset; posturing usually involves the contralateral upper extremity and contraversive head and eye deviation {fencer’s position}
iii.premotor cortex focus (Brodmann area 6) and frontal eye field focus (Brodmann area 8): cause unilateral forced gaze and head deviation {version seizures}, which are reliably contralateral only when the focus is in the frontal eye field
(1)may involve a decrease in the level of consciousness, which would be better classified as a complex-partial seizure
iv.insula cortex focus: typical symptomatic progression involves abnormal laryngeal sensation, followed by dysarthria with perioral and/or contralateral body paresthesias, and finally contralateral convulsions in the face and/or arm (Box 3.3)
v.frontal operculum focus: causes bilateral facial movements accompanied by mastication, salivation, and swallowing after an aura consisting of epigastric sensations, fear, and/or autonomic phenomena; may cause speech arrest or vocalizations with foci in either hemisphere, or a Broca’s-type aphasia with a focus in the dominant hemisphere
vi.dorsolateral prefrontal cortex focus: causes contralateral forced head and gaze deviation
b.simple partial sensory seizures
i.parietal lobe focus: most seizure foci are in the primary somatosensory cortex (Brodmann areas 3-1–2), which causes contralateral tingling and/or numbness according to the sensory homunculus that frequently progresses to frank motor seizure activity after a period of general tremulousness
(1)sensory symptoms can have a Jacksonian march equivalent
(2)negative seizure phenomena with parietal seizures
(a)loss of awareness of a body part {asomatognosia} can occur with nondominant hemisphere seizures
(b)receptiveor conductive-like aphasias can occur with dominant hemisphere seizures
(3)pain or temperature sensations, or bilateral sensations suggest involvement of the secondary somatosensory cortex (Brodmann area 40)
(4)the sensation of vertigo (10%), sinking, choking, or nausea indicates inferior parietal lobe involvement
(5)changes in affect may also be associated with parietal lobe seizures
ii.primary visual cortex focus (Brodmann area 17): causes elementary visual sensations (flashes, scotoma, hemianopia, amaurosis)
(1)seizures often involve tonic eye contraversion, oculoclonic, or nystagmoid eye movements, forced eye closure, eyelid fluttering, sensory hallucinations of eye movements, or even the sensation of eye pain
iii.occipito-parieto-temporal junction focus: causes complex imagery or hallucinations; frequently visualized objects are recognizable but are distorted in size {micropsia, macropsia} and/or shape {metamorphopsia}
iv.temporal lobe focus: two thirds of seizure foci are mesial; one third are lateral (i.e., neocortical)
Box 3.3
Gelastic Seizures
Related to hypothalamic hamartomas but EEG identifies epileptiform activity in frontal or temporal lobes
Laughter begins in childhood; later involves drop attacks, generalized seizures, cognitive impairment, precocious puberty, and behavioral disorders
Treatment: lamictal or clonazepam; surgery, particularly stereotaxic

Table 3–1 Complex Partial Versus Absence Seizures
Feature |
Complex Partial |
Absence |
|
|
|
Aura |
Yes |
No |
Hyperventilation-inducible |
No |
Yes |
Photic stimulation-inducible |
No |
Maybe |
EEG |
Temporal spike & wave |
Diffuse 3 Hz spike & wave |
Postictal confusion |
Yes |
No |
Duration |
Minutes |
Seconds |
Automatisms |
Present |
Only if prolonged |
|
|
|
(1)causes auditory or olfactory hallucinations, emotional, or psychic symptoms (e.g., flashbacks, déjà vu, jamais vu), sensations of vertigo, and autonomic changes (e.g., epigastric rising sensation, GI upset with nausea and hypermotility, respiratory arrest)
(a)olfactory hallucinations are usually unpleasant, indicating medial temporal lobe involvement {uncinate fits}
(b)gustatory sensations suggest involvement of the temporal opercular cortex
(2)rare manifestations include forced thinking (a rapid recollection of past life experiences), depersonalization, extremes in affect, or rage
2.Complex partial seizures: involves some impairment of consciousness without tonic-clonic activity; does not have to involve a complete loss of consciousness (Table 3–1)
a.general pathophysiology: 40% of cases have seizures that are initiated by a temporal lobe focus, either mesial or neocortical
b.general symptoms: can begin with a simple partial seizure that serves as an aura, or with outright impairment of consciousness
i.complex partial seizures often involve coordinated involuntary movements
(1)reactive automatism: a reaction to the environment (i.e., drinking from a cup that is placed in the hands)
(2)perseverative automatism: the persistence during a seizure of a complex action that was being performed before the seizure onset
c.mesial temporal cortex seizure foci: typically begins with auras consisting of epigastric sensations, fear, and/or oro-alimentary automatisms
i.mesial temporal/hippocampal sclerosis
(1)subtypes: do not correlate with seizure severity or responsiveness to treatment
(a)classic mesial temporal sclerosis: cell loss in the hilum of the dentate gyrus, the tip of the CA4 region, and the CA1subiculum interface; the CA2 region is relatively preserved (Box 3.4)
(b)minimal mesial temporal sclerosis: cell loss in the hilum of the dentate gyrus
(c)total mesial temporal sclerosis: cell loss in the dentate gyrus and CA1–4 regions
(2)histology: sclerosis implies neuron loss and gliosis in the affected regions
(a)50% of cases exhibit bilateral sclerosis but usually it is asymmetric
(b)20% of cases coexist with another pathological process (e.g., cortical dysplasia, tumor)
Partial Seizures
Box 3.4
The CA2 region is particularly susceptible to damage from status epilepticus.
85
3 Seizures and Epilepsy
(3)pathophysiology: unknown cause but often disease onset is preceded by febrile seizures
(a)atrophy of the mesial temporal lobes exists at the time of seizure onset, and can be observed even in neonates
(b)mesial temporal sclerosis exhibits familial segregation, so it likely has a genetic predisposition
(4)specific diagnostic testing: EEG demonstrates spike-and-wave complexes in the inferior temporal leads or low-voltage beta activity at seizure onset that remains local for several seconds before spreading; best observed with subdural/depth electrodes
d.neocortical temporal foci: seizures often begin with auditory hallucinations, vertigo, or visual misperceptions; language disturbance can occur with foci in the dominant hemisphere
i.musicogenic seizures: a type of reflex seizure (Box 3.5) that is triggered by music (e.g., a particular composition, the sound from a certain instrument, or discussing or thinking about music)
(1)not the same as a seizure involving the perception of sound or musical notes {musical partial seizure}, wherein the perceived sound tends to be relative unformed (e.g., ringing, whistling)
e.frontal lobe seizure foci: cause complex and bizarre motor automatisms (e.g., eyelid fluttering, repetitive bilateral movements) that occur in clusters, often with minimal post-ictal confusion; frequently are confused with pseudoseizures
i.frontal operculum focus: causes auditory or visceral auras and visceral motor phenomena (salivation, spitting, vomiting)
ii.cingulate cortex or supplementary motor area (Brodmann area 6) focus: cause asymmetric bilateral posturing movements with automatisms; cingulate foci may cause gesturing, simple sexual behavior, or ictal laughter
iii.frontopolar cortex focus: causes forced thinking, aversive head movements, and/or axial clonic jerks
iv.orbitofrontal cortex focus: causes complex motor automatisms and olfactory hallucinations due to involvement of the cortex overlying the septal nuclei that receives the medial olfactory stria; may involve formed vocalizations (e.g., cursing)
v.dorsolateral frontal cortex focus: causes tonic posturing and speech arrest
3.Diagnostic testing: Scalp EEG gives adequate localization in only 50% of cases with temporal foci, and is even worse with frontal lobe foci; a single EEG demonstrates abnormalities in 40% of possible seizure cases, which is increased to 75% with prolonged EEG monitoring
a.ictal EEG patterns of complex partial seizures
i.rhythmic spike and wave activity, which are bilateral in 30% of cases
ii.rhythmic 5–7 Hz activity without spikes
iii.focal attenuation of background activity
Box 3.5
Reflex Seizure Triggers
Visual patterns (as in 3% of all epilepsies)
Tactile stimuli: hot water, body postures
Problem-solving
Reading, writing, or speaking
Specific movements
Startle involving any kind of stimulus
III. Nonsyndromal Generalized Seizures
1.Tonic-clonic seizures
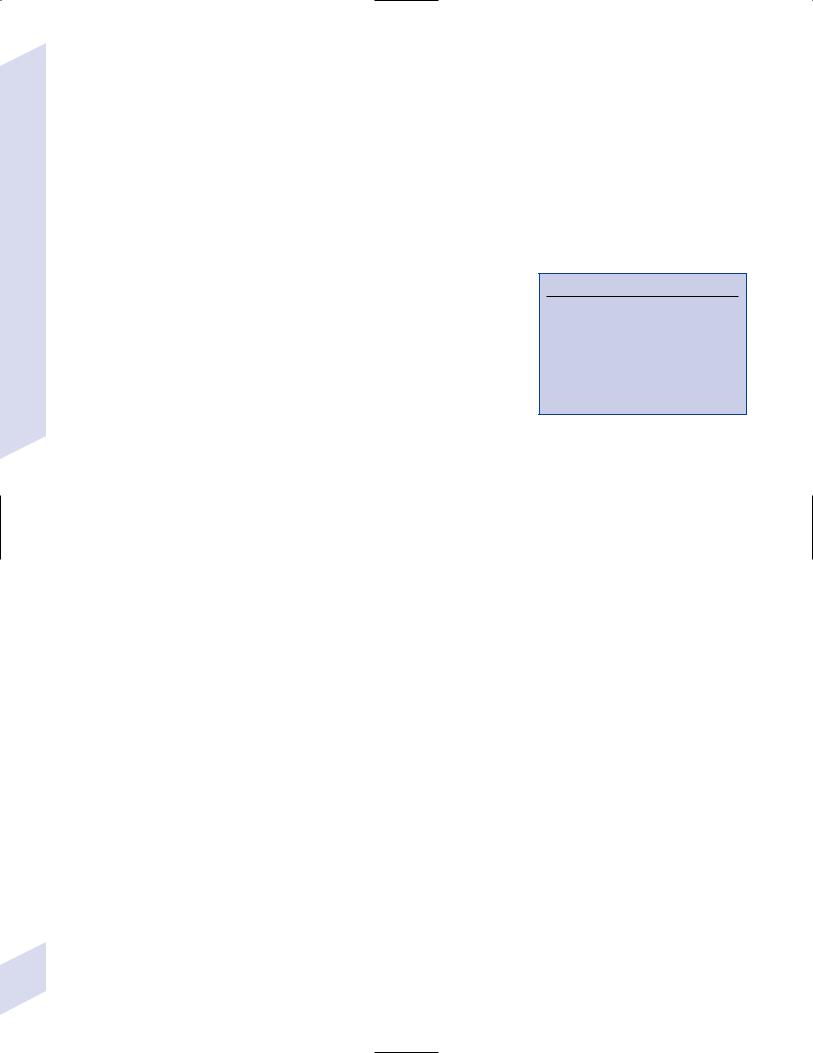
a. symptoms: convulsive activity typically lasts 1 minute; five phrases include
i.premonition phase: involves mood changes (e.g., irritability) and headaches that may last for days prior to the seizure
ii.immediately pre-tonic-clonic phase: involves a few myoclonic jerks or brief clonic seizure activity; occasionally begins with forced eye and
86 |
head deviation {aversive movements} |
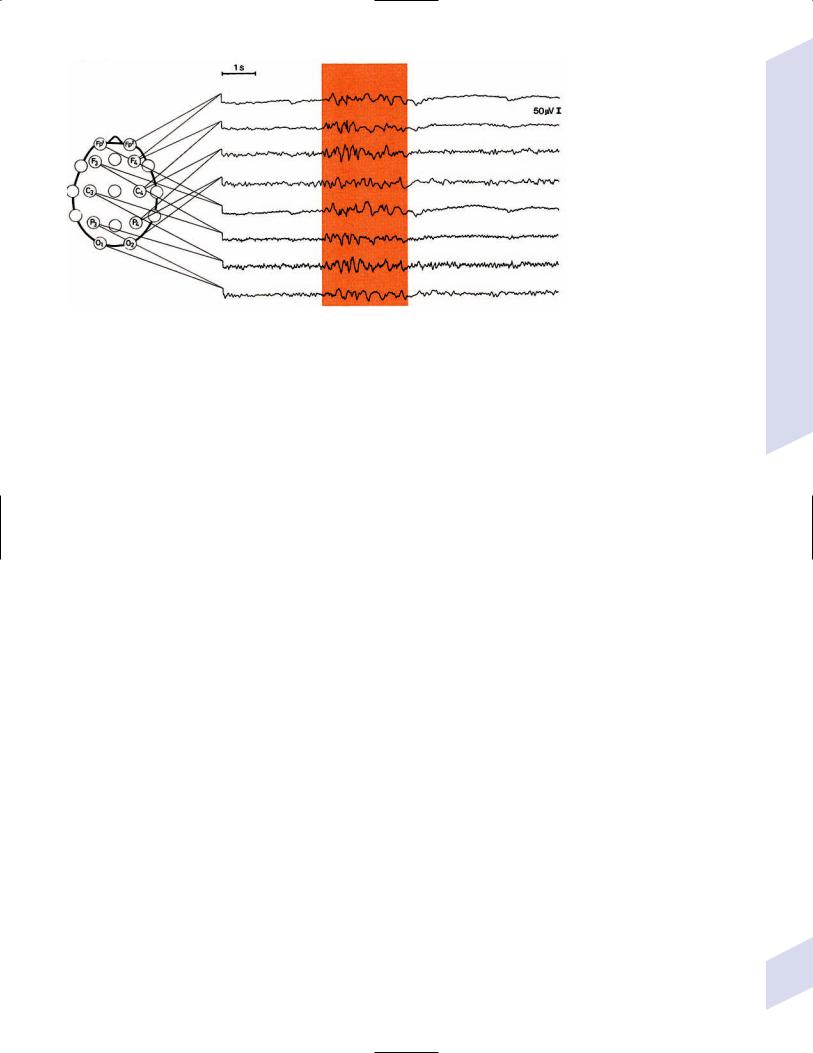
Figure 3–3 Generalized tonic-clonic seizure. (From Mumenthaler M. Neurological Differential Diagnosis. 2nd ed. Stuttgart, Germany: Georg Thieme; 1992, Fig. 1, p. 1. Reprinted by permission.)
iii.tonic phase: contracture of the axial musculature with upward eye deviation, pupillary dilation, and forced expiration of air {epileptic cry}; usually involves some decerebrate posturing
(1)tongue and jaw muscle tonus causes perioral injury, typically lateral tongue biting
(2)frequently patient becomes cyanotic, tachycardic, and hypertensive
iv.clonic phase: starts as low-amplitude, high-frequency ( 8 Hz)
convulsive movements of the extremities thorax and abdomen that progresses to high-amplitude, low-frequency ( 4 Hz) movements
(1)development of atonia breaks the seizure and causes incontinence
(2)bilaterally asynchronous movements may be caused by separate unilateral seizure foci, but this is rare
v.post-ictal phase: patient is poorly responsive and hypotonic; confusion and memory impairment may last a few minutes to hours, occasionally followed by psychiatric changes (depression, psychosis, anxiety, irritability) that can persist for a day
(1)post-ictal phase involves generalized fatigue, soreness, and migraine headaches
(2)laboratory testing during the post-ictal phase demonstrates respiratory-metabolic acidosis, hyperglycemia, and mild cerebrospinal fluid pleocytosis
b.epidemiology: unlike most other seizure types, occurs with similar incidences across all ages
c.diagnostic testing (Fig. 3–3)
i.ictal EEG: bilateral hemispheric involvement from the seizure onset that is bilaterally synchronous; refined computer analysis may identify asynchronous ictal onset, but this is not necessarily exclusive of a primarily generalized seizure
(1)immediate pre-tonic-clonic phase: any myoclonic or clonic activity is represented as a generalized burst of spike or polyspike activity
(2)tonic phase: identified by an abrupt decrease in voltage with diffuse 20–40 Hz activity {desynchronization pattern}; 8–10 Hz sharp waves build in amplitude during the tonic phase
(3)clonic phase: 8–10 Hz sharp waves of the late tonic phase are replaced by polyspike-and-wave pattern that slows to 4 Hz
(4)post-ictal phase: diffuse suppression of EEG activity with a lowvoltage delta frequency predominance
Nonsyndromal Generalized Seizures
87
3 Seizures and Epilepsy
ii.interictal EEG: 50% show epileptiform activity with 5 days of a seizure, and the yield is higher closer to the time of the seizure
(1)subtypes of interictal abnormalities: often multiple subtypes occur in the same patient
(a)multifocal spike complexes
(b)3–5 Hz spike-and-wave complexes (usually are bilateral with frontocentral predominance)
(c)irregular spike-and-wave complexes
(2)anticonvulsants tend to slow the background rhythm, except for barbiturates and benzodiazepines, which characteristically increase the beta activity in the 14–20 Hz range
iii.cerebrospinal fluid: any post-ictal pleocytosis should be assumed to be caused by an inflammatory process until proven otherwise
2.Tonic seizures
a.symptoms: diffuse contraction of the axial musculature, sometimes involving the proximal limbs or proximal and distal limbs; paralysis of respiration causes apnea
i.often results in the patient falling {drop attack}, so tonic seizures are frequently confused with atonic seizures
ii.startle seizures—a type of reflex seizure that is usually tonic in nature, triggered by the sudden or unexpected presentation of a stimulus (Box 3.6)
(1)specific symptoms: the seizure follows a startle response (i.e., eye closure with facial grimacing and flexion of the limbs and trunk) induced by a sudden stimulus of any type
(2)often occurs in children with diffuse cerebral abnormalities (e.g., Down’s syndrome, anoxic encephalopathy)
(3)treatment: benzodiazepines, carbamazepine, lamotrigine
b.diagnostic testing: subtypes of ictal EEG patterns
i.sudden reduction of background activity {electrodecremental response} (Fig. 3–4)
ii.bilaterally synchronous 15–25 Hz activity with increasing voltage
iii.diffuse high-voltage 10 Hz activity
iv.diffuse theta or delta activity
Box 3.6
Hyperekplexia
A hyperactive startle response followed by rigidity leading to falls (no impairment of consciousness)
Caused by glycine receptor mutations; treat with clonazepam
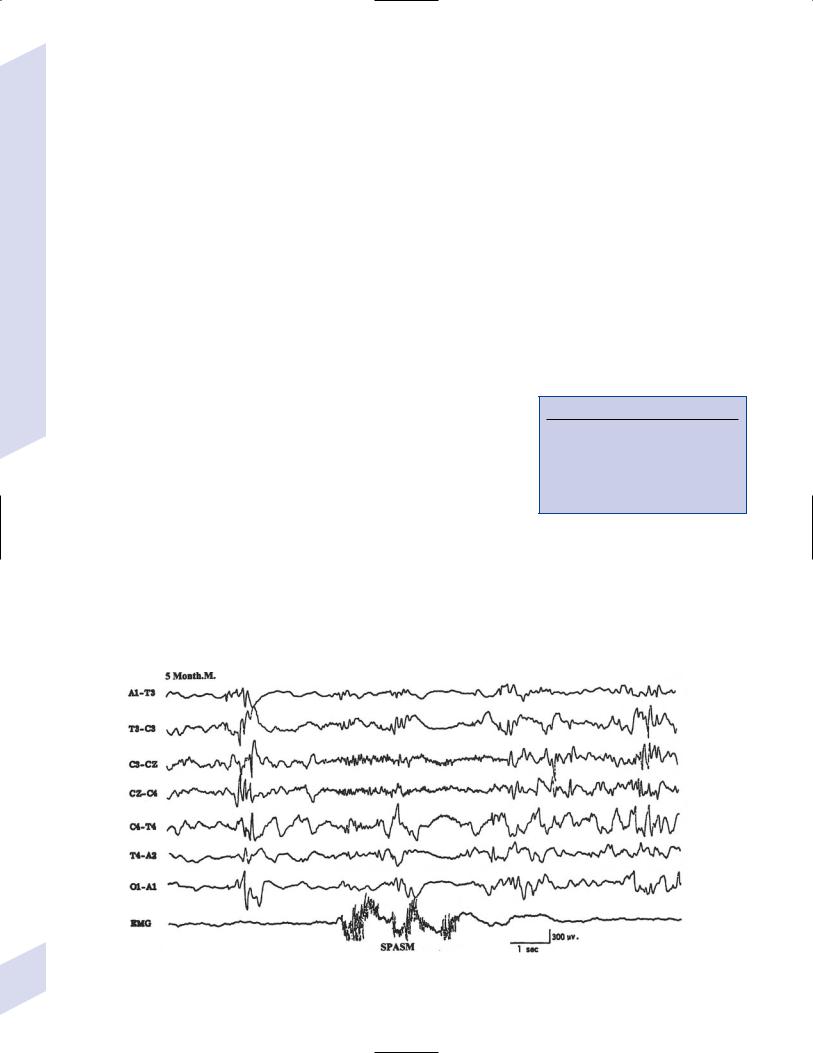
Figure 3–4 Electrodecremental response of an infantile spasm. (From Markand ON. Pearls, perils, and pitfalls in the use of the electroencephalogram. 88 Semin Neurol 2003, 23: 29, Fig. 23. Reprinted by permission.)
3.Atonic seizures
a.symptoms: sudden loss of consciousness and postural muscle tone that can be limited to the head but generally involves the whole body {drop attack} or only the head, usually occurring after a brief myoclonic seizure; lasts several seconds
i.loss of consciousness with preservation of muscle tone {akinetic seizures} can also occur
ii.overall, drop attacks are more likely to be caused by tonic than atonic seizures
b.diagnostic testing: ictal EEG demonstrates polyspike-and-wave discharges followed by diffuse slowing that is maximal over central regions during the atonia
4.Myoclonic seizures
a.symptoms: shock-like ( 50 ms), irregular, and asynchronous contraction of one or a few muscle groups that may be singular or repetitive; affects predominantly the eyelids, facial muscles, and upper more than lower extremities
b.diagnostic testing: ictal EEG demonstrates diffuse polyspike and wave discharges
IV. Status Epilepticus
1.Definition: any type of seizure 30-minute duration or that recurs without the patient fully regaining consciousness; in practice, seizures 5 minutes are treated as status epilepticus
2.Causes (Box 3.7)
a.acute brain injury from infarction, mass lesions, or head trauma
b.metabolic abnormality (particularly non-ketotic hyperglycemia)
c.drug intoxication/withdrawal
d.abrupt reduction or discontinuation of antiepileptic medication
e.idiopathic (30%)
3.Treatment (Table 3–2)
Table 3–2 Treatment of Status Epilepticus
Drug |
|
Time to |
Duration of |
(in order of use) |
Dose |
effect |
action |
|
|
|
|
#1: Benzodiazepines |
Adult: 4–8 mg IV |
|
|
Lorazepam (Ativan) |
6 min |
24 h |
|
|
Pediatric: 1–4 mg IV |
|
|
Diazepam (Valium, |
Adult: 10 mg IV |
2 min |
30 min |
Diastat) |
Pediatric: 5–10 mg IV |
|
|
|
|
|
|
|
or PR |
|
|
Midazolam (Versed) |
Adult: 0.2 mg/kg IM |
15 min |
2 h |
|
Pediatric : 0.1 mg/kg IM |
|
|
#2: Phenytoin (Dilantin)
Phosphenytoin (Cerebyx)
#3: Phenobarbital
#4: Anesthetic coma Propofol drip
Pentobarbital drip
20 mg/kg IV, or same of |
10 min |
24 h |
phenytoin equivalents for |
|
|
phosphenytoin |
|
|
Supplement 10 mg/kg if |
|
|
first dose fails |
|
|
20 mg/kg IV |
20 min |
48 h |
Load 2 mg/kg IV, then 2–10 mg/kg/h titrated
Load 10 mg/kg IV, then 0.5–3 mg/kg/h titrated
Abbreviations: IV, intravenously; IM, intramuscularly; PR , per rectum.
Status Epilepticus
Box 3.7
Epilepsia partialis continua—spontaneous, regular or irregular clonic epileptic movements that last for long periods of time
89

3 Seizures and Epilepsy
90
a.thiamine 100 mg IM 50 mL of D50 IV (adult) or 2 mL/kg D25 IV (pediatric) should be routinely administered to all status epilepticus patients considering the possibility of Wernicke’s encephalopathy
b.continuous EEG monitoring is indicated when the patient becomes overly sedated and is not overtly seizing to evaluate for subclinical status epilepticus
4.Prognosis: 3–30% mortality, depending upon the cause, duration, and patient age
a.outcome is worse with prolonged seizures because of the development of hypoglycemia, hypotension, and/or acidosis
b.cognitive dysfunction is often the residual of status epilepticus
V. Inherited and Pediatric Seizures
A. Neonatal Seizures
1.Subtypes: generalized seizures do not exist in neonates because myelination is incomplete, thereby preventing spread of epileptiform activity
a.focal clonic seizures: usually caused by focal cerebral injury; Jacksonian march does not occur in neonates because of the lack of myelination
b.multifocal clonic seizures: involve random migration of seizure activity across several muscle groups; usually occurs after diffuse cerebral injury, particularly severe hypoxic encephalopathy
c.tonic seizures: extension and stiffening of the body with apnea and upward eye deviation
i.usually caused by focal cerebral injury or by intraventricular hemorrhage in premature infants; very poorly localized with EEG, may involve a brainstem release mechanism that accounts for the majority of symptoms
d.myoclonic seizures: rare in neonates; can be misinterpreted as a generalized seizure; usually are caused by diffuse cerebral injury in the children of drug-addicted mothers
e.“subtle” seizures: involves tonic eye deviations (most diagnostic symptom for this condition), sucking behavior, and/or cycling movements of the extremities
2.Causes (Table 3–3)
a.severe hypoxic encephalopathy: symptoms also include stupor or coma, hypotonia, multiple cranial nerve palsies; EEG demonstrates slow-wave, flat-line, or burst-suppression EEG
i.usually occurs in fetuses with irregular heart rates or meconium aspiration
Table 3–3 Causes of Neonatal Seizures
Seizure onset |
Cause (in order) |
|
|
1 d from birth |
cerebral ischemia |
|
meningitis, sepsis |
|
intracranial hemorrhage |
|
(intraventricular, subependymal, or subarachnoid) |
1–3 d from birth |
intracranial hemorrhage |
|
(intraventricular, subependymal, or subarachnoid) |
|
meningitis, sepsis |
|
hypercalcemia |
3d–1wk from birth |
metabolic diseases (e.g., aminoacidurias, urea cycle disorders) |
|
herpetic encephalitis |
1–4wk from birth |
cerebral malformations |
|
metabolic diseases |
|
|

ii.seizures from severe hypoxic encephalopathy are difficult to control because of elevated intracranial pressure but become manageable with a lowering of the pressure
b.mild hypoxic encephalopathy: symptoms also include lethargy, jitteriness, and sympathetic hyperactivity (tachycardia, dilated pupils); unlike severe hypoxic encephalopathy, the EEG is relatively normal
i.mild hypoxic encephalopathy does not cause seizures except when provoked by hypoglycemia or an electrolyte disturbance
c.trauma: usually occurs in large-for-gestational-age neonates after complicated deliveries; often associated with intracranial hemorrhage
d.intracranial hemorrhage
i.subarachnoid hemorrhage in neonates is more likely caused by ruptured superficial veins than arteries
ii.subdural hemorrhage in neonates is caused by disruption of the veins on the tentorium near the falx due to excessive vertical head molding
e.hypoglycemia: defined as blood glucose 20 mg/dL in premature infants or 30 mg/dL in fullterm infants; occurs in 10% of all neonates
i.symptoms also include apnea, vomiting, hypotonia, seizures; progression to coma during hypoglycemia is associated with long-term neurological impairment
f.hypocalcemia: may exacerbate seizures but is not likely their underlying cause
B.Pediatric Seizures
1.Febrile seizures
a.pathophysiology: convulsions that are not necessarily associated with a rising or peak body temperature; exhibits an autosomal dominant inheritance with incomplete penetrance
i.subtypes
(1)simple febrile seizures (70%): a generalized tonic-clonic seizure that lasts 1 minute; no further seizures occur in the next 24hour period
(2)complex febrile seizures (30%): a seizure with focal or multifocal features that lasts 15 minutes, or that recurs within 24 hours
ii.epidemiology: occur in 3% of children
(1)3% of febrile seizure children will ultimately develop epilepsy
(2)40% of children with a single febrile seizure will have a second febrile seizure, and 50% of children with two febrile seizures will have a third febrile seizure
(a)the risk of further febrile seizures is increased by having
(i)the first seizure at age 1 year
(ii)a seizure with a body temperature 40°C
(iii)an abnormal neurological exam or developmental delays
(iv)prolonged, focal-onset, or multiple episodes of febrile seizures
(v)a family history of seizures
iii.febrile seizures are triggered by the measles–mumps–rubella (MMR) vaccine (that induces a fever), but this does not increase the risk of developing epilepsy over that of febrile seizures not caused by a vaccination
b.diagnostic testing
i.cerebrospinal fluid analysis to rule-out meningitis or encephalitis, particularly in children 6 months old; however, 90% of children with meningitis or encephalitis are obtunded after a seizure, which is rare with simple febrile seizures
(1)lumbar puncture is not necessary if child recovers rapidly and completely after the seizure, and if the fever is readily explained by an extracranial source of infection
Inherited and Pediatric Seizures
91
3 Seizures and Epilepsy
ii.EEG and neuroimaging are indicated only if child has an abnormal neurological exam or atypical febrile seizures
c.treatment: no treatment is indicated after simple febrile seizures; antiepileptic medications are indicated for complex febrile seizures, febrile seizures in patients 1 year of age, and patients with an abnormal neurological exam
2.Generalized myoclonic seizures
a.infantile spasms/West’s syndrome (Box 3.8)
i.pathophysiology: 75% of cases have an underlying structural lesion (e.g., a phakomatosis), acquired cerebral injury (e.g., intracranial hemorrhage or infection), or genetic abnormality (e.g., non-ketotic hyperglycinemia, Down’s syndrome)
ii.symptoms: onset between 4–6 months of age
(1)seizures: classically manifests as clusters of symmetric extensor and/or flexor movements, followed by tonic contractions lasting10 seconds; seizures most commonly occur after waking
(a)abdominal flexions are often mistaken for colic
(2)autism
(3)progressive mental retardation (usually in patients with an underlying malformation)
(4)microcephaly
(5)focal neurological injury (60%): can be cortical or subcortical (e.g., athetosis) in nature
iii.diagnostic testing
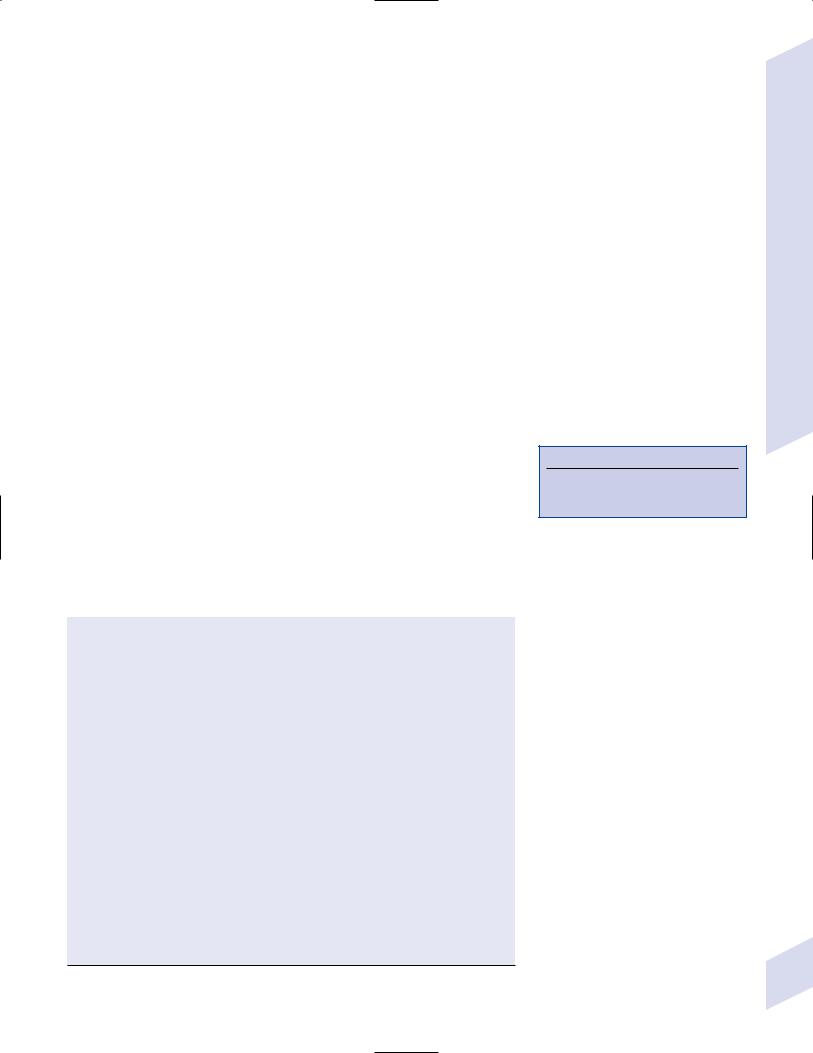
(1)interictal EEG demonstrates a chaotic background with randomlydispersed high-voltage slow waves and spike discharges across all leads {hypsarrhythmia} (Fig. 3–5)
(a)the multifocality of hypsarrhythmia is replaced within a few months by generalized synchronous discharges, which eventually degrade into a slow spike-and-wave discharge similar to Lennox-Gastaut syndrome by 2 years of age
(2)ictal EEG demonstrates background suppression during the convulsive movements {electrodecremental response} (Fig. 3–4), high-voltage generalized slow waves, and/or paroxysmal lowvoltage fast activity
(3)in the absence of an underlying structural lesion on neuroimaging, combined positron emission tomography (PET) scanning and video EEG monitoring may identify an area of cortical abnormality
Box 3.8
Aicardi’s Syndrome
Infantile spasms agenesis of the corpus callosum retinal malformations
Exhibits X-linked dominant inheritance (a hemizygous lethal phenotype in males)
Figure 3–5 Infantile spasms demonstrating hypsarrhythmia. (From Mumenthaler M. Neurological Differential Diagnosis. 2nd ed. Stuttgart, Germany: 92 Georg Thieme; 1992:266, Fig. 6.2. Reprinted by permission.)
iv.treatment
(1)vigabatrin: highly effective, particularly in tuberous sclerosis patients; however, vigabatrin is not approved for use in the United States
(2)adrenocorticotropic hormone (ACTH)
(3)high-dose pyridoxine, presumptively assuming the infantile spasms are due to vitamin deficiency
(4)surgery to remove seizure foci
v.prognosis: 5% mortality
(1)in 65% of survivors, the infantile spasms are replaced by another seizure type such as Lennox-Gastaut or complex partial seizures
(2)only 10% of patients have normal mental and motor development, although the likelihood of normal development is higher in patients with idiopathic/cryptogenic infantile spasms
b.Lennox-Gastaut syndrome
i.pathophysiology: 60% of cases are related to structural brain abnormalities; 20% of cases develop from infantile spasms
ii.symptoms: onset between 3–5 years of age
(1)multiple coexisting seizures
(a)tonic seizures (90%)
(b)atypical absence seizures (65%) that involve repetitive facial motions followed by loss of tone in head and face
(c)myoclonic seizures (20%)
(2)mental retardation
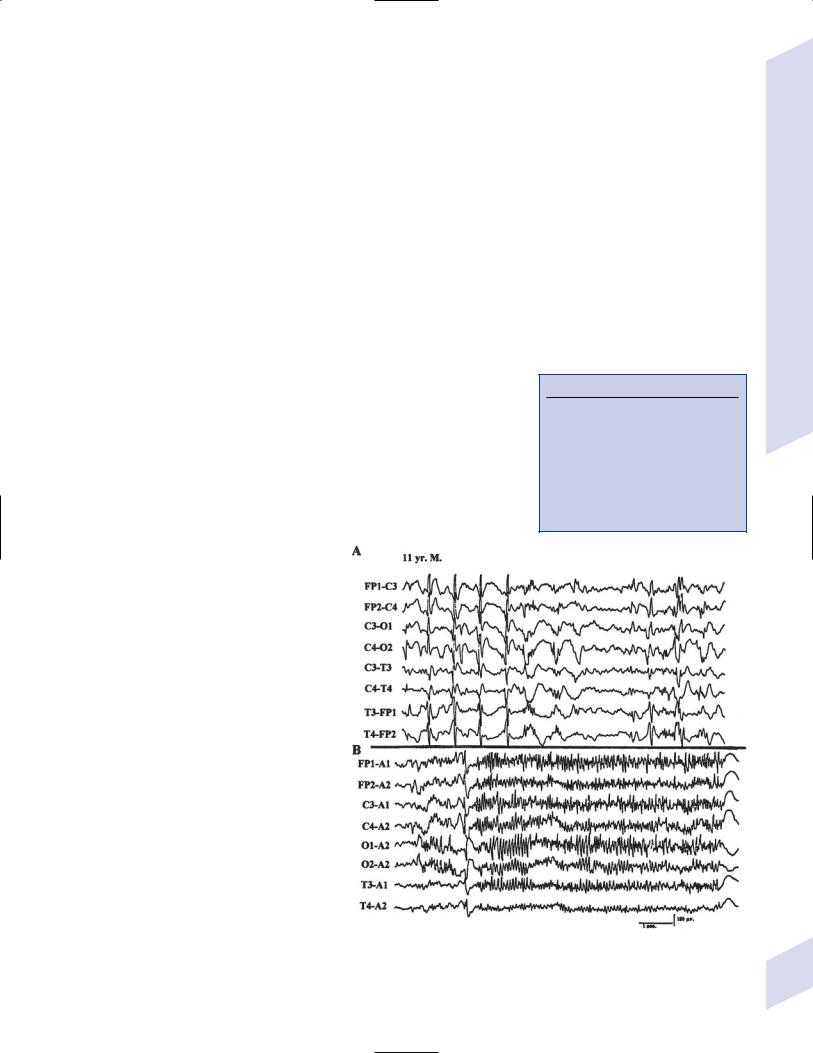
iii.diagnostic testing (Fig. 3–6)
(1)interictal EEG: exhibit 2–2.5 Hz slow spike-and-wave complexes with frontotemporal predominance; paroxysmal fast activity 10 Hz is apparent while sleeping (Box 3.9)
(2)ictal EEG: atypical absence seizures exhibit 2–2.5 Hz spike-and-wave similar to interictal pattern; tonic seizures exhibit synchronous 15–25 Hz activity that is dominant in the frontal and vertex EEG leads
iv.treatment
Box 3.9
Epileptic Pseudoataxia
Episodic ataxic limb movements and gait produced by repeated myoclonus and/or brief periods of akinesis; exhibits confusion but no nystagmus with episodes
Exhibits 2–2.5 Hz spike-and-wave complexes, like Lennox-Gastaut syndrome
Treat with clonazepam or valproate
(1)numerous medications can be used, although they nearly always fail to completely control the seizures
(a)vigabatrin is effective but not approved for use in the United States
(2)ketogenic diet
(3)vagal nerve stimulator; corpus callosotomy
c.benign myoclonic epilepsy benign myoclonus of infancy
i.symptoms: onset between 6 months–2 years of age, with remission within 5 years from onset; involves brief myoclonic jerking without a loss of consciousness that in 15% of cases develops into generalized tonic-clonic seizures by adolescence
Figure 3–6 Lennox-Gastaut syndrome while awake (A) and during sleep (B) during which it demonstrates generalized paroxysmal fast activity. (From Markand ON. Pearls, perils, and pitfalls in the use of the electroencephalogram. Semin Neurol 2000, 23: 31, Fig. 26. Reprinted by permission.)
Inherited and Pediatric Seizures
93

3 Seizures and Epilepsy
(1)does not involve mental retardation
ii.diagnostic testing: interictal EEG is normal; ictal EEG identifies generalized polyspike or spike-and-wave discharges during myoclonic jerks
(1)EEG during sleep exhibits exaggerated discharges that occur with or without myoclonic jerks
iii.treatment: valproate
d.severe myoclonic epilepsy (Box 3.10)
i.pathophysiology: unknown, but appears to have a familial predilection
ii.symptoms: develops by 1 year of age; typically begins as a febrile seizure that then exhibits nonfebrile clonic seizures, with the eventual development of myoclonus within several years
(1)interictal neurological deficits (e.g., gait ataxia, dystonia, weakness) also develop
(2)associated with mental retardation
iii.diagnostic testing: EEG eventually demonstrates spontaneous and photic stimulation-induced bursts of generalized spike-and-wave activity with a reduction in background activity
iv.treatment: resistant to therapy
e.juvenile myoclonic epilepsy
i.pathophysiology: autosomal dominant inheritance of unknown genetic factors
ii.symptoms: seizures involve bilateral but asymmetric flexor movements of the upper extremities or rarely of the lower extremities that develop most commonly after awakening; no loss of consciousness occurs with seizures
iii.diagnostic testing: interictal EEG demonstrates bilateral, symmetric 3.5–6 Hz spike-and-wave and polyspike discharges; ictal EEG exhibits diffuse polyspike activity followed by 1–3 Hz slow waves (Fig. 3–7)
(1)photic stimulation provokes discharges
iv.treatment: valproate
Box 3.10
Ohtahara’s Syndrome/Infantile
Epileptic Encephalopathy
Symptoms: onset shortly after birth, up to 3 months of age, rarely begins in utero; involves tonic spasms, erratic motor clonic movements, and/or hemiconvulsions
Diagnostic testing: clusters of burstsuppression
Treatment: seizures are poorly responsive; may attempt ACTH, vigabatrin, valproate, or benzodiazepines; avoid carbamazepine, which increases seizure frequency
Figure 3–7 Juvenile myoclonic epilepsy. (From Markand ON. Pearls, perils, and pitfalls in the use 94 of the electroencephalogram. Semin Neurol 2000, 23: 27, Fig. 21. Reprinted by permission.)

Table 3–4 Myoclonic Seizure Syndromes
Syndrome |
Mutated gene/protein |
Age of onset |
Specific clinical sign |
Notes |
|
|
|
|
|
Neuronal ceroid |
Tripeptidyl peptidase |
9 |
Macular degeneration, |
Neuronal curvilinear bodies |
lipofuscinosis |
|
|
dementia, dystonia |
(infant form) enlarged |
(infant form) |
|
|
|
axons & dendrites |
Unverricht- |
EPM1/cystatin B (see Box 3.11) |
6–15 |
Cerebellar signs, startle |
Diffuse atrophy, particularly |
Lundborg disease |
|
|
myoclonus |
of cerebellar Purkinje cells |
Lafora disease |
EPM2A/laforin |
6–20 |
Occipital seizures |
Perinuclear polyglucosan |
|
|
|
causing visual |
inclusion bodies |
|
|
|
hallucinations, |
|
|
|
|
blindness, scotomata |
|
Sialidosis type 1 |
NEU1/neuraminidase |
6–15 |
Retinal cherry-red spot |
Urine oligosaccharides |
Myoclonic epilepsy |
Mitochondrial tRNA |
3–65 |
Short stature, muscle |
Ragged red muscle fibers; |
with ragged red |
|
|
weakness |
lactic acidosis |
fibers (MERRF) |
|
|
|
|
|
|
|
|
|
f.myoclonic seizure syndromes: all exhibit a progressive course (Table 3–4)
3.Absence seizures (Box 3.12)
a.childhood and juvenile absence seizures
i.pathophysiology: autosomal dominant inheritance; slow wave discharges on EEG may relate to cyclic activity of T-type calcium channels and repolarizing potassium currents in reticular thalamic nucleus
(1)childhood-onset form is self-limited, whereas the juvenile-onset form is more likely to persist into adulthood
ii.symptoms
(1)5–10 second long unresponsive staring spells (Box 3.13) rhythmic facial movements or picking behaviors; does not have an aura or post-ictal state
(a)occur on a daily basis in the childhood-onset form, more infrequent in the juvenile-onset form
(b)50% also have generalized tonic-clonic seizures, more commonly in the juvenile-onset form; these are infrequent and well controlled with medication
(2)mental retardation, focal neurological dysfunction (in 15% of cases)
iii.diagnostic testing: EEG demonstrates bilateral spike-and-wave complexes at 3 Hz that are reliably activated by hyperventilation (Fig. 3–8)
(1)interictal EEG is usually normal; abnormal interictal EEGs are associated with mental retardation
iv.treatment: ethosuximide, valproate, acetazolamide
(1)avoid carbamazepine, which increases seizure frequency (as in severe myoclonic epilepsy) and may induce absence status epilepticus
4.Partial seizures
a.simple partial seizures
i.acquired epileptiform aphasia/Landau-Kleffner syndrome
(1)pathophysiology: an idiopathic disorder involving simultaneous hypoperfusion and epileptiform activity in one or both temporal lobes
(2)symptoms: onset between 2–11 years of age
Box 3.11
Cystatin C mutated in Icelandic-type hereditary cerebral amyloidosis
Box 3.12
Eyelid Myoclonus with Absence Seizures (Jeavon’s Syndrome)
Rapid blinking of the eyelids with upward eye deviation, typically occurring immediately after eye closure; does not occur in the dark
Associated with brief bilateral spike- and-wave activity
Unclear if this is a type of seizure or a means by which the patient induces seizures
Box 3.13
Staring spells are most likely to be due to complex-partial seizures at this age.
(a)receptive aphasia progressing to global aphasia over a period of several days
(i)the patient must have normal language and cognitive development prior to seizure onset to rule out autism
Inherited and Pediatric Seizures
95

3 Seizures and Epilepsy
96
(ii)progression of aphasia not related to frequency of seizures, and is fluctuant
(iii)auditory agnosia may occur with involvement of the nondominant hemisphere
(b)focal or generalized seizures, usually atonic (70%)
(c)behavioral changes: hyperactivity, aggression
(3)diagnostic testing: EEG demonstrates multifocal cortical spike discharges predominantly in the temporal and parietal lobes that are bilateral in 90%; these EEG abnormalities are minimal or even absent during waking, but become so severe during sleep (particularly at sleep onset) as to be nearly continuous
(4)treatment
(a)glucocorticoids often improve seizures and aphasia
(b)antiepileptic drugs control seizures but has an unclear effect on aphasia
(c)subpial transection to isolate seizure focus, or have surgical resection of seizure focus improve seizures and aphasia
(5)prognosis: seizure always remit by age 15; aphasia may persist into early adulthood before improving
ii.acquired epileptiform opercula syndrome
(1)pathophysiology: exhibits autosomal dominant inheritance with anticipation
(2)symptoms: oral dysphasia and an inability to perform complex facial movements {facial apraxia} associated with focal or generalized seizures
(3)diagnostic testing: ictal EEG demonstrates centrotemporal discharges
(4)treatment: completely unresponsive to treatment
iii.autosomal dominant nocturnal frontal lobe epilepsy
(1)pathophysiology: mutation of the presynaptic nicotinic acetylcholine receptor 4 subunit causing a decreased calcium conductance of the channel; exhibits autosomal dominant inheritance with 70% penetrance
(2)symptoms: seizures begin with vocalization or a psychic prodrome that wakes the patient from sleep, followed by tonic-clonic movements without loss of consciousness; occur only during sleep, often in clusters
(a)previously was considered a movement disorder (“nocturnal paroxysmal dystonia”)
(3)diagnostic testing: normal interictal EEG in 85%; requires continuous video monitoring to distinguish from REM sleep behavior disorder
(4)treatment: carbamazepine
iv.benign occipital epilepsy
(1)pathophysiology: unknown; frequently occurs in patients with migraine; not associated with structural lesions
(2)symptoms: seizures involve unformed visual hallucinations, visual illusions (micropsia, macropsia, metamorphopsia), vision loss (either complete or hemianopic), vomiting and drooling, and/or forced blinking and forced gaze deviation; some patients will have seizures followed by a loss of consciousness lasting several hours
(a)seizures usually develop after a migraine headache, and after the patient will typically have a migraine headache
(b)seizures usually occur at sleep onset
(3)diagnostic testing: interictal EEG demonstrates unilateral or bilateral high-amplitude occipital spike-and-wave discharges at 1.5–2.5 Hz that are inhibited by eye opening; ictal EEG is similar except that the spike-and-wave discharges are at higher frequencies
(a)epileptiform activity is reliably induced by photic stimulation
v.Rolandic epilepsy/centrotemporal epilepsy
(1)pathophysiology: two subtypes based on inheritance patterns
(a)autosomal dominant inheritance: 10% of cases with this subtype have only a single seizure, and the rest have very infrequent seizures; 70% of seizures occur during sleep
(b)autosomal recessive inheritance: linked to chromosome 15
(2)symptoms: perioral paresthesias followed by twitching of face and mouth with speech arrest and drooling but without impairment of consciousness; convulsions may develop in an upper extremity or generalize, usually in patients 5 years of age
(a)in the autosomal recessive subtype, seizures occur in conjunction with paroxysmal dystonias (e.g., writer’s cramp)
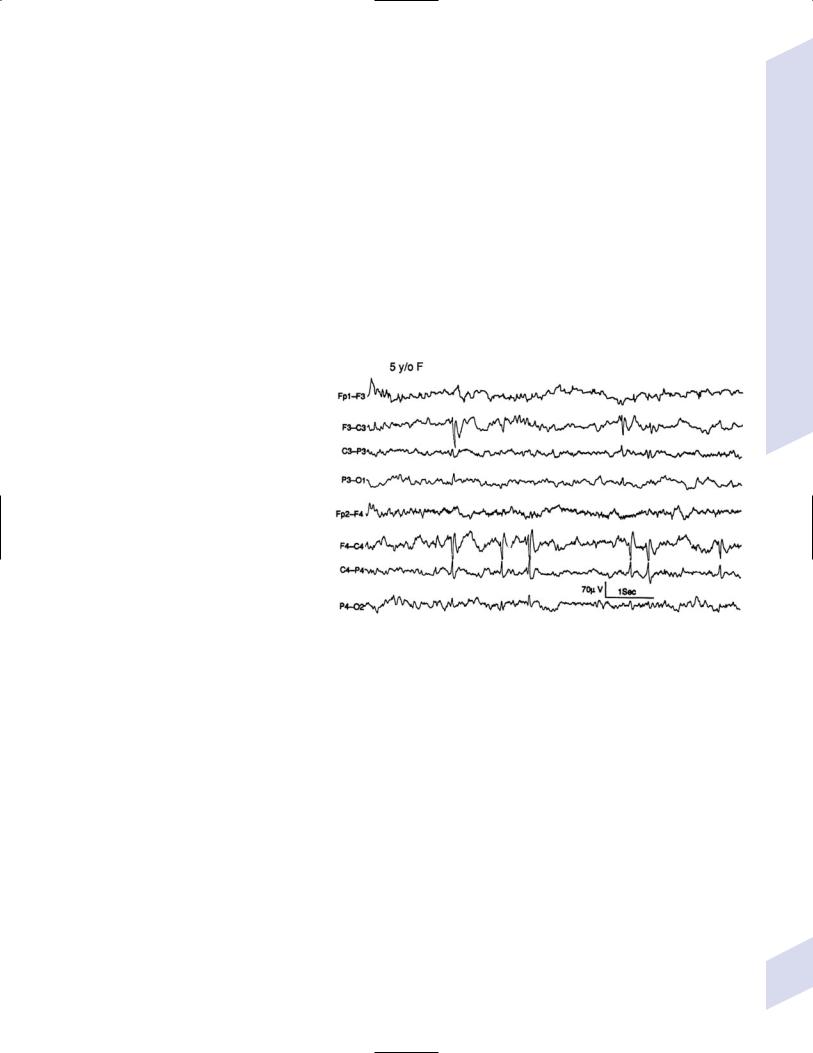
(3)diagnostic testing: EEG demonstrates unilateral or bilateral high-voltage spike discharges in central or centrotemporal region (Fig. 3–8) that are increased during sleep
(a)frequency of spike discharges does not correlate with seizures
(b)neuroimaging is warranted only in patients with atypical features or treatment-refrac- tory seizures
(4)treatment: phenobarbital or trileptal only if the seizures are frequent
(5)prognosis: spontaneous resolution of seizures by age 14; withdrawal of antiepileptic medication after 2 years leaves 80% seizurefree
Figure 3–8 Rolandic epilepsy. Note that sharp waves occur independently in the central regions. (From Panteliadis CP, Darras BT. Encyclopaedia of Paediatric Neurology: Theory and Practice, 2nd ed. Stuttgart, Germany: Georg Thieme;1999:415, Fig. 6. Reprinted by permission.)
vi.Rasmussen’s syndrome/hemiconvulsion-hemiplegia-epilepsy syndrome
(1)pathophysiology: inflammatory degeneration of the cortex and supratentorial subcortical structures involving lymphocyte and macrophage invasion, microglial nodule formation, and neuronophagia that is initially limited to a single hemisphere or part thereof; inflammatory degeneration does not involve the contralateral hemisphere or posterior fossa structures
(a)resembles the histological changes of a rare Russian tick-born encephalitis; however, there is no reproducible evidence of an infectious agent causing Rasmussen’s syndrome
(b)autoantibodies against GluR3 subunit of glutamate receptors are infrequent and do not clearly mediate the inflammatory degeneration, although their levels may indicate disease activity
(2)symptoms: focal motor seizures that spread to contiguous muscle groups; 60% develop focal status epilepticus {epilepsia partialis continua} before the disease burns out in 2–10 years
Inherited and Pediatric Seizures
97

3 Seizures and Epilepsy
98
(3)diagnostic testing
(a)EEG demonstrates continuous focal spike discharges that spread to contiguous areas of cortex, and eventually mirror foci on the contralateral hemisphere
(b)neuroimaging: may be normal initially, but hemispheric atrophy and ipsilateral hydrocephalus ex vacuo develop within 6 months
(c)PET and single photon emission computed tomography (SPECT) scanning demonstrate hypometabolism and decreased cerebral blood flow over large areas of the affected hemisphere
(d)cerebrospinal fluid analysis is usually normal
(4)treatment: refractory to antiepileptic medications, immunosuppressive agents, and antiviral therapy; early hemispherectomy can be curative
b.complex partial seizures: epileptogenic focus is usually in the temporal frontal or parietal lobes
i.general symptoms: staring spells, automatic behaviors (e.g., facial grimacing, hand wringing), posturing, or loss of body tone lasting 1–2 minutes
(1)post-ictal confusion and lethargy can occur with or without secondary generalization
(2)30% of patients exhibit an aura consisting of a nondescript unpleasant feeling, auditory hallucination, or abdominal discomfort; auras are particularly common with mesial temporal foci
ii.subtypes
(1)autosomal dominant partial epilepsy with auditory features
(a)genetics: caused by mutations of leucine-rich glioma- inactivated-1 gene (LGI-1), which has an unknown function (Box 3.14)
(b)specific symptoms: seizures involve unformed sounds (e.g., clicks, buzzing), changes in sound perception (e.g., deafness, muffling, increase in volume), or complex sounds, then usually progresses to a psychic symptom (e.g., déjà vu)
(i)30% of patients have unilateral symptoms
(ii)some patients with familial seizures exhibit a Wernicke’s- type aphasia at the seizure onset
(iii)seizures may be triggered by sounds
(2)benign familial neonatal convulsions
(a)genetics: caused by mutations in the voltage-gated potassium channel KCNQ-2 gene (chromosome 20) and KCNQ-3 gene (chromosome 8); abnormal channels reduce the repolarizing potassium current, allowing prolonged neuronal depolarization
(b)specific symptoms: clonic or apneic seizures that occur in clusters within the first few postnatal days
(c)prognosis: seizures resolve by a few months of age
(3)benign familial infantile convulsions
(a)genetics: most cases are linked to poorly defined abnormalities on chromosome 16; cases associated with familial hemiplegic migraine are caused by mutations of ATP1A2 sodium-potassium ATPase gene on chromosome 1
(b)specific symptoms: seizure onset between 3–12 months of age; otherwise normal development, and seizures remit by adulthood
(i)can be associated with familial hemiplegic migraine
(4)benign familial neonatal-infantile convulsions
Box 3.14
LGI-1 is located in the loss of heterogeneity region on chromosome 10 in glioblastoma multiforme (see p. 120)

(a)genetics: caused by mutations in the SCN2A gene for voltagegated sodium channel
(b)specific symptoms: clonic seizures developing within first few postnatal weeks, which is later than benign familial neonatal convulsions but earlier than benign familial infantile convulsions
iii.treatment: carbamazepine, phenytoin, primidone, valproate; surgical removal of medically refractory seizure foci
C. Nonseizure Paroxysmal Disorders of Children
1.Pediatric apnea syndromes
a.periodic breathing—irregular respiratory pattern with intermittent pauses of 3–6 seconds duration, followed by 10–15 seconds of hyperventilation without associated autonomic changes; seen mostly in premature infants due to immaturity of the brainstem respiratory centers
b.true apnea—cessation of breathing 15 seconds, or 15 seconds if associated with bradycardia
i.can be considered normal if the patient is 3 months of age and if the apnea is not associated with tonic eye deviation, limb stiffening, or clonic movements
(1)seizure-induced apnea rarely is associated with bradycardia
2.Jitteriness—an excessive response to stimulation characterized by a lowfrequency, high-amplitude shaking of the limbs and jaw; can be confused with myoclonic seizures but jitteriness is more rhythmic
a.jitteriness occurs in the newborns of drug-abusing mothers (particularly those using cocaine and methadone) or mothers using phenothiazines or propoxyphene, in newborns with perinatal hypoxia, or in newborns with metabolic disorders, therefore it has a high coincidence with seizures
b.identify episodes of jitteriness by a normal EEG, the absence of eye movements, and the lack of change in respiration; jittery neonates also have a hyperactive Moro reflex
c.treatment: paregoric; phenobarbital
3.Breath-holding spells
a.pathophysiology: a type of syncope thought to be related to immaturity of the brainstem respiratory centers; exhibits autosomal dominant inheritance with incomplete penetrance
i.breathing stops after expiration (not after inspiration) in response to an adverse stimuli
b.subtypes
i.pallid breath-holding spells—breathing stops in response to a painful stimulus; the child often falls asleep after the breath-holding spell but is otherwise normal
ii.cyanotic breath-holding spells—breathing stops during a crying episode; long episodes can exhibit tonic posturing, extremity trembling, and upward eye deviation; recovery is rapid without any post-ictal confusion
(1)EEG during the breath-holding spell is low-voltage and nonepileptiform
VI. Pseudoseizures/Nonepileptiform
Seizures (Box 3.15)
1.Pathophysiology a. subtypes
i.posttraumatic pseudoseizures: develop in response to physical, psychological, or sexual abuse
Box 3.15
Pseudo-Pseudoseizures
Short duration ( 30 seconds) repetitive episodes with bizarre behavior (e.g., complex automatisms) and large movements (e.g., abduction of upper extremities)
Involve the epileptiform activity developing out of mesial frontal lobes or mesial parietal lobes
Epileptiform activity is EEG inaccessible; may require high-frequency filters to demonstrate theta activity in parasagittal leads
Pseudoseizures/Nonepileptiform Seizures
99

3 Seizures and Epilepsy
ii.developmental pseudoseizures: develop in response to poor coping with increasingly complex developmental challenges or failure to achieve developmental milestones
b.other risk factors
i.comorbid psychiatric disorders: anxiety; depression; histrionic or borderline personality disorders
(1)pseudoseizures may represent the presentation of a conversion disorder
ii.family history of pseudoseizures
iii.head trauma
iv.pending disability claims
v.abrupt elimination of epileptiform seizures, as after seizure surgery
vi.epileptiform seizures
2.Epidemiology: more common in late adolescence and early adulthood as well as in women; rare at 5 years of age and 55 years of age
a.present in upwards of 30% of patients in specialty seizure clinics and 20% of adolescents with new-onset seizures
b.30% coincidence with epileptiform seizures
3.Symptoms: Seizure-like episodes without electroencephalographic epileptiform activity
a.symptoms differentiating pseudoseizures from epileptiform seizures: none are sufficiently accurate to ensure the diagnosis
i.clonic activity in which the limbs are out-of-phase with each other
ii.events occurring while the patient appears to be asleep but has EEG activity indicating wakefulness {preictal pseudosleep}
iii.pelvic thrusting
iv.eye movements or fluttering eyelids during the event
v.preserved recall of the event
vi.complex, well-formed vocalizations at the beginning of the event
(1)vocalizations are simple (e.g., grunting) and occur more commonly during the convulsive movements in epileptiform seizures
vii.incontinence: incontinence early in the course of pseudoseizures is rare, it often develops later after the patient is repeatedly questioned about it
viii.lip biting or biting the tip of the tongue is suggestive of pseudoseizures
(1)biting the side of the tongue is suggestive of epileptiform seizures
ix.typical events are inducible by suggestion (e.g., saline injection)
b.symptoms that are unable to differentiate pseudoseizures from epileptiform seizures: the presence of an aura, the duration of event, post-ictal confusion, physical trauma from events, prolonged course of events (i.e., there is a “pseudo-status epilepticus”
4.Diagnostic testing
a.continuous video EEG monitoring, which must capture some of the suspect events
b.prolactin level: generally does not increase from baseline after a pseudoseizure
c.neuropsychiatric testing: generally is not helpful in distinguishing pseudoseizures from epileptiform seizures
5.Treatment: no proven treatment; divulging diagnosis in a supportive manner may improve pseudoseizure frequency in 80% of cases that developed as an acute or situation-dependent reaction
6.Prognosis: with multidisciplinary treatment, one third completely resolve and
100 |
one third have a reduction in the frequency of the events; as with epilepsy, |
quality of life improves only with complete resolution of pseudoseizures |

Figure 3–9 Treatments for primary generalized seizures.
VII. General Treatment of Seizures
1.Medical treatment: Quality of life improves only with complete seizure control (Fig. 3–9; Fig. 3–10)
a. prophylactic antiepileptic medication use in patients with neurological disorder but without seizures
i.severe head trauma (prolonged loss of consciousness, amnesia, depressed skull fracture, contusion, or hematoma): prophylaxis does not reduce seizure rate
ii.brain tumor: prophylaxis does not seem to reduce development of seizures in patients with primary or metastatic tumors
iii.stroke: no indication, although it is routinely used short-term in subarachnoid hemorrhage patients because of the fear of increased intracranial pressure that accompanies seizures
b.discontinuing medication therapy
i.predictive factors for successful seizure remission (Box 3.16)
(1)the patient will most likely be seizure free if he or she has
(a)a single type of seizure
(b)had no seizures for 3.5 years
(c)a normalized EEG on antiepileptic treatment
(d)a normal neurological exam and intelligence quota (IQ)
i.general rules
(1)hepatic-inducing medications (e.g., lower other drug levels): phenytoin, carbamazepine, phenobarbital, primidone
(2)hepatic-suppressing medications (e.g., raise other drug levels): valproate
(3)albumin-binding medications (e.g., raise other drug levels): phenytoin, valproate
d.management of medication failures
i.if maximal doses of an antiepileptic medication fail to control the seizures, there is only a 10% likelihood of developing control over seizures by adding a second antiepileptic medication
ii.there is 5% likelihood of developing control over seizures by adding a third antiepileptic medication or by using multiple antiepileptic drugs after failing a second antiepileptic medication
Box 3.16
All four factors achieve 70% remission in children, and 60% remission in adults.
General Treatment of Seizures
Figure 3–10 Treatments for partial seizures. |
101 |
3 Seizures and Epilepsy
2.Surgical treatment
a.preoperative evaluation involves
i.video-EEG monitoring
ii.neuroimaging: CT, MRI; ictal studies with SPECT or PET scanning
iii.invasive continuous monitoring with subdural electrodes
iv.Wada test evaluation of hemispheric dominance for language function and verbal memory
b.procedures
i.multiple subpial transections: vertical incisions that interrupt intracortical connections; can be used to isolate a seizure focus in eloquent cortex, leaving minimal neurological deficits (as in Landau-Kleffner syndrome where it is effective in 55% of cases)
ii.focal cortical resection: removal of a small block of epileptogenic cortex; 80% effective with temporal lobe foci, 50% effective with extratemporal foci
iii.temporal lobectomy: eliminates seizures in 70% of mesial temporal sclerosis cases
iv.hemispherectomy: for large blocks of epileptogenic cortex, as in Rasmussen’s syndrome where it is effective in 90%
v.corpus callosotomy: for multifocal seizures or unidentified epileptogenic foci that cause seizures with a high risk of physical injury (e.g., the drop attacks of tonic or atonic seizures)
c.vagal nerve stimulation: reduces frequency by 30% of either generalized or partial seizures; chief side effect is hoarseness during activation
i.likely acts by desynchronization of brain activity and/or by inhibition of epileptogenic foci by monoaminergic modulation
Appendix 3–1 Antiepileptic Drugs
Drug |
Mechanism of action |
Specific side effects |
Notes |
carbamazepine |
Slows recovery of voltage- |
Hyponatremia, transient |
(Tegretol, Carbatrol) |
gated Na channels |
diplopia or visual blurring |
|
|
after dosing, asymptomatic |
|
|
leukopenia |
Also used for trigeminal neuralgia, bipolar disorder; avoid in absence, myotonic, atonic seizures
Target range 8–12 g/mL
|
clonazepam, |
Increases GABAA receptor |
Respiratory depression, |
Other benzodiazepines are not approved for long- |
|
clorazepate |
currents |
somnolence |
term seizure treatment; generally tolerance devel- |
|
|
|
|
ops within 6 months |
|
|
|
|
|
|
ethosuximide (Zarontin) |
Inhibits T-type voltage- |
Nausea, exacerbation of behav- |
Useful only in absence seizures; no effect on other |
|
|
gated Ca channels in |
ioral disorders |
generalized seizures |
|
|
thalamus |
|
|
|
|
|
|
|
|
gabapentin (Neurontin) |
Unknown |
None in particular |
As effective as carbamazepine for partial seizures |
|
|
|
|
Used for neuropathic pain |
|
|
|
|
|
|
lamotrigine (Lamictal) |
May decrease glutamate |
Ataxia, diplopia, rash (1%) |
Effective in Lennox-Gastaut syndrome and juvenile |
|
|
release |
|
myoclonic epilepsy |
|
|
|
|
Synergistic effect when used with valproate but |
|
|
|
|
also increases its toxicity |
|
|
|
|
Lowest teratogenic risk of the antiepileptic drugs |
|
|
|
|
Nonsedating |
|
|
|
|
|
|
levetiracetam (Keppra) |
Unknown |
Psychosis |
|
|
|
|
|
|
|
oxcarbazepine |
Unknown |
Hyponatremia |
Equivalent dose is 1.5 that of carbamazepine |
|
(Trileptal) |
|
|
Converted to bioactive monohydroxy-derivative |
|
|
|
|
|
|
|
|
|
Does not affect levels of other medications |
|
|
|
|
|
|
phenobarbital |
Activates GABAA receptor; |
Respiratory depression, somno- |
Target range 0 15–40 g/mL |
|
|
inhibits voltage-gated Ca |
lence, cognitive slowing with |
|
102 |
|
channels |
chronic use |
|
|
|
|
|

Appendix 3–1 |
(Continued) |
|
|
|
|
|
|
Drug |
Mechanism of action |
Specific side effects |
Notes |
|
|
|
|
phenytoin (Dilantin) |
Slows recovery of voltage- |
Gum hyperplasia, hirsutism, |
Half-life depends upon dose |
fosphenytoin (Cerebyx) |
gated Na channels |
coarse facies, pseudolymphoma, |
Dilantin by IV precipitates in glucose solution |
|
skin rash (5%), purple glove |
||
|
|
Dilantin brand is sustained release qd dose when |
|
|
|
syndrome (IV route); |
|
|
|
osteoporosis |
given PO fosphenytoin loaded 3 faster |
|
|
|
not useful in absence seizures |
|
|
|
Target range 1–2 g/mL free level |
|
|
|
|
primidone (Mysoline) |
As per phenobarbital |
As per phenobarbital |
Converted to phenobarbital and PEMA |
|
|
|
Not useful in absence seizures |
|
|
|
Used for essential tremor |
|
|
|
|
tiagabine (Gabitril) |
Inhibits GABA reuptake |
Nonconvulsive status epilepticus |
|
|
|
(contraindicated in generalized |
|
|
|
seizures) |
|
|
|
|
|
topiramate (Topamax) |
Inhibits voltage-gated Na |
Transient aphasia/anomia, |
Useful in infantile spasms, Lennox-Gastaut syn- |
|
channels; inhibits AMPA |
nephrolithiasis, acute angle |
drome, atonic and myoclonic seizures |
|
receptors; activates GABAA |
closure glaucoma |
Inhibits oral contraceptives |
|
receptors |
|
|
|
|
Causes weight loss |
|
|
|
|
|
|
|
|
|
valproate (Depakote, |
Slows recovery of voltage- |
Weight gain, hair loss, tremor; |
Depakote ER can be divided bid |
Depacon) |
gated Na channels; inhibits |
thrombocytopenia; |
Also used for migraine, bipolar disorders |
|
T-type voltage-gated |
hyperammonemia; carnitine |
|
|
Can cause status when given with clonazepam |
||
|
Ca channels in thalamus; |
depletion, which can be |
|
|
increases GABA synthesis |
fatal in the malnourished |
Target range 30–100 g/mL |
|
|
|
|
|
|
|
|
zonisamide (Zonegran) |
Inhibits T-type voltage- |
Nephrolithiasis; weight loss |
Also used for migraine |
|
gated Ca channels |
|
|
|
slows recovery of voltage- |
|
|
|
gated Na channels |
|
|
|
|
|
|
felbamate* |
Antagonizes the glycine site |
Rarely used due to high incidence of aplastic anemia |
|
|
on NMDA receptors |
|
|
|
|
|
|
vigabatrin* |
Inhibits GABA metabolism |
Effective against infantile spasms, particularly those caused by tuberous sclerosis; not |
|
|
|
approved for use in US because of high rates of peripheral visual field loss |
|
|
|
|
|
*Not available in the United States. Abbreviations: bid, twice a day; IV, intravenously.
General Treatment of Seizures
103