

Краевое государственное бюджетное образовательное учреждение
среднего профессионального образования
«Красноярский медицинский техникум»
Электронное пособие
По дисциплине «Генетика человека с основами
медицинской генетики»
Специальность: 060501 Сестринское дело,
060102 Акушерское дело
По теме: «Наследственность и патология»
Составила: преподаватель Панова н.А.
Красноярск 2011г.
Содержание:
1.Конспект лекции.
2.Методическое пособие для студентов практического занятия.
3.Методическое пособие для самоподготовки студентов.
4.Контроль знаний.
5. Презентация по теме.
6.Литература.
Лекция № 5
Наследственность и патология
План
1.Классификация наследственной патологии
2Особенности клинических проявлений наследственных заболеваний
3.Генные заболевания
-аутосомно-доминантные
-аутосомно-рецессивные
-сцепленные с половыми хромосомами
-врожденные заболевания
Разделяют пять групп наследственных болезней:
1.Генные болезни – причина генные мутации.
2.Хромосомные болезни –причина хромосомные и геномные мутации.
3.Болезни с наследственной предраспложенностью (мультифакториальные) – возникают в результате соответствующей генетической конституции и наличия определенных факторов внешней среды.
4.Группа генетических болезней – возникают в результате мутаций в соматических клетках (опухоли, аутоиммунные заболевания…)
5.Болезни генетической несовместимости матери и плода –
Возникают в результате иммунологической реакции организма матери на антиген плода.
Генные заболевания – причина – поражение генетического материала на уровне молекулы ДНК, повреждается один только ген. (моногенные болезни). – более 40000. Первичное действие мутантного гена направлено на определенные клеточные структуры (митохондрии, лизосомы, мембраны…). Лизосомное заболевание – мукополисахаридозы…
Митохондриальные – адренолейкодистрофия.
Моногенные болезни подразделяют по типам наследования:
-аутосомно-доминантные
-аутосомно-рецессивные
-Х-сцепленные доминантные
-Х-сцепленные рецессивные
-У-сцепленные рецессивные
-У-сцепленные (голандрические)
-митохондриальные.
Моногенные болезни наследуются по законам Менделя.
Болезни обмена веществ около 600, они затрагивают изменения аминокислотного, углеводного, липидного состава клетки.
признак |
Доминантный характер наследования |
Рецессивный характер наследования |
Обмен веществ:
Аминокислотный
Углеводный
Липидный |
|
Альбинизм Фенилкетонурия
Галоктоземия
Мукополисахаридозы
|
1.Аутосомно- доминантные генные заболевания.
-По неполно-доминантному типу наследуются гемоглобинопатии (серповидно-клеточная анемия и талассемия.)
-по доминантному типу – патология зрения, патологии скелета, патология соединительной ткани, нейрофиброматоз.
Серповидная –клеточная анемия – зависит от доминантного гена- S, который ответственен за синтез гемоглобина HвS. Эритроциты при этом имеют форму – О.
Р. ♀SS х ♀ ss
F1 Ss Ss Ss Ss
Если у организма гены Ss. Ss – гетерозиготы, они либо не болеют, либо болеют в более легкой форме. Особенно тяжело эта анемия протекает у гомозигот – SS, так как аномальные эритроциты распадаются, наблюдается кислородное голодание, приступы лихорадки.., закончиться заболевание может смертью. Причиной этой анемии является замена глютаминовой кислоты на валин в шестой паре нуклеотидов, кодирующей гемоглобин человека.
Талассемия – тоже гемогобинопатия, за нее отвечает ген Т в гомозиготном состоянии-ТТ, вызывая развитие очень мелкиз эритроцитов – микроцитарную анемию. Симптомы: башенный череп, деформированные кости в виде иголок ежа. Гетерозиготы практически здоровы (Тт).
Патология зрения:
-катаракта и глаукома – наследственные заболевания, проявляющиеся в наклонном возрасте. Аутосомно- доминантные. Катаракта – помутнение хрусталика. Глаукома – следствие повышенного внутриглазного давления. Оба заболевания приводят к слепоте.
Патология скелета:
-Брахидактилия – короткопалость-недоразвитие дистальных фаланг пальцев.
-полидактилия – многопалость
-синдактилия – сращение мягких и костных тканей двух и более пальцев.
-ахондропластическая карликовость – общее заболевание костной системы(нарушение роста трубчатых костей, деформация основания черепа и носовых костей) При нормальном тулувище и головы короткие конечности, выпкулый лоб, седловидная переносица..
Моногенные синдромы множественных пороков развития:
-Синдром Морфана – мутация гена фибриллина, в 15-й хромосоме. (высокий рост, конечности удлинены, длинные тонкие пальцы(арахнодактилия), деформация грудной клетки… психическое и умственное развитие не отличаются от нормы
-.Нейрофиброматоз – множественная мутация гена в 17-й хромосоме.(На спине, груди, животе – пигментные пятна. Мелкие опухоли на коже и слизистых, изменение костной системы, умственная отсталость не глубокая.
2.Аутосомно-рецессивные заболевания:
-немоглухота – отсутствие слуха с рождения, что приводит к немате.
-болезни обмена веществ (энзимопатии)-600., большинство из них с умственной отсталостью, кроме альбинизма.
Нарушение белкового обмена:
- альбинизм – отсутствие фермента тирозиназы, он превращает белок тиразин в пигмент меланин.( больные светлые без пигмента, глаза красные…)
-фенилкетонурия – ее ген расположен в 12-й хромосоме, аминокислота фенилаланин полностью не превращается в тирозин, его накопления превращаются в пировиноградную кислоту, которая является нейротропным ядом, токсичного для клеток головного мозга. У больных выражена умственная отсталость, снижено количество меланина(голубоглазые блондины). Проводят экспресс – диагностику при рождении каждого ребенка – проба Фелленга – 2-5 мл свежей мочи+10 капель 10% раствора треххлористого железа при появлении сине-зеленого окрашивания говорят о наличии заболевания.
С раннего возраста больных детей применяю диету , исключающая фенилаланин.
Нарушение липидного обмена:
-Амавротическая идиотия – (Болезнь Тея-Сакса) –связанс с отсутствием фермента гексозаминидазы в печени и селезенке, но с резким увеличением его в мозге. Нарушается зрение, отстает в физическом и интеллектуальном развитии, … к 4-5 годам ребенок гибнет.
-афибриногенемия – дефицит фибриногена, что приводит к нарушению свертываемости крови.
-ихтиоз – наследственное злокачественное заболевание. Кожа покрыта ороговевшими пластинами(чешуя рыбы), невозможно кожное дыхание, умирает вскоре после рождения.
Нарушение углеводного обмена:
-галоктоземия – в тканях и крови накапливается галактоза. Отставание в развитии физическом и умственном , поражение печени, Н.С., глаз… Проявляется с первых дней жизни – расстройство пищеварения, рвота… Без лечения погибают на первом году. Диагноз – биохимическим методом – повышен уровень в моче и в крови фермент галактоза.
Из пищи исключают молоко.
-мукополисахаридоз – нарушается расщепление углеводов. Продукты полураспада откладываются в соединительной ткани, в печени, селезенке, роговице глаз, ЦНС. Отмечается башенный череп, грубые черты лица, укороченное туловище, затем развивается уродливое телосложение. Срок жизни 30 лет.
Нарушение обмена гормонов:
Адреногенитальный синдром – нарушается биосинтез коры надпочечников., что приводитк нарушению развития многих систем организма.
Гипотериоз – понижение функции щитовидной железы.
Генные заболевания, сцепленные с Х- половой хромосомой.
Рецессивные генные заболевания, сцепленные с Х-хромосомой.
-гемофелия, дальтонизм, облысение, мышечная дистрофия Дюшенна, несахарный диабет, агаммаглобулинемия.
Гемофелия – наследственное заболевание, связанное с резким снижением свертываемости крови. Больная Х-хромосома достается сыновьям от мате6ри, а дочери являются носителями этого рецессивного гена.
Агаммаглобулинемия – дефицит защитного белка плазмы крови-гаммаглобулина, заканчивается смертью в раннем возрасте.
Несахарный диабет – наследственная гипофункция гипофиза – приводит к резкому обезвоживанию организма, иногда заканчивается летально.
Дальтонизм – нарушение цветного зрения.
Мышечная дистрофия – дегенеративные изменения в поперечно-полосатой мускулатуре, из-за нарушения синтеза белка дистрофина. Проявляется у детей 3-5 лет. Признаком является уплотнекние икроножных мышц в объеме. Погибают на третьем десятилетии, а к 15-16 годам –они обездвижены.
В случае сцепления с Х-хромосомой доминантного гена:
Гипофосфатемический рахит, отсутствие резцов челюсти.
В У-хромосоме кроме генов, определяющих мужской пол, есть гены перепонок между пальцами и волосатых ушей.
Врожденные пороки развития подразделяются по срокам возникновения и развития:
-гаметопатии – нарушение гамет и зиготы
-бластопатии – патология дробления яйца-первые 15 дней.
-эмбриопатии – с 16дня до 10 недель, наиболее часты и гибельны.
-фетопатии – от 11 недель, смертельны.
Хромосомные болезни. Мультифакториальные и приобретенные болезни.
План
-общая характеристика хромосомных болезней
-аномалии числа половых хромосом
-аномалии числа аутосом
-аномалии строения хромосом
Мультифакториальные болезни
-приобретенные болезни
Общая характеристика хромосомных болезней.
Хромосомные болезни – заболевания, основанные на мутациях колическтва и структуры хромосом. Патологическая роль хромосомных аномалий начинает уже проявляться со стадии зиготы.
Клинически почти все хромосомные болезни нарушением интеллектуального развития, множественными врожденными пороками. Частота хромосомных болезней 7:1000. среди мертворожденных и детей, умерших до года – 22:1000.
Хромосомные болезни делятся:
-аномалии числа половых хромосом
-аномалии числа аутосом
-аномалии строения хромосом.
Аномалии числа половых хромосом:
Они происходят в мейозе и митозе во время первых делений зиготы.
Синдром Шерешевского0Тернера - причина –нерасхождение половых хромосом в мейозе у мужчин. Кариотип больного
45,ХО.-не хватает одной половой хромосомы, но женский организм развивается нормально. Частота рождений 1:3000, болеют только девочки. Их рост 114-145см, крыловидная складка на боковых поверхностях шеи.
Аномалии: короткая шея, широкая грудь, укорочение 4,5 пальцевна руках, много пигментных пятен, птоз, нарушение зрения…Больные бесплодны. При исследовании в буккальном эпителии нет полового хроматина. Психическое развитие страдает в меньшей степени Лечебные мероприятия с целью стимулирования роста до полового созревания и уменьшения полового инфантилизма дают положительные результаты.
Синдром Клайнфельтера – кариотип 47, ХХУ. Обнаруживается половой хроматин, частота 2:1000. Болеют мальчики. Проявление заболевания после полового созревания.. Наблюдается гипоплазия гонад(недоразвитие семенников), Высокий рост, скелет развит по женскому типу, ожирение, бесплодны. Умственное развитие отстает от легких до тяжелых стадий., но могут быть с нормальным интеллектом., чем больше число Х-хромосом, тем больше выражена умственная отсталость., вплоть до идиотии..
Синдром 47, ХУУ.
У этих мужчин отмечается массивность скелета и мышц, снижение интеллекта, в поведении агрессивность, жестокость, намативированные поступки.
Сндром трисомии Х, кариотип 47,ХХХ.
На слизистой рта обнаружено три тельца Бара. Частота 1:1000 девочек. Физическое и психическое развитие не имеет отклонений, , иногда отмечается нарушение со стороны репродуктивной функции. У женщин с тетрасомией Х снижен интеллект. Высокий рост с мужским телосложением.
Мозаицизм по половым хромосомам типа 48ХХ/ХУможет возникать:
-при оплодотворении ооцита двумя различными спермиями, при слиянии двух оплодотворенных яйцеклеток. Проявляется фено-типически в виде гермафродитизма-интерсекса.
Аномалии числа аутосом:
Синдром Дауна – трисомия по 21 хромосоме, с кариотипом 47,ХУ,21+, частота 1:700-800 новорожденных. Голова меньших размеров,скошенный затылок, лицо плоское с косым разрезом глаз, маленький нос, невмещающийся во рту увеличенный яык, рот полуоткрыт, пороки развития внутренних органов, рост низкий, у мужчин бесплодие, женщины могут рожать, но 50% их детей тоже Дауны.
Синдром Патау – гетероплоидия по 13-15 Хромосоме, кариотип 47,ХХ,13+
Множемтвенные пороки развития головного мозга, ССС, почек, ранняя смертность3-4 мес. Частота – 1:6000. Характерно: аномалии черепа и лица –микроцефалия.скошенный лоб, узкие глазные щели низко расположенные, деформированные уши, расщелина верхней губы и неба. На первом году жизни умирают 95%. Синдром Эдварса – трисомия 18 Хромосомы, кариотип -47,ХХ,18+.Дефекты жизненно важных органов, до 1 года доживают 70%, чаще рождаются девочки.
Аномалии строения хромосом.
Причиной является делеция от 1/3 до ½ длины короткого плеча одной из 5-й пары хромосом (5р-) Характерно: спецефический плач, микроцефалия, низко расположенные дерформированные уши, лунообразное лицо, умственная отсталость
Мультифакториальное наследие.
Наследственной предрасположенностью страдают 95% от общей патологии человека. Вызываются они чаще всего изменением нескольких генов. Это: ревматизм, ишемическая болезнь сердца, гипертоническая болезнт, язвенная болезнь, сахарный диабед, псориаз.
Болезни, возникающие при несовместимости матери и плода по антигенам
Болезни, возникающие при несовместимости матери и плода по антигенам, развиваются в результате иммунной реакции матери на антигены плода. Кровь плода в небольшом количестве попадает в организм беременной. Если плод унаследовал от отца такой аллель антигена (Аг+), которого нет у матери (Аг-), то организм беременной отвечает иммунной реакцией. Антитела матери, проникая в кровь плода, вызывают у него иммунный конфликт. Наиболее типичное и хорошо изученное заболевание этой группы - гемолитическая болезнь новорождённых, возникающая в результате несовместимости матери и плода по Rh-Ar. Болезнь возникает в тех случаях, когда мать имеет Rh- группу крови, а плод унаследовал Rh+ аллель от отца.
Практическое занятие №5
Тема: Наследственность и патология
Цели: Изучить аномальные фенотипы и клинические проявления генных заболеваний по фотографиям больных.
Самостоятельная работа
Алгоритм работы
-рассмотреть иллюстрации
-Определить название
-Написать кариотип или тип наследования признака
-Сиптомы заболевания или вражденного уродства
-Записать в тетрадь.
1.
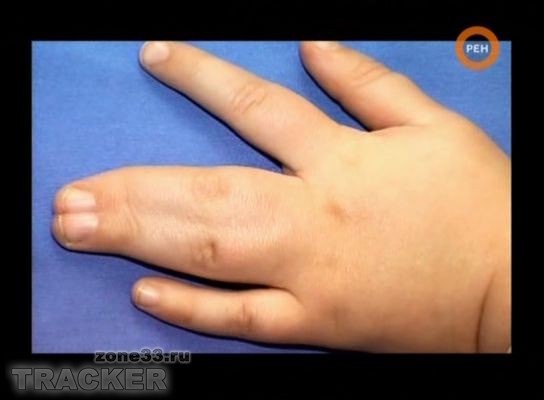
При этой патологии в утробе не происходит разделения тех частей руки...
Вопросы:
-Название патологии
-Тип наследования
-Причины
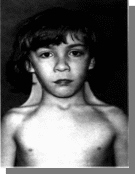
2.

Вопросы:
-Название патологии
-Тип наследования
-Причины
3.
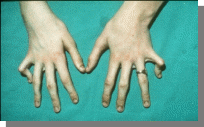
Вопросы:
-Название синдрома
-Тип наследования
-Причины
-Остальные симптомы
4.
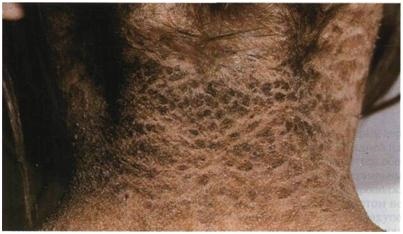 Чешуйки
крупные и темные, почти черные….
Чешуйки
крупные и темные, почти черные….
Вопросы:
-Название патологии
-Тип наследования
-Причины
5.
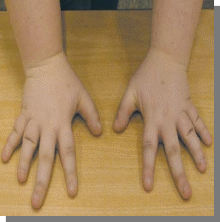
Вопросы:
-Название патологии
-Тип наследования
-Причин
6.
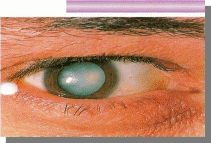
Вопросы:
-Название патологии
-Тип наследования
-Причины
- симптомы.
7.
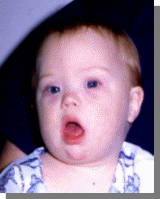
Характерно:типичное плоское лицо, монголоидный разрез глаз, эпикант, открытый рот….
Вопросы:
-Название синдрома
-Кариотип
-Причины
-Остальные симптом
8.
Характерно:аномалии развития конечностей: верхних - сгибательные деформациипальцев, перекрывание пальцев, сжатые пальцы рук, гипоплазия ногтей (особенно V пальца); нижних - короткий и широкий палец стопы, типичная форма стопы в виде качалки, кожная синдактилия стоп.
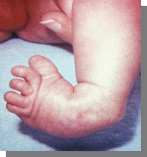
Вопросы:
-Название синдрома
-Кариотип
-Причины
-Остальные симптомы
10
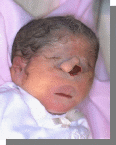
Характерно:Микроцефалия, расщелина верхней губы и неба, низко посаженные деформированные ушные раковины…..
Вопросы:
-Его кариотип.
-Название синдрома.
-Причина.
-Остальные симптомы
11
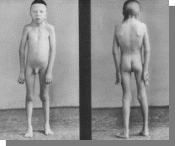
Характерно -больные высокого роста с непропорционально длинными конечностями, выраженной гинекомастией и оволосением поженскому типу…
. Вопросы:
-Его кариотип.
-Название синдрома.
-Причина.
-Остальные симпто
12.
Характерно: нанизм, крыловидные кожные складки на шее, короткая шея с низкой линией роста волос…
Вопросы:
-Его кариотип.
-Название синдрома.
-Причина.
-Остальные симптомы

Контроль знаний
Задания в тестовой форме
Наследственность и патология (2вариант)
1.По-неполно-доминантному типу наследуются болезни:
А) серповидно-клеточная анемия
Б) фенилкетонурия
В) катаракта
Г) альбинизм
2. … заболевание, обусловленное нарушением цветного зрения
3.Врожденные пороки подразделяются на:
А) гаметопатии
Б) эмбриопатии
В) бластопатии
Г) энзимопатии
4.Критические периоды беременности:
А) Конец первой недели
Б) начало второй недели
В) роды
Г) три месяца
5.По какому типу наследуется альбинизм?
А) аутосомно-доминантный
Б) сцепленный с полом доминантный
В) аутосомно-рецессивный
Г) сцепленный с полом рецессивный
6.Гемофелия - это наследственное заболевание, связанное с:
А) гипофункцией гипофиза
Б) дефицитом гаммаглобулина
В) резким снижением свертываемости крови
Г)отсутствием фермента ДНК-полимеразы
7.Основной причиной болезни Дауна является нарушение процесса:
А) митоза
Б) мейоза
В) цитокинеза
Г) транскрипции
8.К какому типу болезней относится синдром Клайфельтера?
А) ненаследственные
Б) моногенные
В) хромосомные
Г) мультифакториальные
9. К хромосомным болезням относятся болезни:
А) фенилкетонурия
Б) галактоземия
В) синдром Патау
Г) альбинизм
10. К генным болезням относятся болезни:
А) фенилкетонурия
Б) синдром кошачьего типа
В) синдром Патау
Г) альбинизм
11.Хромосомные болезни проявляются нарушением всегда:
А) множественными пороками развития
Б) интеллектуального развития
В) слуха
Г) зрения
12.Хромосомные болезни подразделяются на:
А) аномалии числа половых хромосом
Б) аномалии количества хромосом
В) аномалии строения хромосом
Г) все одинаковы
13.Болезни обмена веществ называются……
14.Группа генетических болезней возникают в результате мутаций в:
А) соматических клетках
Б) половых
В) генеративных
Г) мышеных
15. Соответствие синдрома кариотипу:
а) Патау 1) 47xx + 18
б) Дауна 2) 47xx + 13
в) Эдварса 3) 47xxy
г) Клайнфельтера 4) 47ху + 21
5) 45 хо
16. Соответствие заболевания типу наследования:
а) галоктоземия 1) сцепленное с У-хромосомой
б) повышенное оволосение ушных раковин 2) сцепленный с Х-хромосомой
в) гемофилия 3) аутосомно-доминантный
г) синдром Люрфана 4) аутосомно – рецессивный
5) цитоплазматический
6) мозаичный
17. Соответствие кариотипа синдрому:
а) 47 хх + 13 1) Шерешевского - Тернера
б) 45 хо 2) Дауна
в) 47 хху 3) Патау
г) 47 хх + 21 4) Клайнфельтера
5) «Кошачьего крика»
18. Наследованные заболевания, вызванные мутацией в пределах одного гена:
а) хромосомные
б) моногенные
в) мультифакториальные
г) генеративные
19. …– заболевание, обусловленное биохимическим дефектом превращения фенилаланина.
20.. Полуоткрытый рот с высунутым языком и выступающей нижней челюстью характерен для синдрома:
а) Клайнфельтера
б) Дауна
в) Шерешевского – Тернера
г) Эдварса
21. …. - генетическая природа синдрома Клайнфельтера.
22. ….- по 13 хромосоме - это генетическая природа синдрома Патау.
23. ….- отсутствие пигмента меланина в коже радужной оболочке глаза.
24..Положительная реакция Фелинга выявляется при заболевании ….
25..Основные задачи клинико-генеалогического метода:
а).установление наследственного характера заболевания
б).установление типа наследования
в).асчет риска для потомства
г).пределение членов семьи, нуждающихся в детальном обследовании
26. Для диагностики моногенных заболеваний используются методы:
а).исследование полового хроматина
б).биохимический
в).функциональной диагностики
г).цитогенетический
27. Для фенилкетонурии характерно:
а). аутосомно-рецессивный тип наследования
б). аутосомно-доминантный тип наследования
в). гиперфенилаланинемия
г). слабоумие
28. Для диагностики фенилкетонурии используют:
а). молекулярно-генетическую диагностику
б). скрининг новорожденных
в). определение содержания фенилаланингидроксилазы
г). определение содержания фенилаланина в крови
29.. Аутосомно-доминантно наследуются:
а). ахондроплазия (дистрофическая карликовость)
б). хорея Гентингтона
в). серповидно-клеточная анемия
г). гемофилия
30.. Наследуются сцепленно с Х-хромосомой:
а). миодистрофия Беккера
б). муковисцидоз
в). гипертрофическая миодистрофия Дюшена
г). цветовая слепота (дальтонизм)
31... Мутации, которые приводят организм к внутриутробной гибели называются…
32.. Для аутосомно-доминантного типа наследования характерно:
а). наличие болезни у родителей
б). передача из поколения в поколение
в). проявление в гетерозиготном состоянии
г). проявляется только у мужчин
33. Для аутосомно-рецессивного типа наследования характерно:
а). родители фенотипически здоровы
б). родители облигатные гетерозиготные носители
в). проявляется в каждом поколении
г). женщины болеют тяжелее
34. Скрининг новорожденных должен удовлетворять следующим требованиям:
а). быть дешевым
б). давать минимальное количество ложно-отрицательных результатов
в). использоваться для диагностики наиболее редких заболеваний
г). должен осуществляться законодательно во всех родильных домах
35. Для лечения фенилкетонурии используют:
а). диету без кетоновых тел
б). диету без фенилаланина
в). психолого-педагогическую коррекцию
г). лекарственные препараты