6.1.2. Строение карбонильной группы
С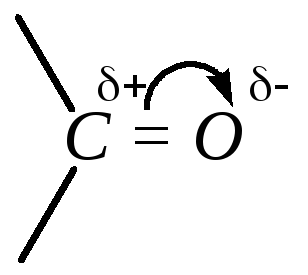 вязь
между атомами углерода и кислорода в
карбонильной группе — двойная, образована
двумя парами валентных электронов, то
есть кроме-связи
C–О
имеется -связь,
обусловленная боковым перекрыванием
негибридных р-орбиталей
этих атомов. Атом углерода образует три
-связи
за счёт sp2-гибридных
орбиталей, оси которых лежат в одной
плоскости.
вязь
между атомами углерода и кислорода в
карбонильной группе — двойная, образована
двумя парами валентных электронов, то
есть кроме-связи
C–О
имеется -связь,
обусловленная боковым перекрыванием
негибридных р-орбиталей
этих атомов. Атом углерода образует три
-связи
за счёт sp2-гибридных
орбиталей, оси которых лежат в одной
плоскости.
Двойная связь C=О сильнополярна, поэтому на атомах углерода и кислорода карбонильной группы локализуются соответственно значительные положительный и отрицательный заряды.
Электроны -связи подвижны, поэтому возможно изображение карбонильной группы в виде одной из двух граничных структур:

6.1.3. Физические свойства
Альдегиды и кетоны — полярные соединения из-за наличия сильнополярной связи C=О. Поэтому простейшие представители этих соединений хорошо растворяются в воде. По этой же причине, ввиду наличия диполь-дипольных взаимодействий, температуры кипения альдегидов и кетонов выше, чем у углеводородов с такими же молекулярными массами, но ниже, чем у соответствующих спиртов. Это объясняется отсутствием водородных связей между молекулами карбонильных соединений в отличие от спиртов. Кроме того, кетоны кипят при более высокой температуре, чем изомерные им альдегиды.
Альдегиды и кетоны — бесцветные соединения; многие из них обладают резким запахом.
6.1.4. Химические свойства
Наличие в молекулах альдегидов и кетонов карбонильной группы является причиной проявления ими общих свойств, связанных с особенностями строения этой функциональной группы.
1. Высокая полярность связи С=О вызывает на карбонильном атоме углерода значительный дефицит электронной плотности (C+), и по этому атому углерода возможна нуклеофильная атака. А так как в этой группе присутствует -связь, энергия которой существенно меньше энергии -связи, то наиболее характерным типом реакции альдегидов и кетонов должны быть реакции нуклеофильного присоединения (AdN).
2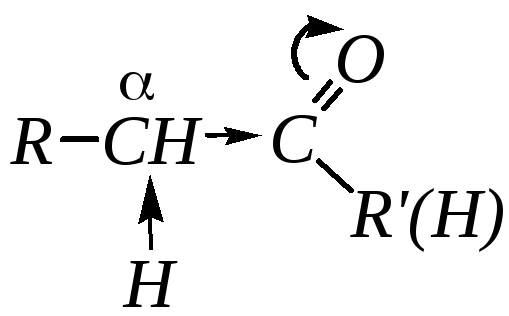 .
Кроме того, высокая полярность связиС=О
вызывает на атоме углерода, соседнем с
карбонильной группой (-углеродном
атоме), значительный электроноакцепторный
эффект и, как следствие этого, повышенную
полярность связи С–Н
-углеродного
атома. Это характеризует данные соединения
как СН-кислоты,
анионы которых могут проявлять сильные
нуклеофильные свойства.
.
Кроме того, высокая полярность связиС=О
вызывает на атоме углерода, соседнем с
карбонильной группой (-углеродном
атоме), значительный электроноакцепторный
эффект и, как следствие этого, повышенную
полярность связи С–Н
-углеродного
атома. Это характеризует данные соединения
как СН-кислоты,
анионы которых могут проявлять сильные
нуклеофильные свойства.
Здесь будут рассмотрены также другие свойства альдегидов и кетонов, а также реакции, связанные с особенностями строения некоторых из них.
6.1.4.1. Реакции нуклеофильного присоединения
Реакции нуклеофильного присоединения — большая группа реакций альдегидов и кетонов. В этих реакциях атака может начинаться либо нуклеофилами по атому углерода, либо электрофилами по атому кислорода. Однако первичная электрофильная атака по кислороду происходит редко, за исключением атаки протонами и кислотами Льюиса. При этом быстрое и обратимое протонирование
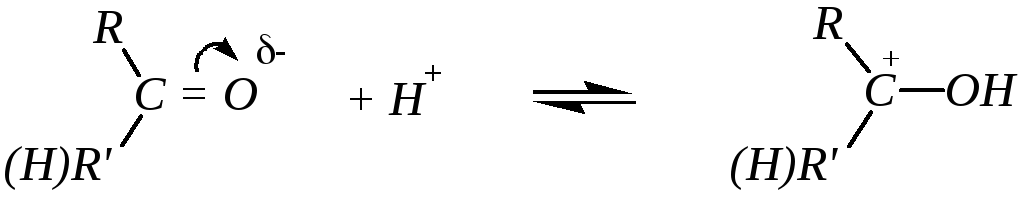
предшествует, как правило, более медленной нуклеофильной атаке по атому углерода:
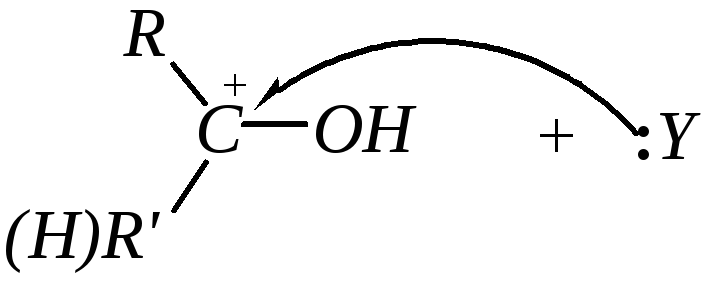
То есть реакции присоединения по карбонильной группе будут катализироваться кислотами. Однако многие активные нуклеофилы являются анионами слабых кислот, а в кислой среде диссоциация этих кислот, а значит, и образование анионов будет подавлено, например:
HCN
![]() Н+
+ СN¯
Н+
+ СN¯
В этом случае в кислой среде концентрация анионов СN¯ будет ничтожна, а молекулы HCN практически не обладают нуклеофильными свойствами.
Для нуклеофилов, представленных молекулами аммиака и его производных, применение кислотных катализаторов вообще может быть неэффективно, так как сильные кислоты превращают молекулу в катион, не обладающий нуклеофильными свойствами:
R-NH2 + Н+ R-NH3+
Поэтому взаимодействие с такими нуклеофилами проводят в нейтральной или слабокислой среде, в которой концентрация нейтральных молекул реагента остаётся достаточной для осуществления нуклеофильной атаки.
Взаимодействие карбонильных соединений с нуклеофилами в отсутствие кислотных катализаторов осуществляется по схеме:
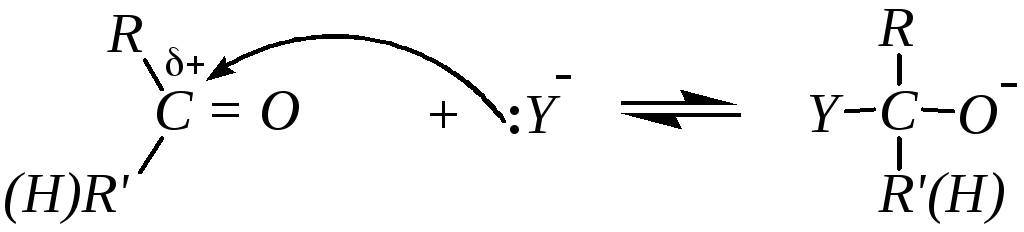
Дальнейшее превращение с образованием продукта присоединения требует наличия в среде хотя бы слабой кислоты, то есть в данном случае соединения, способного отщепить протон:
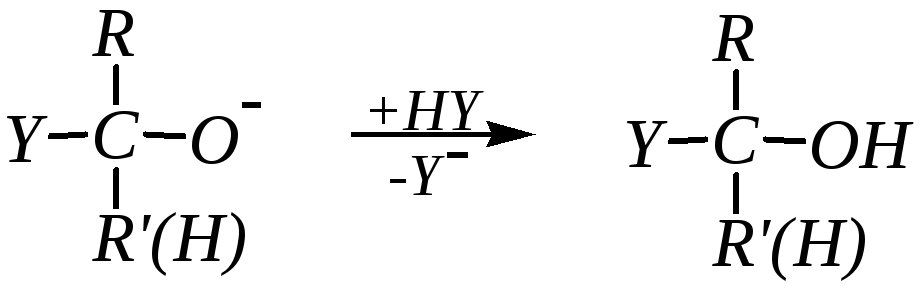
Таким образом, нуклеофильное присоединение к альдегидам и кетонам в отсутствие кислотных катализаторов включает две стадии:
1) взаимодействие карбонильного соединения с нуклеофилом с образованием аниона, протекающее через переходное состояние:
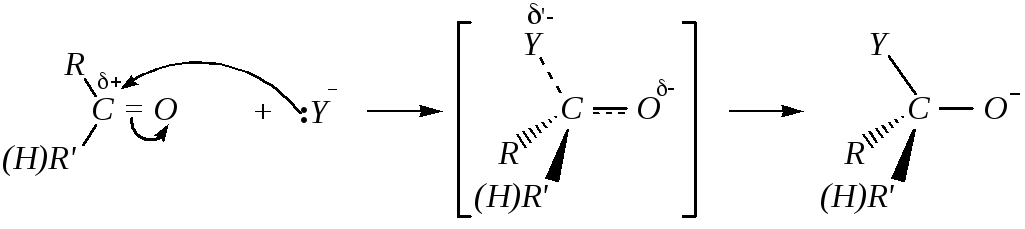
2) превращение аниона в нейтральную молекулу:
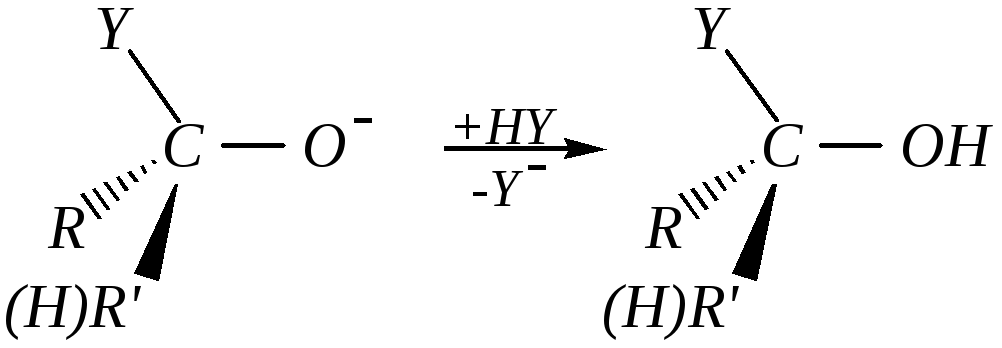
При кислотном катализе на первой стадии происходит быстрое протонирование с образованием карбокатиона, который при взаимодействии с нуклеофилом (лимитирующая стадия) превращается в продукт присоединения:
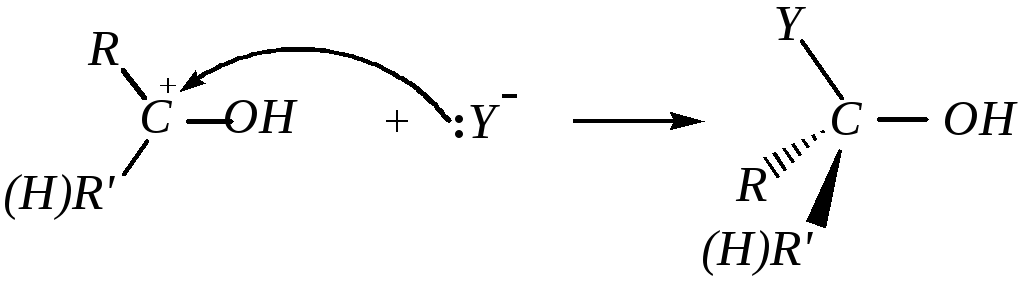
Можно сравнить альдегиды и кетоны между собой по способности вступать в AdN-реакции. Так, кетоны реагируют обычно в более жёстких условиях, чем альдегиды, а в некоторые реакции не вступают вовсе. Для этого есть две причины: электронная и пространственная.
Первая причина связана с величиной эффективного положительного заряда на карбонильном атоме углерода и влиянием на него электронодонорных эффектов заместителей R и R :
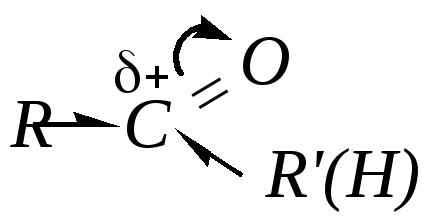
В молекулах кетонов на карбонильный атом углерода действуют индуктивные эффекты двух углеводородных радикалов Rи R, а в молекулах альдегидов (R = Н) — только одного (R), поэтому в случае кетонов величина положительного заряда на этом атоме меньше. Легкость взаимодействия субстрата с нуклеофилом должна зависеть от величины эффективного положительного заряда на карбонильном атоме углерода. Поэтому электронодонорные группы при реакционном центре будут снижать, а электроноакцепторные, наоборот, — повышать реакционную способность карбонильных соединений. Поэтому взаимодействие кетонов с нуклеофилами протекает труднее.
Вторая причина связана с пространственными размерами заместителей R и R и влиянием этого на доступность реакционного центра (C+) нуклеофилу. Так как в молекулах альдегидов вместо одного из углеводородных радикалов (R) — атом водорода, то карбонильная группа оказывается более доступна для нуклеофильной атаки, нежели в кетонах. И по этой причине нуклеофильные реакции кетонов протекают труднее.
Наиболее типичными примерами нуклеофильного присоединения по карбонильной группе являются реакции взаимодействия с водой, со спиртами, с аммиаком и его производными, с синильной кислотой, с гидросульфитом натрия, с реактивом Гриньяра.
1. Гидратация альдегидов и кетонов протекает в водных растворах карбонильных соединений по схеме:
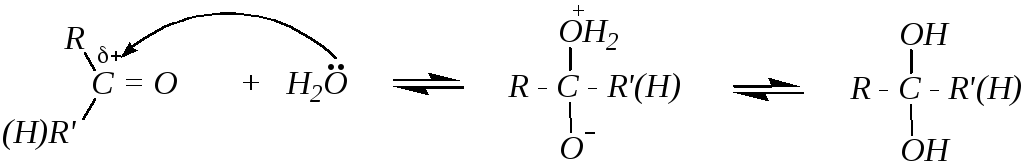
Способность присоединять молекулу воды во многом определяется структурой исходного соединения. Так, формальдегид в обычных условиях практически полностью гидратирован (на 99.99%), тогда как для ацетальдегида эта величина составляет 58 %, а для ацетона концентрация гидрата пренебрежительно мала. Однако гидратация кетонов в значительной степени катализируется кислотами и основаниями:
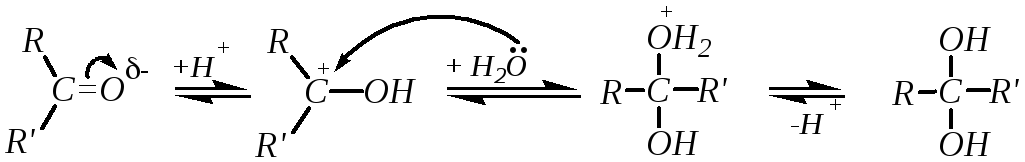
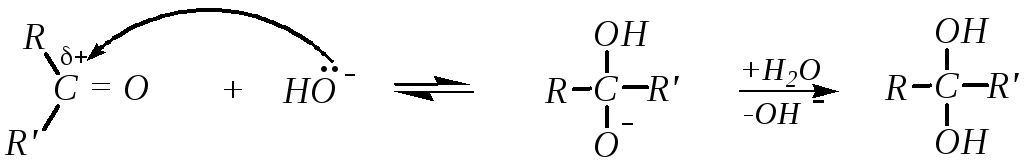
Электроноакцепторные заместители в алкильных группах облегчают гидратацию и стабилизируют образующиеся гидраты. Так, например, трихлоруксусный альдегид (хлораль) гидратирован практически полностью. Продукт реакции, хлоральгидрат Cl3C-CH(OH)2, представляет собой устойчивое кристаллическое соединение, способное отщеплять воду только при нагревании с водоотнимающими веществами (конц. H2SO4 и др.).
2. Присоединение спиртов приводит к образованию полуацеталей и полукеталей. Эта реакция аналогична гидратации альдегидов и кетонов. Например:

метилкеталь ацетона
Реакция катализируется как кислотами, так и основаниями (аналогично гидратации). Однако в присутствии кислотных катализаторов реакция идёт дальше, образующийся полуацеталь (полукеталь) вступает в нуклеофильное замещение и образуются полные ацетали и, соответственно, кетали. Механизм превращения полуацеталя (полукеталя) в ацеталь (кеталь) мономолекулярный, так как образующийся промежуточный карбокатион стабилизирован сопряжением с участием неподелённой электронной пары соседнего атома кислорода:
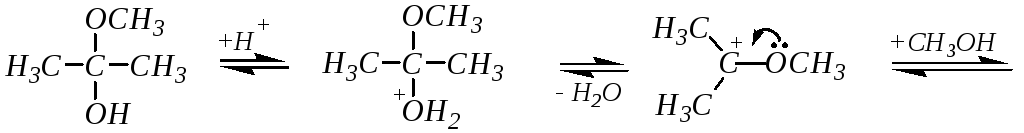
метилкеталь ацетона
(полукеталь)
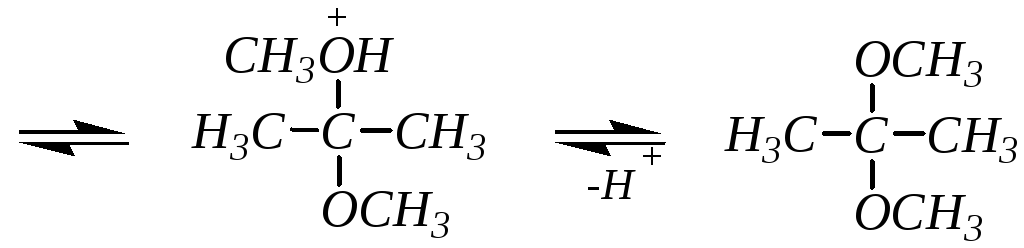
диметилкеталь ацетона
Эти реакции обратимы — ацетали и кетали гидролизуются под действием кислоты. Основания не катализируют нуклеофильное замещение (превращение полуацеталей в ацетали) ни в прямом, ни в обратном направлениях, поэтому в щелочной среде образуются только полуацетали.
Большинство альдегидов легко превращается в ацетали. Для кетонов процесс протекает медленнее настолько, что провести реакцию часто не удаётся.
3. Взаимодействие с аммиаком и его производными (аминами, гидразином NH2-NH2 и замещёнными гидразинами, гидроксиламином NH2-ОH и т.д.) обычно не требует катализатора, хотя часто реакцию проводят в слабокислой среде (рН = 3 5). Если центром нуклеофильности атакующей молекулы является вторичный атом азота (например, во вторичных аминах), то сначала протекает обычное присоединение с образованием аминоспирта, который затем отщепляет воду, и образуется непредельный амин винильного типа (енамин), стабилизированный р--сопряжением, например:
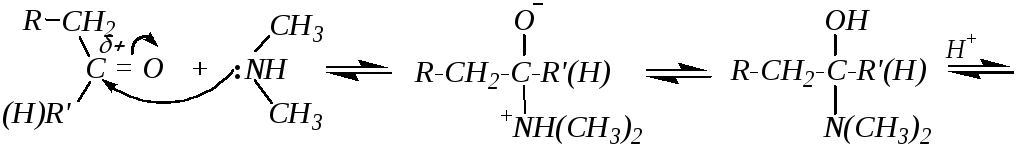
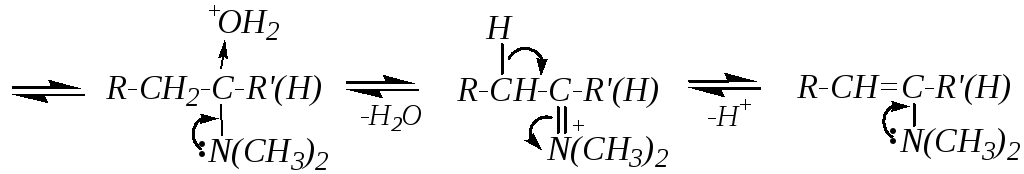
Если же нуклеофильная атака производится первичным атомом азота, то присоединение также сопровождается последующим отщеплением, но депротонированию в этом случае подвергается атом азота. При взаимодействии с первичными аминами это приводит к образованию иминов, а при взаимодействии с гидроксиламином — к образованию оксимов. Для этих случаев можно привести общую схему:
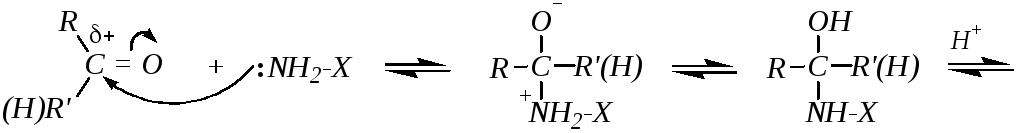
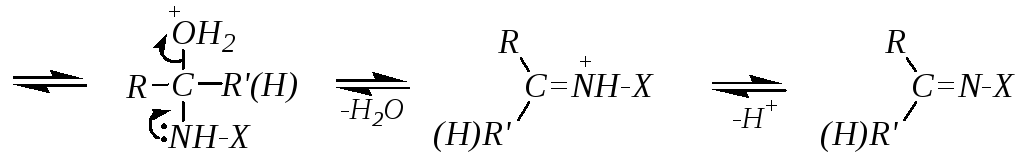
Лимитирующей стадией может быть либо первая стадия (нуклеофильная атака), либо последняя (отщепление воды). Это в значительной мере зависит от рН среды. В щелочной среде протонирование промежуточного продукта присоединения будет осуществляться в очень незначительной степени, а, значит, отщепление молекулы воды (и тем более гидроксид-иона) будет затруднено. Лимитирующей стадией будет последняя. А при низких значениях рН последняя стадия проходит легко и быстро (за счёт протонирования гидроксогруппы), но первичное образование продукта присоединения замедлится из-за превращения реакционноспособного нуклеофила NH2-X в нереакционноспособный катион +NH3-X. Компромиссом является слабокислая среда, в которой концентрация непротонированной формы нуклеофила остается максимальной. В этих условиях первая стадия, то есть первичная атака карбонильной группы нуклеофилом, может быть лимитирующей стадией всего процесса, но скорость реакции в целом будет достаточно высокой.
При взаимодействии карбонильных соединений с одним из производных аммиака — гидразином — образуются гидразоны, широко использующиеся в органическом синтезе.

Их нагревание в присутствии катализатора приводит к разложению с образованием алканов. Катализаторами служат гидроксид калия в присутствии платины, или гидроксид натрия, или этилат натрия (реакция Кижнера–Вольфа). Механизм реакции при использованииКОН иPtможно представить в следующем виде:
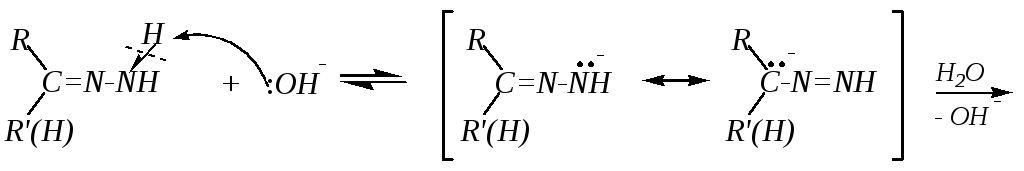
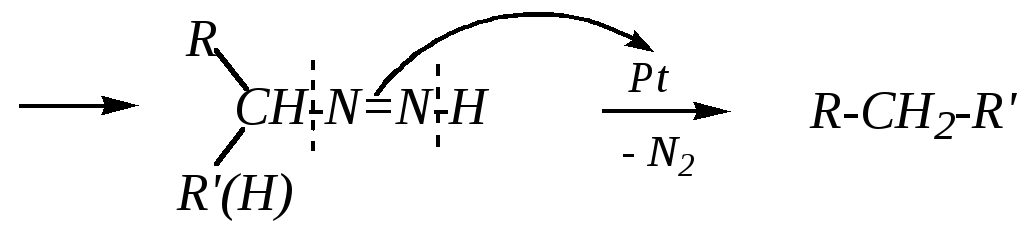
Данная реакция может применяться как способ получения предельных углеводородов (гл. 2.5).
4. Присоединение синильной кислоты приводит к образованию циангидринов. Например:
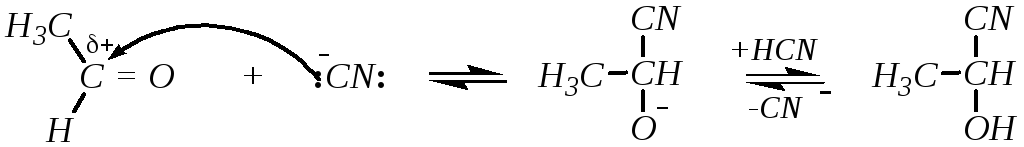
циангидрин ацетальдегида
Эта реакция обратима. Присоединение катализируется основаниями, лимитирующей стадией является атака цианид-ионами.
5. Присоединение гидросульфита натрия даёт бисульфитные производные альдегидов и кетонов. Бисульфитные производные образуются при взаимодействии гидросульфита преимущественно с альдегидами и метилкетонами. Большинство других кетонов в реакцию не вступает по пространственным причинам. Механизм присоединения:
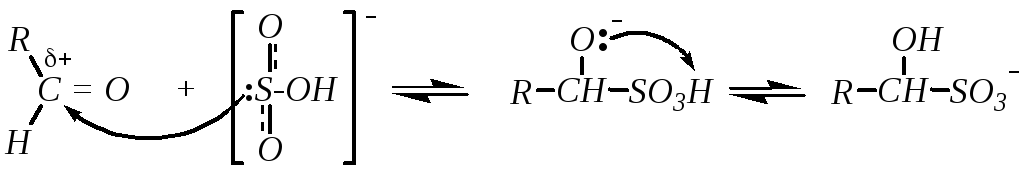
Реакция обратима. В обратную сторону она протекает при обработке продукта либо кислотой, либо основанием.
6. Взаимодействие с реактивами Гриньяра, называемое реакцией Гриньяра, приводит к образованию алкоголятов, а последующий гидролиз — к получению первичного, вторичного или третичного спирта в зависимости от структуры исходного карбонильного соединения.
Реактивы Гриньяра (галогениды алкилмагния — гл. 3.2.2.3) обладают нуклеофильностью за счёт полярной связи С–Mg и, таким образом, локализацией на атоме углерода частичного отрицательного заряда. Присоединение к альдегидам и кетонам, например, бромида метилмагния к ацетальдегиду, протекает через образование четырёхцентрового переходного состояния:
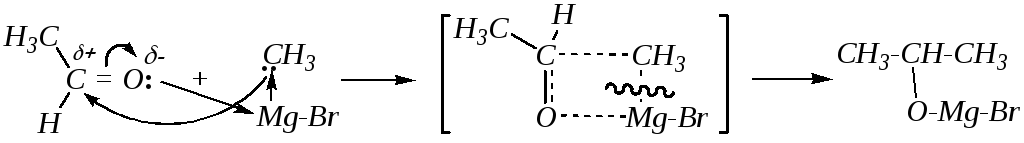
бромид-изопропилат
магния
Гидролиз продукта присоединения даёт изопропиловый спирт:
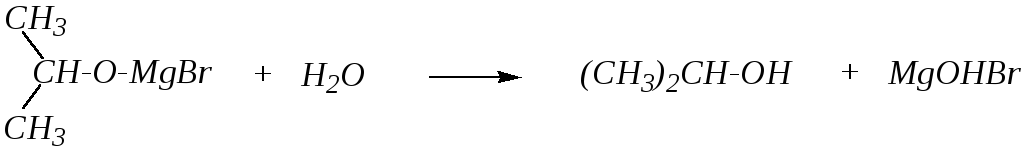
Данная реакция может рассматриваться как способ получения спиртов из альдегидов и кетонов.
7. Восстановление гидридами металлов. Наиболее типичными реагентами, применяемыми для восстановления карбонильной группы, являются комплексные соединения — тетрагидроалюминат лития (алюмогидрид лития, Li[AlH4]) и тетрагидроборат натрия (борогидрид натрия, Na[BH4]).
В качестве примера приведём взаимодействие алюмогидрида лития с кетонами. От молекулы алюмогидрида отщепляется гидрид-ион, проявляющий нуклеофильные свойства:
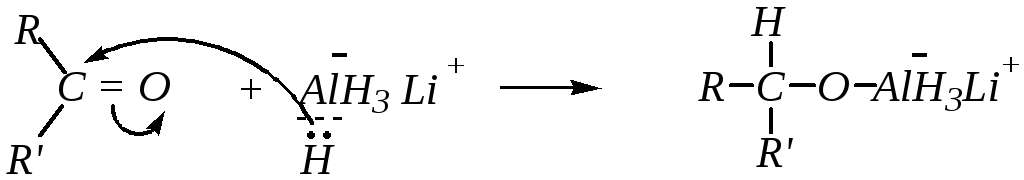
За счёт оставшихся трёх атомов водорода у алюминия также может происходить нуклеофильная атака
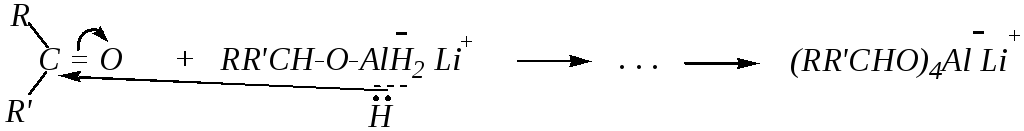
И затем при гидролизе образовавшегося комплексного алюмината лития получается вторичный спирт
(RRCHO)4Al¯
Li+
![]() RRCHOH
RRCHOH
Если в качестве исходного карбонильного соединения был взят альдегид, то при восстановлении алюмогидридом лития (или борогидридом натрия) получится первичный спирт.
8. Взаимодействие с пентахлоридом фосфора. Пентахлорид фосфора PCl5 димеризован, и его можно условно представить в виде ионной пары [PCl4]+ [PCl6]¯. Первой стадией реакции является электрофильное содействие нуклеофильной атаке в виде взаимодействия атома кислорода альдегида или кетона с катионом [PCl4]+
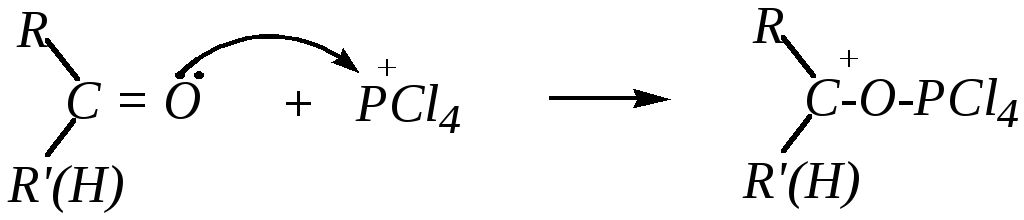
Образовавшийся карбокатион подвергается нуклеофильной атаке анионом [PCl6]¯
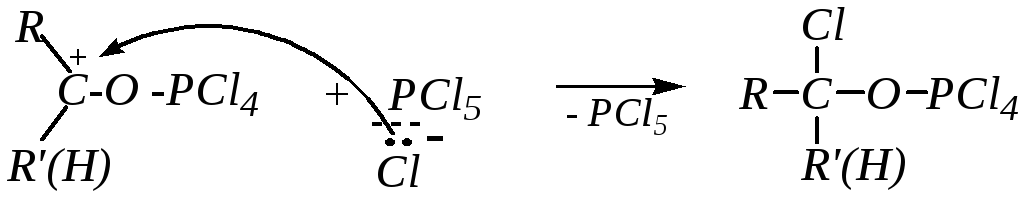
Продукт присоединения малоустойчив, и далее происходит внутримолекулярное нуклеофильное замещение, механизм которого похож на механизм бимолекулярного замещения, но атака нуклеофила осуществляется с той же стороны, где находится нуклеофугная группа.

Конечный продукт представляет собой геминальное дигалогенопроизводное алкана, а рассматриваемая реакция может служить способом получения этих соединений. Вместо PCl5могут быть использованы, например,PBr3,SF4и др.
9. Присоединение ацетилена и его металлических производных. В свойствах ацетиленовых углеводородов отмечалось, что замещённые ацетилениды металлов являются эффективными нуклеофилами (гл. 4.4.2). Поэтому взаимодействие альдегидов и кетонов с ацетиленидами — это также один из примеров реакций нуклеофильного присоединения. Продуктами реакций являются непредельные спирты:
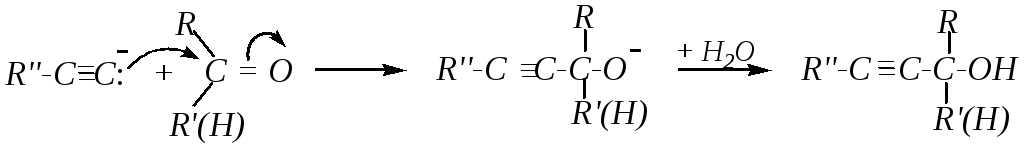
Однако эта реакция возможна не только с алкинидами металлов, но и с самими ацетиленовыми углеводородами в присутствии щёлочи. Например:
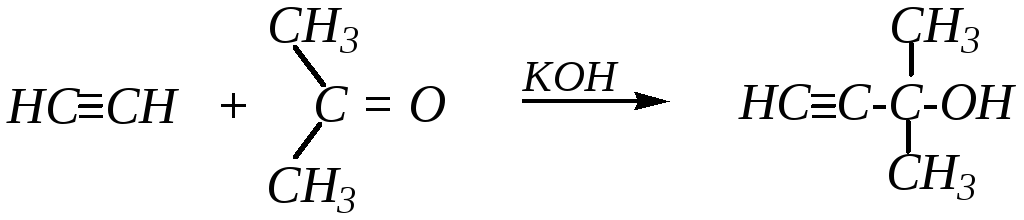
2-метилбутин-3-ол-2
Механизм реакции тот же, так как нуклеофилом является алкинид-анион.
Другим примером может служить взаимодействие ацетилена с формальдегидом в присутствии хлорида меди (I):
HCCH
![]() HCC¯
HCC¯
![]() HCC–CH2OH
HCC–CH2OH
пропаргиловый спирт
10. Альдольная и кротоновая конденсации характерны для альдегидов и кетонов, имеющих -водородные атомы. При альдольной конденсации происходит присоединение одной молекулы карбонильного соединения к другой молекуле; образуется альдегидоспирт (альдоль) или кетоноспирт (кетол). Например:
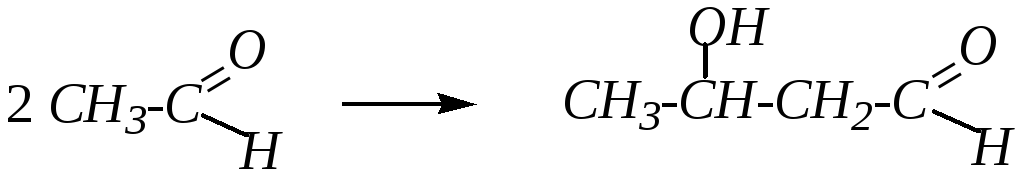
3-гидроксибутаналь
При кротоновой конденсации из двух молекул карбонильного соединения образуется молекула непредельного альдегида или кетона; процесс сопровождается выделением молекулы воды. Так как в наиболее простом случае конденсации (уксусного альдегида) образуется кротоновыйальдегид, то и конденсация получила такое же название:
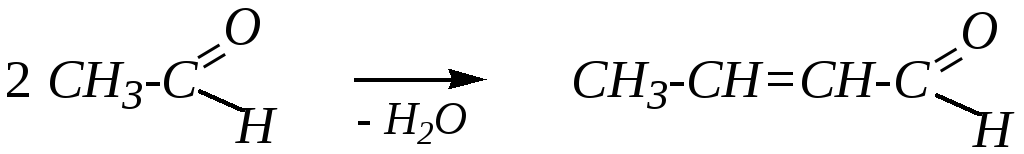
бутен-2-аль (кротоновый альдегид)
Реакции обычно проводят в присутствии оснований, однако возможен и кислотный катализ. В качестве оснований для катализа используют 5—10%-й раствор щёлочи. Реакция начинается с отрыва протона от -углеродного атома:
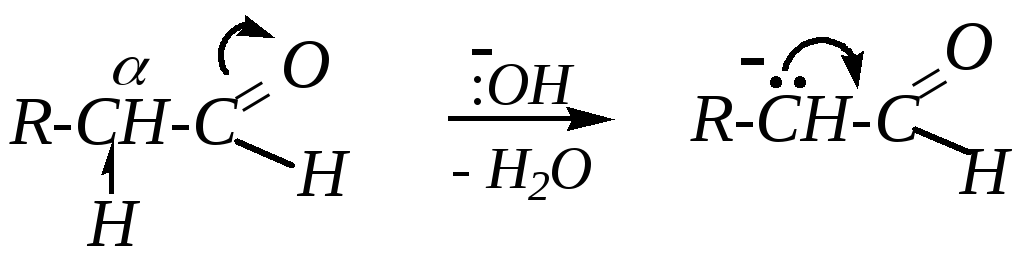
При этом образуется карбанион, стабилизированный р--сопряжением. Дальнейшее превращение происходит за счёт проявления им нуклеофильных свойств по отношению ко второй молекуле альдегида:

Образовавшийся анион отрывает протон от молекулы воды, сам превращаясь в молекулу альдоля:
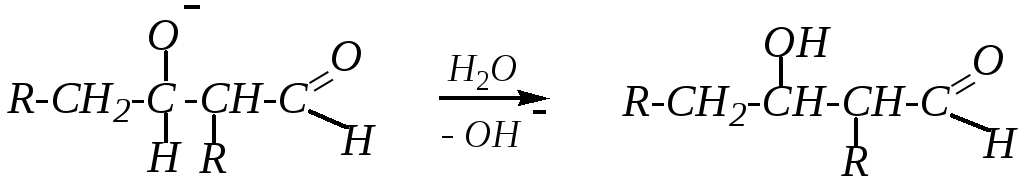
При повышенной температуре может идти дальнейшее превращение альдоля, которое также связано с отрывом -водородного атома от того же атома углерода (находящегося между функциональными группами) под действием щёлочи, а затем происходит отщепление гидроксид-иона от образовавшегося карбониевого аниона:
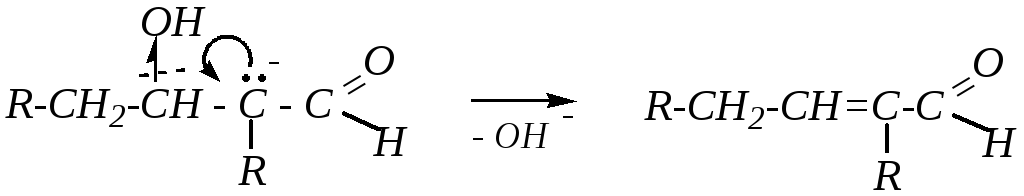
Из кетонов по аналогии получаются кетолы, однако при применении оснóвного катализатора происходит депротонирование по наиболее полярной C–Н-связи:
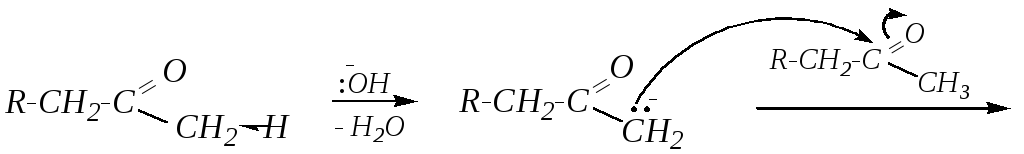

(Возможно также дальнейшее превращение кетола в непредельный кетон.)
При применении кислотного катализатора промежуточным соединением является термодинамически более стабильный енол, образованный за счёт более разветвлённого -углеродного атома:
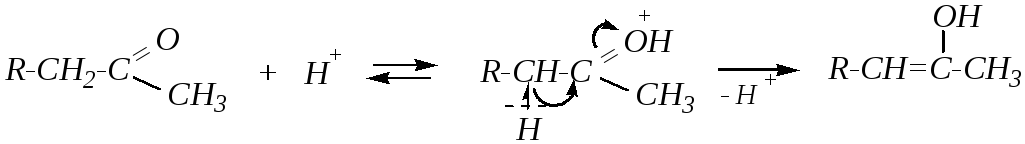
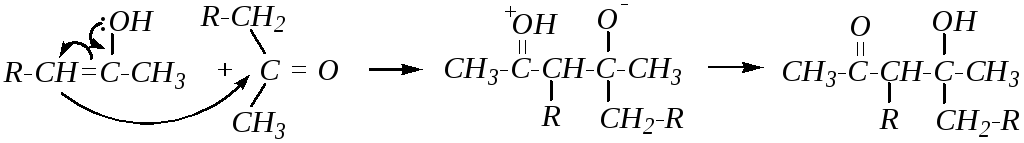
Таким образом, использование того или иного катализатора приводит к разным изомерным непредельным кетонам.
11. Олиго- и полимеризация. Между молекулами альдегидов возможны реакции нуклеофильного присоединения, в которых нуклеофильной частью реагента является карбонильный атом кислорода каждой из реагирующих молекул. Такие реакции осуществляются в кислой среде, и они обычно приводят к тримеризации, тетрамеризации или полимеризации исходных веществ. Например:
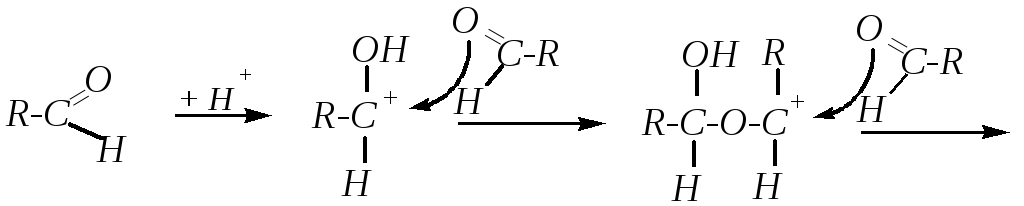

В частности, такая реакция характерна для ацетальдегида. При этом в зависимости от условий может получиться паральдегид (тример) или метальдегид (тетрамер):
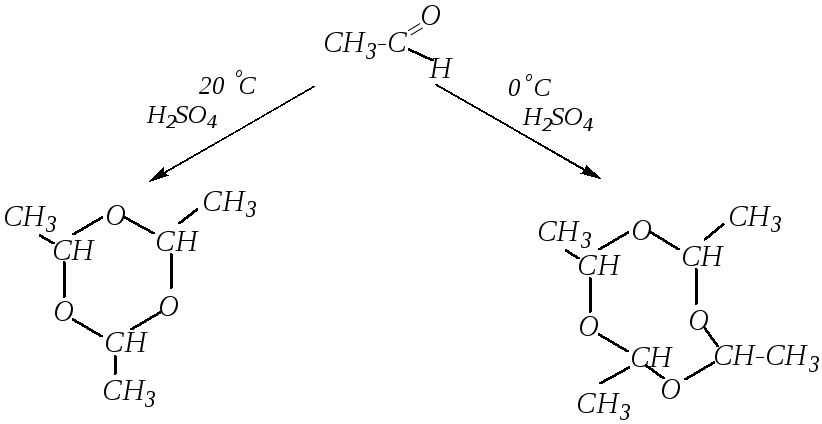
паральдегид метальдегид
Формальдегид легко образует не только три- и тетрамеры, но и линейные полимеры:
n
CH2=O
![]() HO-(-CH2-O-)n-H
HO-(-CH2-O-)n-H
полиформальдегид
При упаривании водных растворов формальдегида в вакууме образуется параформ(илипараформальдегид)(CH2O)n(n= 812).
6.1.4.2. Реакции замещения в радикале
Примером реакций замещения, протекающих по углеводородному радикалу, является галогенирование. Эти реакции осуществляются в присутствии как кислотного, так и оснóвного катализатора и протекают по -углеродному атому (в случае кетонов — по одному из -углеродных атомов).
В присутствии основания происходит отщепление протона от молекулы альдегида или кетона. При этом в случае кетонов разрывается более полярная C–Н-связь (часто наименее разветвлённого -углеродного атома), образуется карбониевый анион, который затем подвергается электрофильной атаке молекулой галогена:
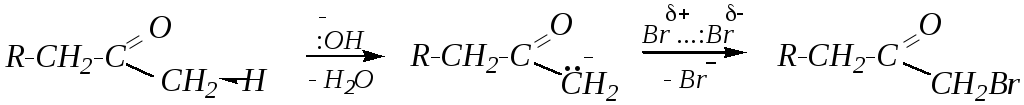
В избытке галогена реакция протекает дальше, причём реакция введения каждого последующего атома брома протекает с нарастающей скоростью, так как депротонирование галогенозамещённых кетонов происходит легче из-за акцепторного влияния галогена:

При этом реакция протекает по одному и тому же -углеродному атому до полного, исчерпывающего галогенирования. При использовании в качестве карбонильного соединения алкилметилкетона (как в приведенном примере) после образования тригалогенозамещённого кетона происходит его щелочной гидролиз:
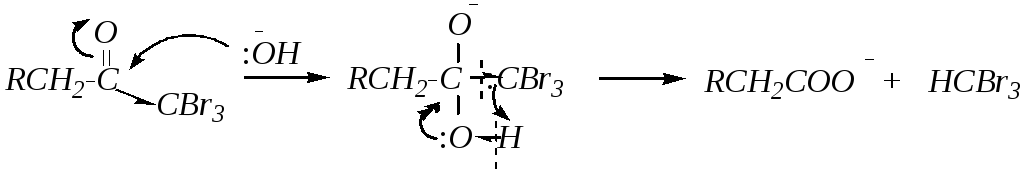
В результате образуются тригалогенометаны — галоформы(в данном случае — бромоформ). Поэтому реакция называетсягалоформной пробойи является качественной реакцией на метилкетоны.
В кислой среде (кислотный катализ) повышается активность электрофила (молекулы галогена), который может взаимодействовать с енольной формой карбонильного соединения:
![]()
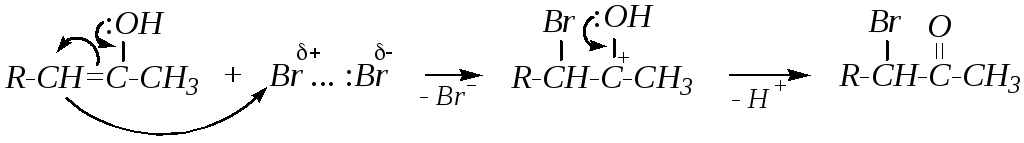
В отличие от основно-катализируемой реакции в кислой среде происходит галогенирование, как правило, по более разветвлённому -углеродному атому, так как доля именно такой енольной формы (см. схему) в среде выше ввиду её большей стабильности.
В общем случае важной особенностью галогенирования альдегидов и кетонов является независимость скорости процесса от природы галогена. Скорость реакции зависит только от концентрации катализатора, определяющего скорость лимитирующей стадии процесса.
6.1.4.3. Реакции окисления-восстановления
Окисление альдегидов до соответствующих кислот (или их солей) протекает легко за счёт наличия подвижного атома водорода в альдегидной группе. В качестве окислителей обычно применяют влажный оксид серебра, раствор перманганата калия или фелингову жидкость. Качественной реакцией на альдегиды (для отличия их от кетонов) обычно считается реакция серебряного зеркала— взаимодействие альдегидов с гидроксидом диамминсеребра (реактивом Толленса):
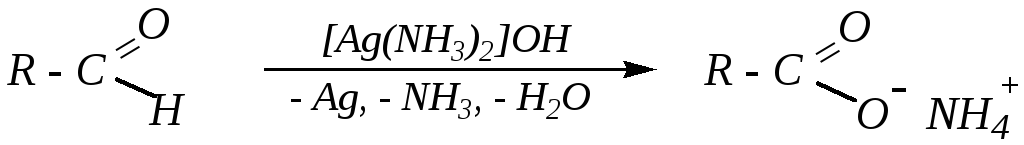
Этот реактив является настолько мягким окислителем, что при его применении не происходит окисления ни каких-либо других функциональных групп, ни кратных связей. Поэтому окисление реактивом Толленса непредельных альдегидов приведёт к соответствующим непредельным кислотам.
Аналогично этому окисление некоторых альдегидов происходит и свежеосаждённым гидроксидом меди (II) (иногда для этих целей используютреактив Фелинга*— комплекс катионаCu2+ с винной кислотой):
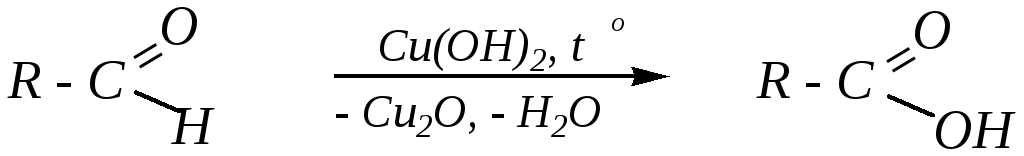
Однако эту реакцию не даёт, например, уксусный альдегид.
В отличие от альдегидов кетоны подвергаются действию окислителей в жёстких условиях. Так, диоксид селена окисляет -метиленовую группу до карбонильной:
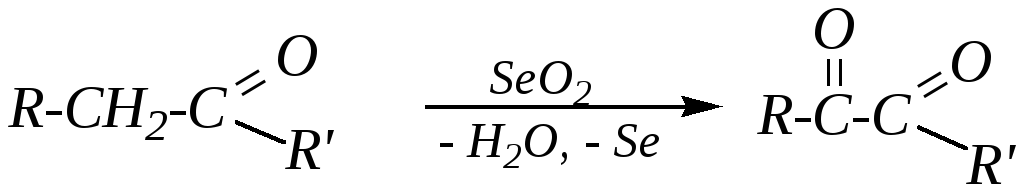
А действие сильных окислителей (например, хромовой смеси) при нагревании приводит к окислению одного из -углеродных атомов и разрывуC–C-связи по обе стороны от карбонильной группы:
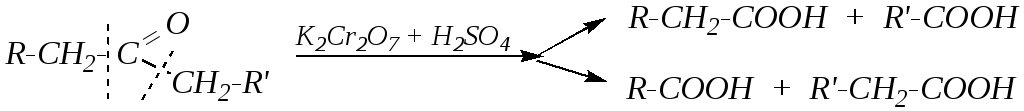
При этом из каждого осколка образуется молекула карбоновой кислоты, и в общем случае получается смесь четырёх кислот.
Карбонильные соединения могут быть восстановлены до соответствующих спиртов или углеводородов. Восстановление гидридами металлов было рассмотрено выше.
В реакциях каталитического гидрирования альдегиды и кетоны превращаются в спирты:
![]()
Катализаторами обычно являются никель, палладий, платина или комплексные соединения переходных металлов. Восстановление протекает в более жёстких условиях, чем гидрирование двойной связи в алкенах.
При восстановлении металлическим натрием образуются двухатомные спирты (гликоли) с удвоенным количеством атомов углерода. Механизм реакции радикальный:
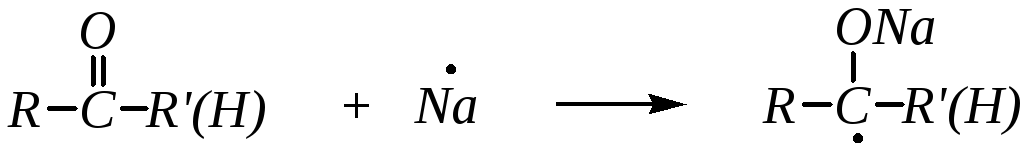
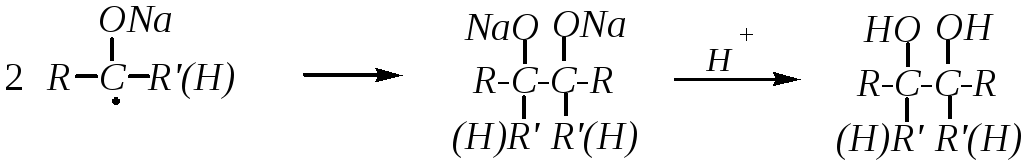
Восстановление альдегидов и кетонов амальгамой цинка в соляной кислоте (реакция Клемменсена*) приводит к получению алканов:
R–CO–R
![]() R–CH2–R
R–CH2–R
Альдегиды вступают в реакцию диспропорционирования (реакция Канниццаро*), в которой одна молекула окисляется до кислоты, а другая восстанавливается до спирта:
2
R–CHO
![]() R–COONa + R–CH2OH
R–COONa + R–CH2OH
Реакция протекает при участии концентрированного (40—50%-го) водного раствора щёлочи и характерна для альдегидов, не содержащих -водородных атомов. В противном случае в этих условиях будет проходить альдольная (и кротоновая) конденсация с участием-водородных атомов (см. выше).
Реакция начинается с обратимого присоединения гидроксид-иона по двойной связи C=О, а затем от образовавшегося аниона происходит отрыв гидрид-иона и его взаимодействие с карбонильным атомом углерода другой молекулы альдегида:
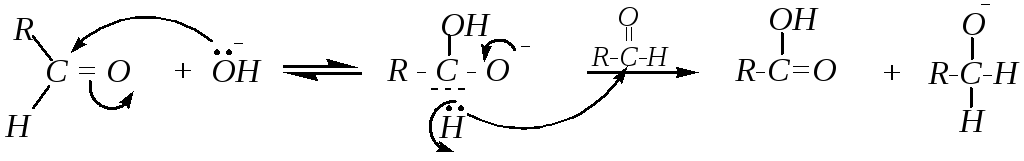
По завершении нуклеофильной атаки происходит быстрая миграция протона от молекулы карбоновой кислоты к алкоголят-иону:
R–COOH + R–CH2O¯ R–COO¯ + R–CH2OH
Реакция Канниццаро может протекать и в смеси двух альдегидов. При этом окисляться будет альдегид, более реакционноспособный по отношению к нуклеофилам, так как именно этот альдегид подвергается атаке гидроксид-анионом на первой стадии (см. механизм). Так, если одним из альдегидов является метаналь H2С=O, то гидроксид-ион взаимодействует с карбонильным атомом углерода именно этого альдегида за счёт значительного положительного заряда на этом атоме:
H2С=О + ¯OH НО-Н2C–O¯
Гидрид-ион отщепляется именно от этого аниона, и в итоге формальдегид превращается в анион муравьиной кислоты, а молекула второго альдегида присоединяет гидрид-ион к карбонильному атому углерода:
НО-Н2C–O¯ + R3C–CH=O HO-HС=О + R3C–CH2–O¯
HCOO¯ + R3C–CH2-OH
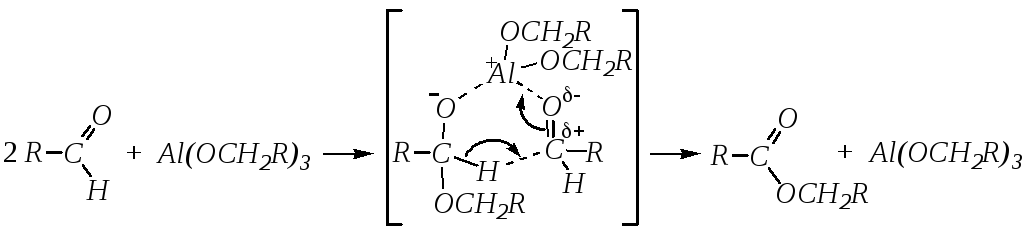
Реакция Тищенко*, или сложноэфирная конденсация Тищенко, — это тоже окислительно-восстановительная реакция, в которой одна молекула альдегида окисляется до кислоты, а другая восстанавливается до спирта, но в результате образуется молекула сложного эфира. Реакция протекает при участии алкоголята алюминия в безводной среде. В роли катализатора используется алюминиевое производное того же спирта, который должен получиться в результате восстановления исходного альдегида. В отличие от реакции Канниццаро в эту реакцию вступают в том числе и альдегиды, имеющие -водородные атомы. Механизм включает гидридный перенос в шестичленном циклическом комплексе: