

3 курс / Патологическая физиология / Баранов_А_А_,_Намазова_Баранова_Л_С_ред_Атлас_редких_болезней_2016
.pdfАтлас редких болезней
•ранняя младенческая (от 3 месяцев до 2 лет): гепатоспленомегалия, мышечная гипотония, задержка психомоторного развития, мозжечковые расстройства;
•поздняя младенческая (2–6 лет): вертикальный надъядерный паралич взора, мышечная гипотония, утрата ранее приобретенных психомоторных навыков, мозжечковые расстройства (атаксия, дизартрия, дисфагия), дистония, судороги, снижение когнитивных функций, гепатоспленомегалия;
•юношеская (6–15 лет): вертикальный надъядерный паралич взора, катаплексия, утрата ранее приобретенных психомоторных навыков, мозжечковые расстройства (атаксия, дизартрия, дисфагия), дистония, судороги, гепатоспленомегалия, снижение когнитивных функций;
•подростковая/взрослая (старше 15 лет): психические расстройства (психоз, депрессия, нарушения поведения, снижение когнитивных функций), вертикальный надъядерный паралич взора, катаплексия, утрата ранее приобретенных навыков, мозжечковые расстройства (атаксия, дизартрия), дистония, судороги, гепатоспленомегалия.
Диагностика
•Характерные висцеральные проявления (гепатоспленомегалия);
•неврологические симптомы (вертикальный надъядерный паралич взора, катаплексия, мозжечковые нарушения);
Неонатальный |
Системные симптомы |
|
холестаз |
|
(Гепато) спленомегалия |
Многоводие, |
|
|
|
Может отсутствовать в ~ 15% случаев |
|
желтуха |
|
|
|
Возраст появления варьирует |
|
новорожденных |
|
|
|
Часто предшествует неврологическим симптомам |
|
|
|
|
|
|
Возможна регрессия с возрастом |
|
(Гепато) |
|
Неонатальная |
спленомегалия |
|
|
|
|
0 |
1 |
2 |
3 |
|
6 |
10 |
20 |
|
|
30 |
40 |
50 |
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
Ранняя младенческая |
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
Поздняя младенческая |
|
|
|
|
|
|
||
Моторные |
|
|
|
|
|
|
|
||||||
|
|
|
|
|
Юношеская |
|
|
|
|||||
нарушения |
|
|
|
|
|
|
|
|
|||||
|
|
|
Нарушение |
|
|
|
|
|
|
Взрослая |
|
||
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
походки, |
|
Проблемы с обучением, |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|||||
|
|
|
неуклюжесть, |
атаксия, эпилептические |
|
|
|
|
|||||
|
|
|
Психиатрическая |
|
|||||||||
|
|
|
нарушение речи |
приступы, катаплексия |
|
||||||||
|
|
|
|
|
|
|
|||||||
симптоматика, деменция, атаксия, мышечная дистония
Вертикальный надъядерный офтальмопарез
Неврологические симптомы
Клинические проявления болезни НП-С. (По: Vanier. Orphanet Journal of Rare Diseases. 2010, 5: 16)
60

Болезнь Ниманна–Пика
Алгоритм диагностики болезни НП-С
61

Атлас редких болезней
•положительный филиппиновый тест (накопление неэтерифицированного холестерина в фибробластах кожи);
•выявление мутаций в одном из двух генов — NPC1 или NPC2.
Для пренатальной диагностики проводят филиппиновый тест в культуре
амниотических фибробластов или генетический анализ (в зависимости от вида диагностики у сибсов).
В настоящее время разработан алгоритм клинической диагностики болезни Ниманна–Пика тип С «Индекс вероятности болезни НП-С» (см. стр. 43).
Дифференциальный диагноз:
•GM2-ганглиозидоз, тип I;
•GM1-ганглиозидоз, тип I;
•болезнь Волмана;
•болезнь Гоше;
•болезнь Ниманна–Пика типы А и В;
•другие лизосомные болезни накопления.
Лечение. Разнообразное симптоматическое лечение может частично облегчать неврологические проявления и улучшить качество жизни пациентов. Расширяется клинический опыт применения миглустата (Завеска, Актелион Фармасьютикалз Лтд, Швейцария), который в настоящее время является единственным зарегистрированным специфическим препаратом для патогенетической терапии болезни НП-С у детей и взрослых. Миглустат был зарегистрирован для лечения прогрессирующих неврологических проявлений у детей и взрослых с болезнью НП-С в Европе и России в 2009 году.
Прогноз. Без специфической терапии прогноз неблагоприятный. Все пациенты с болезнью НП-С умирают преждевременно, хотя степень прогрессирования заболевания и средняя продолжительность жизни значительно различаются. Большинство пациентов умирает в возрасте между 10 и 25 годами. Как правило, у пациентов с ранним началом неврологических проявлений заболевание прогрессирует быстрее, и смерть наступает раньше.
Список рекомендованной литературы
1.Patterson M.C. et al. Recommendations for the diagnosis and management of Niemann–Pick disease type C: an update. Mol. Genet. Metab. 2012. Doi10.1016/j. ymgme.2012.03.012.
2.URL: http:// www.npc-info.ru
3.Wraith J.E., Baumgartner M.R., Bembi B., Covanis A., Levade T., Mengel E., Pineda M., Sedel F., Topcu M., Vanier M., Widner H., Wijburg F., Pattersonm M. Рекомендации по диагностике и лечению болезни Ниманна-Пика тип С. Педиатрическая фармакология. 2010; 7 (1): 16–24.
62

БОЛЕЗНЬ ОТТО–ХРОБАКА
(OTTO-CHROBAK DISEASE)
МКБ-10: M24.7; ОМIМ 177050
Определение. Болезнь Отто–Хробака — протрузионная деформация таза, при которой происходит сужение тазового кольца; всегда двустороннее поражение. Встречается у лиц женского пола. Болезнь Отто–Хробака описана немецким анатомом и патологом A.W. Otto (1786–1845) и австрийским гинекологом R. Chrobak (1843–1910).
Синонимы: таз Отто-Хробака, артрокатадиз тазобедренного сустава, дисплазия вертлужной впадины.
Эпидемиология. Не известна.
Тип наследования: аутосомно-доминантный. Описаны семейные формы.
Этиология, патогенез. В основе заболевания лежит мутация гена CBFA1/RUNX2, в результате чего развивается нарушение хондрогенеза Y-образ- ного хряща и, как следствие, деформация костей таза. Под влиянием нагрузок неполноценно сформированный хрящ обладает низкими прочностными качествами и не в состоянии удерживать в одном узле три тазовые кости. Кости таза начинают постепенно деформироваться в полость малого таза, головка бедренной кости погружается глубоко в вертлужную впадину, формируя так называемый таз Отто–Хробака.
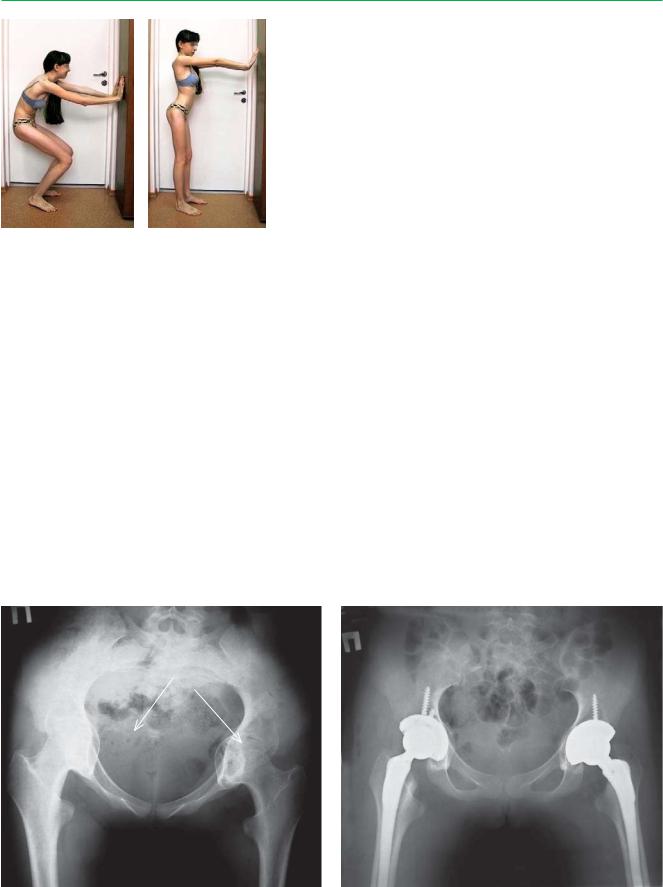
А |
Б |
В |
А–В. Варианты деформации при болезни Отто–Хробака.
А — нормальный тазобедренный сустав: впадины достаточно покрывают головку бедренной кости. Б — таз profunda, головка бедра расположена более медиально по отношению к малому тазу.
В — при протрузии головка бедренной кости близко и медиальнее расположена к полости малого таза
63
Атлас редких болезней
|
|
Клинические проявления |
|
|
|
Болезнь Отто–Хробака протекает с выра- |
|
|
|
женной клинической картиной, ведущи- |
|
|
|
ми симптомами которой являются болевой |
|
|
|
синдром, нередко выраженный даже в покое; |
|
|
|
ограничение амплитуды движений в тазобе- |
|
|
|
дренных суставах; сгибательно-приводящие |
|
|
|
контрактуры; костный или костно-фиброз- |
|
|
|
ный анкилоз тазобедренных суставов; выра- |
|
А |
Б |
женный гиперлордоз поясничного отдела |
|
А, Б. Характерный внешний вид больной. |
позвоночника; нарушение походки. Как след- |
||
ствие, отмечается снижение качества жизни. |
|||
Отмечается гиперлордоз поясничного |
|||
Рентгенологическое исследование и дан- |
|||
отдела позвоночника, ограничение |
|||
ные компьютерной томографии: дляболезни |
|||
амплитуды движений в тазобедренных |
|||
суставах |
|
Отто–Хробака характерно углубление дна |
|
|
|
вертлужной впадины в полость малого таза, |
|
|
|
которое значительно сужает его объем |
|
и определяется над пограничной линией таза (linea terminals pelvis), вследствие чего контуры «слезы» (kohler) деформируются или полностью исчезают.
Диагностика
При тщательном обследовании пациента постановка диагноза обычно не вызывает затруднений.
Дифференциальный диагноз:
•болезнь Пертеса;
•повреждения вертлужной впадины при ювенильном идиопатическом артрите;
•посттравматический центральный вывих.
Рентгенограмма тазобедренных суставов, |
Рентгенограмма тазобедренных суставов, |
протрузия головок бедренных костей в полость |
прямая проекция (после оперативного лечения) |
малого таза |
|
64

Болезнь Отто–Хробака
Трудности возникают на ранних стадиях патологического состояния, когда симптомы могут быть нечеткими. В большинстве таких случаев диагноз может быть поставлен только после комплексного обследования. При поражении вертлужной впадины, развившейся на фоне ювенильного артрита, отмечаются эрозивные, иногда кистозные изменения сустава с неровными контурами. На последствия посттравматического центрального вывиха в анамнезе указывает факт травмы нижних конечностей. Характерным признаком болезни Пертеса является деструкция и субхондральные переломы головки бедренной кости, что определяется по данным лучевых методов исследования.
Лечение. На сегодняшний день консервативного лечения Болезни Отто– Хробака не существует.
Основная рекомендация пациентам — снижение физической нагрузки. Но, несмотря на щадящий режим, развивается и прогрессирует прободение дна вертлужной впадины вместе с головкой бедренной кости в полость малого таза.
Хирургическое лечение. Основным принципом оперативного вмешательства является предотвращение дальнейшей деформации вертлужных впадин путем изменения их нагрузки по отношению к костям малого таза. При незначительной степени деформации выполняется корригирующая остеотомия бедренной кости, которая позволяет устранить деформацию проксимального отдела бедра и временно снизить нагрузку на дно вертлужной впадины.
При прогрессировании болезни с целью устранения выраженных и тяжелых деформаций, болевого синдрома единственным хирургическим методом на сегодняшний день является тотальное эндопротезирование тазобедренного сустава.
Прогноз. Тотальное эндопротезирование тазобедренного сустава позволяет предотвратить развитие дальнейшей деформации тазового кольца и повысить качество жизни.
Список рекомендованной литературы
1.Шапошников Ю. Г. Руководство по ортопедии. Том. 3: Медицина. Москва. 1997. С. 71.
2.Dongying X., Shi L., Xiong Z., Fei H., Lixin L., Chunxian W., Jincai Z. Mutations in the RUNX2 Gene. Annals of Clinical & Laboratory Science. 2008; 38 (1): 15–24.
3.Cary N.A., Barnard L. Arthrokatadysis of the hip joint. Clin Orthop Relat Res. 2007; 465: 4–5.
65

БОЛЕЗНЬ ПЕЛИЦЕУСА–МЕРЦБАХЕРА
(PELIZAEUS–MERZBACHER DISEASE)
МКБ-10: G37.0; ОМIМ 312080
Определение. Болезнь Пелицеуса–Мерцбахера (БПМ) — это Х-сцепленное, прогрессирующее, дегенеративное заболевание. Характеризуется замедлением и затем полной преждевременной остановкой миелинизации.
Синонимы: лейкодистрофия суданофильная.
Эпидемиология. Частота встречаемости 1:500000 новорожденных.
Тип наследования: X-сцепленный рецессивный.
Этиология, патогенез. Заболевание обусловлено мутациями гена PLP, кодирующего синтез протеолипида протеина 1 (PLP1), участвующего в синтезе миелина и дифференцировке олигодендроцитов. Ген картирован на длинном плече хромосомы Хq21.33–22. Тяжесть и начало появления клинических симптомов зависит от типа мутации. У 30% больных с врожденной формой БПМ имеется точковая мутация (point mutation) в экзоне гена PLP, которая вызывает апоптоз олигодендроцитов. У 60–70% пациентов с классической формой БПМ обнаруживается дупликация PLP гена, что приводит к нарушению структуры миелина, но не вызывает смерть олигодендроцитов.
Клинические проявления
Чаще всего первые признаки заболевания появляются в младенчестве в виде нистагма с ротаторным компонентом, блуждающего взгляда, тремора головы по типу spasmus nutans (судорога кивательная), стридора. Младенцы обычно гипотоничны, отмечается выраженная задержка психомоторного развития. С возрастом присоединяется пирамидная симптоматика: атетоз, хореоформные движения, атаксия, интенционный тремор, частые рвоты и трудности с кормлением. Характерными признаками БПМ также являются атрофия глазного нерва (развивается к 6–7 годам) и эпилепсия.
Внешний вид больного с болезнью Пелицеуса– Мерцбахера
Диагностика
Клиническая симптоматика:
характерная клиническая картина, отсутствие патологии периферических нервов.
Молекулярно-генетический анализ: мутации в гене PLP.
Магнитно-резонансная томография: диффузная лейкоэнцефалодистрофия на МРТ, гипогенезия мозолистого тела.
66

Возможна перинатальная и предимплантационная диагностика заболевания.
Дифференциальный диагноз:
•адренолейкодистрофия;
•болезнь Александра;
•болезнь Канаван;
•болезнь Кокейна;
•врожденный нистагм;
•метахромная лейкодистрофия;
•рассеянный склероз.
Лечение. Специфического лечения болезни Пелицеуса–Мерцбахера в настоящее время нет. В 2009 году началась первая стадия клинических исследований по трансплантации невраль-
ных стволовых клеток детям, страдающим болезнью Пелицеуса–Мерцбахера. Паллиативная терапия включает в себя реабилитационную и ортопедическую помощь, антиспастические препараты (интратекальные инфузии баклофена, инъекции ботулотоксина).
Прогноз. При врожденной форме смерть наступает в первой декаде жизни от респираторных осложнений.
При классической форме заболевания средняя продолжительность жизни составляет 20–30 лет.
Список рекомендованной литературы
1.Perlman S.J., Mar S. Leukodystrophies. Adv Exp Med Biol. 2012; 724: 154–71. Review.
2.David R. Clinical pediatric neurology. Demos medical. 2007.
3.Hobson G.M., Garbern J.Y. Pelizaeus-Merzbacher disease, Pelizaeus-Merzbacher-like disease 1, and related hypomyelinating disorders. Semin Neurol. 2012; 32 (1): 62–67. Epub 2012 Mar 15. Review.
4.Van der Knaap M.S., Smit L.M., Barth P.G., Catsman-Berrevoets C.E., Brouwer O.F., Begeer J.H., de Coo I.F., Valk J. Magnetic resonance imaging in classification of congenital muscular dystrophies with brain abnormalities. Ann Neurol. 1997; 42 (1): 50–59.
67
БОЛЕЗНЬ ФАБРИ
(FABRY DISEASE)
МКБ-10: Е75.2; ОМIМ 301500
Определение. Болезнь Фабри (Fabry’s disease или Fabry disease) — тяжелое прогрессирующее наследственное заболевание, относящееся к лизосомным болезням накопления. Она сопряжена с нарушениями метаболизма сфинголипидов и является одной из форм сфинголипидозов.
Синонимы: другой, менее распространенный вариант названия — болезнь Андерсона–Фабри. Причиной двух названий является тот факт, что данное заболевание было впервые описано в 1898 г. независимо друг от друга двумя дерматологами — Джоном Фабри в Германии и Вильямом Андерсоном в Англии.
Эпидемиология. Частота встречаемости, по разным источникам, составляет от 1:40000 до 1:117000 живых новорожденных. Однако, данные массового скрининга новорожденных, проведенного в Италии, показали, что частота этого заболевания намного выше и составляет 1:3100 детей.
Тип наследования: Х-сцепленный, в связи с чем клинические проявления чаще встречаются у лиц мужского пола. В отличие от других заболеваний с подобным типом наследования женщины-носительницы мутантного гена болезни Фабри могут иметь клиническую картину заболевания, в некоторых случаях не менее тяжелую, чем у гемизиготных мужчин. Эта особенность болезни Фабри, возможно, связана с феноменом инактивации «здоровой» Х-хромосомы.
Этиология, патогенез. Причиной заболевания являются разнообразные мутации в гене GLA. Недостаточность лизосомального фермента -галактозидазы А, кодируемого GLA, вызывает аккумуляцию нейтрального сфинголипида глоботриаозилцерамида (Gb3) в лизосомах эндотелиальных, периваскулярных, гладкомышечных клеток кровеносных сосудов, ганглиоцитов вегетативной нервной системы, эпителиальных клеток почечных клубочков и канальцев, кардиомиоцитов, гистиоцитарных и ретикулярных клеток соединительной ткани, а также в роговице, что приводит к нарушениям функций сердечно-сосудистой системы, почек, нервной системы, желудочно-кишечного тракта, кожи, органов зрения и слуха.
Клинические проявления
Нервная система: невропатические боли в виде хронических акропарестезий и/или «кризов Фабри», ангидроз и гипогидроз (отсутствие потоотделения), реже гипергидроз, гипертермия; у взрослых пациентов — ишемические и геморрагические инсульты, транзиторные ишемические атаки.
Почки: в дебюте — микропротеинурия и микроальбуминурия; постепенное прогрессирование поражения почек с развитием нефросклероза, хронической почечной недостаточности, во многом определяющей прогноз болезни.
68

Болезнь Фабри
Сердечно-сосудистая система: нарушения ритма сердца (суправентрикулярная тахикардия, желудочковая экстрасистолия и пр.), повышение артериального давления, проявления гипертрофической кардиомиопатии, сердечная недостаточность.
Кожа: диффузная ангиокератома (мелкие, выступающие над поверхностью кожи безболезненные элементы темно-красного цвета, не исчезающие при надавливании) с преобладанием высыпаний в области бедер, пупка, ягодиц, нижней части живота и промежности. Реже встречаются лимфедема нижних конечностей, изменения плотности волос на теле.
Глаза: «мутовчатое» помутнение роговицы (так называемая вортексная кератопатия, или cornea verticillata) — изменение в роговице, напоминающее пучок листьев или лепестков на конце стебля; помутнение хрусталика в виде радиальной задней субкапсулярной катаракты (катаракта Фабри) и двусторонней передней капсулярной и подкапсулярной катаракты; конъюнктивальные аневризмы, отек зрительной сетчатки и/или диска зрительного нерва, атрофия зрительного нерва, а также расширение ретинальных сосудов.
Слух: нейросенсорная тугоухость, шум (звон) в ушах (одноили двусторонний), головокружение.
Желудочно-кишечный тракт: схваткообразные боли в животе, вздутие живота, неустойчивый стул, тошнота, рвота снижение аппетита и снижение веса.
Первые клинические проявления болезни Фабри чаще всего возникают в подростковом возрасте, реже — на первом десятилетии жизни. Ранними симптомами являются акропарестезии, гипертермия, гипогидроз, боли в животе. Поражение сердечно-сосудистой, центральной нервной систем; почечная недостаточность, диффузная ангиокератома развиваются позже и характерны для взрослых пациентов.
Молекулярно-генетическая диагностика. Доступна ДНК-диагностика: ген GLA
картирован на длинном плече Х-хромосомы (участок Xq22). Дефекты гена GLA чрезвычайно разнообразны (описано более 585 мутаций), большинство из которых являются семейными.
Диагностика
Наличие типичной симптоматики и/или отягощенного генеалогического анамнеза.
Объективные лабораторные методы диагностики:
•энзимодиагностика (исследование активности -галактозидазы А в крови (лейкоциты), культуре кожных фибробластов или других биологических средах) и молекулярно-генетический анализ в мужской популяции;
•молекулярно-генетический анализ в женской популяции (уровень фермента-галактозидазы в крови у девочек и женщин не является информативным).
Дифференциальный диагноз:
•ревматоидный артрит, ревматизм, системная красная волчанка, болезнь Рейно;
•«боли роста»;
•GM1-ганглиозидоз, гликопротеинозы (аспартатгликозаминурия, фукозидоз,-маннозидоз, сиалидоз тип II);
69