

3 курс / Патологическая физиология / Баранов_А_А_,_Намазова_Баранова_Л_С_ред_Атлас_редких_болезней_2016
.pdfАтлас редких болезней
МУКОПОЛИСАХАРИДОЗ ТИП III
(MUCOPOLYSACCHARIDOSIS TYPE III)
Определение. Мукополисахаридоз тип III — лизосомальная болезнь накопления, характеризующаяся прогрессирующей умственной отсталостью, нарушением сна, умеренными изменения скелета.
Синонимы: синдром Санфилиппо.
Описан американским педиатром Сильвестром Санфилиппо (S.J. Sanfilippo) в 1963 г.
Эпидемиология. Частота 1 на 60000–300000 новорожденных. Является третьим по частоте среди всех известных в настоящее время мукополисахаридозов.
Тип наследования: аутосомно-рецессивный.
Этиология, патогенез. МПС тип III является генетически гетерогенным заболеванием.
Возможен дефицит разных ферментов, но во всех случаях в лизосомах накапливается один тип гликозаминогликанов — гепарансульфат.
Различают четыре типа, характеризующихся разными биохимическими дефектами:
•А (дефицит фермента N-сульфоглюкозамин-сульфогидролазы);
•В (дефицит фермента N-ацетил-D-глюкозаминидазы);
•С (дефицит альфа глюкозаминид-N-ацетилтрансферазы);
•D (дефицит N-ацетилглюкозамин-6-сульфатазы).
МПС III А (OMIM 252900) — наиболее распространенный. Течение заболева-
ния при этой форме наиболее тяжелое, с ранним началом, наиболее быстрым прогрессированием симптомов и короткой продолжительностью жизни.
Болезнь обусловлена недостаточностью гeпapaн-N-сульфатазы, за синтез которого ответственен ген SGSH, он был идентифицирован в 1995 г. Ген кодирован на 17 q25.3. В литературе описано около 70 различных мутаций гена SGSH.
Chr 17 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
p13.3 |
p13.2 |
p13.1 |
p12 |
p11.2 |
p11.1 |
q11.1 |
q11.2 |
q12 |
q21.2 |
q21.31 |
q21.32 |
q21.33 |
q22 |
q23.2 |
q23.3 |
q24.1 |
q24.2 |
q24.3 |
q25.1 |
q25.3 |
Ген SGSH
МПС III B (OMIM 252920). Ген NAGLU, кодирующий альфа-N- ацетилглюкозаминидазу, был идентифицирован в 1996 г. Он локализуется в сегменте q21.1 хромосомы 17. В литературе описано около 100 различных мутаций гена NAGLU.
180

Мукополисахаридозы
Chr 17 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
p13.3 |
p13.2 |
p13.1 |
p12 |
p11.2 |
p11.1 |
q11.1 |
q11.2 |
q12 |
q21.2 |
q21.31 |
q21.32 |
q21.33 |
q22 |
q23.2 |
q23.3 |
q24.1 |
q24.2 |
q24.3 |
q25.1 |
q25.3 |
Ген NAGLU
МПС III C (OMIM 252930). Ген HGSNAT, кодирующий ацетил-СоА: альфа- глюкозаминид-N-ацетилтрансферазу, был идентифицирован в 2006 г. Он локализуется в сегменте р11.1 хромосомы 8.
Chr 8 |
|
|
|
|
|
|
|
|
|
|
|
|
|
p23.3 p23.2 |
p23.1 |
p22 |
p21.3 p21.2 |
p12 |
p11.21 p11.1 q11.1 q11.21 q11.23 q12.1 |
q12.3 |
q13.2 q13.3 q21.11 |
q21.13 q21.2 q21.3 |
q22.1 q22.2 q22.3 q23.1 |
q23.3 |
q24.12 q24.13 q24.21 |
q24.22 q24.23 |
q24.3 |
Ген HGSNAT
МПС III D (OMIM 252940). Ген GNS, кодирующий N-ацетилглюкозамин- 6-сульфатазу, был идентифицирован в 1988 г. Он локализуется в сегменте q14 хромосомы 12. С тех пор было установлено только 5 мутаций.
Chr 12 |
|
|
|
|
|
|
|
|
|
|
|
p13.33 p13.32 p13.31 p13.2 p13.1 p12.3 |
p12.1 |
p11.22 p11.21 p11.1 q11 |
q12 |
q13.11 q13.12 q13.13 |
q14.1 q14.2 q14.3 q15 q21.1 |
q21.2 |
q21.31 |
q21.32 q21.33 q22 |
q23.1 q23.2 q23.3 q24.11 q24.13 q24.21 q24.23 q24.31 |
q24.32 |
q24.33 |
Ген GNS
Отличительной чертой является менее выраженное накопление продуктов нарушенного обмена в соединительной ткани, но более значительное отложение их в тканях мозга.
Клинические проявления
Внешний вид. Умственная отсталость, умеренная тугоподвижность в суставах, легкое огрубение лица.
Заболевание манифестирует в основном на 2-м году жизни ребенка. Отмечаются отставание в росте, небольшая тугоподвижность в суставах, иногда увеличение печени и селезенки. Самым важным признаком является задержка темпов психоречевого развития. К 3-му году жизни развитие прекращается, ребенок посте-
181
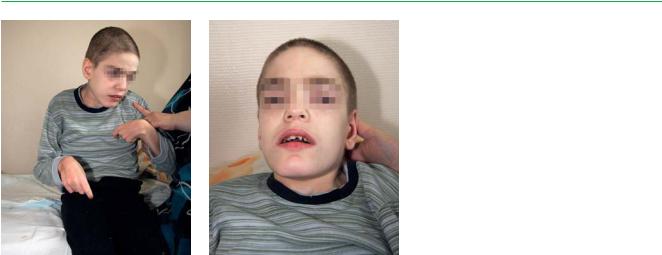

Мукополисахаридозы
Упациентов наблюдаются пищевые расстройства, которые выражаются
втяге к несъедобным веществам. Рецидивирующие пупочные и паховые грыжи. Склонность к эпизодической или хронической диарее.
Диагностика
•Характерный фенотип.
•Повышенная экскреция гепарансульфата с мочой.
•Метахромазия лейкоцитов и фибробластов.
•Пренатальный диагноз с помощью амниоцентеза.
Лечение. Трансплантация стволовых клеток (примеры пациентов в США). Трансплантация костного мозга неэффективна.
В настоящее время не существует патогенетического лечения МПС III. Пациенты получают симптоматическую терапию в соответствии с жалобами и выявленными нарушениями.
Прогноз. Больные умирают в возрасте до 30 лет от присоединившихся инфекций.
Список рекомендованной литературы
1.Коннет Л. Джонс. Наследственные синдромы по Дэвиду Смиту. М.: Практика. 2011.
2.Neufeld E.F., Muenzer J. The mucopolysaccaridoses. The Metabolic Bases of inherited Disease. New York: McGraw Hill. 2001; 3421–2352.
3.Alroy J., Haskins M., Birk D.E. Altered corneal stromal matrix organization is associated with mucopolysaccharidosis I, III and VI. Exp Eye Res. 1999; 68: 523.
4.Siciliano L., Fiumara A., Pavone L. et al. Sanfilippo syndrome type D in two adolescent sisters. J. Med.Genet. 1991; 28: 402–405.
183

Атлас редких болезней
МУКОПОЛИСАХАРИДОЗ ТИП IV
(MUCOPOLYSACCHARIDOSIS TYPE IV)
МКБ-10: E 76.2; OMIM 253000, 253010
Определение. Мукополисахаридоз тип IV — лизосомальная болезнь накопления, характеризуется значительной деформацией скелета, особенно грудной клетки, отставанием в росте.
Синонимы: болезнь Моркио, спондилоэпифизарная дисплазия, хондроостеодистрофия, деформирующая остеохондродистрофия, Morquio–Brailsford синдром, Morquio–Ulrich синдром, KS-мукополисахаридоз, эксцентрохондроплазия, Duggve–Melchior–Clausen синдром.
Моркио тип A впервые описал уругвайский педиатр Луис Моркио (L. Morquio) в 1929 г.
Эпидемиология. Частота — 1: 40000–100000 новорожденных.
Тип наследования: болезнь Моркио передается по аутосомно-рецессивному типу.
Chr 16 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
p13.3 |
p13.2 |
p13.13 |
p13.12 |
p13.11 |
p12.3 |
p12.2 |
p12.1 |
p11.2 |
p11.1 |
q11.1 |
q11.2 |
q12.1 |
q12.2 |
q21 |
q22.1 |
q22.2 |
q23.1 |
q23.2 |
q23.3 |
q24.1 |
q24.2 q24.3 |
Ген GALNS
Этиология, патогенез. Болезнь обусловлена дефицитом лизосомальных гидролаз: галактозамин-6-сульфат-сульфатазы (тип А) или b-галактозидазы (тип В) с отложением в соединительной ткани кератансульфата.
Известно 2 типа заболевания: подтип А — тяжелая форма (ген GALNS локализован в сегменте 16q24.3) и подтип B — легкая форма (ген в сегменте 3q21.33). Важно отметить, что мутация гена b-галактозидазы вызывает также ганглиозидоз типа I.
Клинические проявления
В отличие от других типов мукополисахаридозов IV тип характеризуется отсутствием снижения интеллекта, помутнения роговицы, гепатоспленомегалии и гротескных черт лица.
Внешний вид. Значительные деформации скелета, особенно грудной клетки. Дети рождаются без признаков болезни. Первые симптомы появляют-
ся в возрасте 1–3 лет, и к 7–8 годам клиническая картина уже полностью выражена.
184

Мукополисахаридозы
Выраженное отставание в росте. Лицо обычное, но могут наблюдаться широкий рот, короткий нос, широко поставленные зубы. Руки уродливой формы, шея короткая. Дети пониженного питания. Аномалия грудной клетки (бочкообразная, килевидная, «куриная грудь»), общая слабость мышц, Х-образная деформация ног, дисплазия бедер;
интеллект сохранен. А Б В
Костная система. |
Внешний вид пациента 8,5 лет с мукополисахаридозом тип IV |
|
Болезнь характеризует- |
||
|
||
ся карликовостью (рост |
|
взрослого больного около 80–115 см) и непропорциональным телосложением (относительно короткое туловище, микроцефалия, короткая шея). Деформация скелета, особенно грудной клетки (куриная, бочкообразная, килеобразная).
До и после успешной операции
185

Атлас редких болезней
Кифосколиоз грудного и поясничного отделов позвоночника. Тугоподвижность в суставах и вместе с тем расслабление сумочно-связочного аппарата в мелких суставах, шея укорочена, гипоплазия отростков I и II шейных позвонков. Возникают контрактуры в локтевых, плечевых, коленных суставах, отмечается вальгусная деформация нижних конечностей, плоскостопие.
Рентгенологически видны характерные изменения в позвоночнике. Во всех отделах отмечается платиспондилия (типичная для болезни Моркио деформация позвонков): уплощение и расширение тел позвонков, чем объясняется характерное укорочение туловища и необычно короткая шея. Может также возникать ступенеобразная деформация тела, похожая на башмачок, кифоз. Эпифизы длинных трубчатых костей недоразвиты. Окостенение метафизов также нерегулярное и позднее.
Кисти укорочены, пальцы утолщены, с конической формой эпифизов. Кости предплечья укорочены; локтевая кость не достигает лучезапястного сустава, отмечается вывих ее головки в локтевом суставе; эпифизы треугольной формы. Дистальные эпифизы костей голени скошены, стопы деформированы.
Изменяются кости таза: вертлужные впадины плоские и широкие, их крыша скошена, крылья подвздошных костей неправильной формы; контуры всех костей неровные; головки бедренных костей уплощены. Грибовидная головка бедренной кости внедряется в углубленную вертлужную впадину, шейка бедра укорачивается, поражение суставов двустороннее, в результате чего уменьшается поперечник таза. Часто наблюдаются остеоартриты, особенно в тазобедренных суставах. Уплощение эпифизов бедренной и большеберцовой кости, сглаживание межмыщелковых бугорков сопровождаются специфической деформацией коленных суставов.
Орган слуха. Нередко отмечается снижение слуха. Почти у всех больных, доживших до 20 лет, развивается глухота.
Орган зрения. Иногда отмечается помутнение роговицы.
Центральная нервная система. Интеллект не нарушен или умеренно снижен. В случае компрессии спинного мозга помимо мышечной гипотонии отмечается поражение пирамидной системы, что может привести к параплегии.
Грубые изменения в позвоночнике создают условия для компрессии каудального отдела спинного мозга, которые нередко реализуются после легкой травмы позвоночника, при этом развиваются вялые парезы ног вследствие поражения поясничного утолщения, или синдром конуса-эпиконуса.
У некоторых больных возможны перемежающаяся хромота, нарушение тазовых функций, что является следствием хронического нарушения кровообращения спинного мозга. Большинство больных страдают пояснично-крестцовым радикулитом, рецидивирующей люмбоишиалгией.
Сердечно-сосудистая система. Поражение сердца наблюдается довольно часто. В поздний срок болезни появляются более выраженные изменения со стороны сердечно-сосудистой системы. Типична недостаточность аортального, реже митрального клапанов. Кардиомегалия обычно носит вторичный характер. Иногда наблюдается поражение миокарда при отсутствии клапанной дисфункции.
Желудочно-кишечная система: отсутствует гепатоспленомегалия! Часто выявляются пупочные и паховые грыжи, расхождение прямых мышц живота.
186

Мукополисахаридозы
Диагностика
•Характерные внешние признаки.
•Повышенная экскреция кератансульфата с мочой.
Примечание: описана клинически сходная форма синдрома Моркио В без
отклонений в активности ферментов и без кератансульфатурии.
•Оценка активности ферментов в плазме или лейкоцитах крови, культуре фибробластов кожи.
•Исследование ДНК.
•Для пренатальной диагностики используют амниоциты и клетки ворсин хориона.
Дифференциальный диагноз:
•различные варианты нанизма, при которых отсутствуют специфические изменения скелета.
Лечение. Одним из перспективных считается метод генной терапии. В настоящее время проводится ферментозаместительная терапия элосульфазой альфа. В России этот препарат находится на стадии регистрации. При развитии гидроцефалии, сдавлений спинного мозга, нестабильности атланто-аксиального сочленения и туннельных невропатий показано хирургическое вмешательство. Очень важно симптоматическое лечение.
Прогноз. Летальный исход наступает до 20 лет вследствие сердечно-легочной недостаточности, развивающейся на фоне интеркуррентных заболеваний. Возможна внезапная смерть в результате смещения атланто-окципитального сочленения и повреждения ствола мозга.
Список рекомендованной литературы
1.Коннет Л. Джонс. Наследственные синдромы по Дэвиду Смиту. М.: Практика. 2011.
2.Couprie J., Denis P., Guffon N. et al. Ocular manifestations in patients affected by Morquio syndrome (MPS IV). J Fr Ophtalmol. 2010; 33: 617–22.
3.Bargal B., Avidan N., Olender T. et al. Mucopolysaccharidosis type IV: novel MCOLN1 mutations in Jewish and nonJewish patients and the frequency of one disease in the Ashkenasi Jewish population. Hum. Mut. 2001; 17: 397–402.
4.Beck M., Glossl J., Grubisic A., Spranger J. Heterogeneity of Morquio disease. Clin. Genet. 1986; 29: 325–331.
5.Hechit J.T., Scott C.I., Smith T.K., Williams J.C. Mild manifestations of the Morquio syndrome. Am. J. Med. Genet. 1984; 18: 369–371.
187

Атлас редких болезней
МУКОПОЛИСАХАРИДОЗ ТИП VI
(MUCOPOLYSACCHARIDOSIS TYPE VI)
МКБ-10: E 76.2; OMIM 253200
Определение. Мукополисахаридоз тип VI — наследственная лизосомальная болезнь накопления, которая характеризуется отставанием в росте, огрубением черт лица, снижением слуха, тугоподвижностью в суставах, гепатоспленомегалией, постепенным развитием сердечно-сосудистой и дыхательной недостаточности.
Синонимы: синдром Марото–Лами.
Впервые описан в 1960 г. французскими врачами Марото (Р. Maroteaux) и Лами (М.Е.J. Lamy).
Эпидемиология. МПС VI типа встречается с популяционной частотой 1:300000.
Тип наследования: аутосомно-рецессивный.
Этиология, патогенез. В основе лежит недостаточность фермента N-ацетилгалактозамин-4-cульфатазы (арилсульфатазы В), что приводит к нарушению ступенчатой деградации гликозаминогликана дерматансульфата. ГАГ накапливается внутри лизосомы и обусловливает клиническую картину тяжелого хронического прогрессирующего заболевания. Эта недостаточность обнаруживается во всех тканях, в том числе в культуре фибробластов. Ген картирован на 5q13-q14.
Chr 5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
p15.33 |
p15.31 |
p15.2 |
p15.1 |
p14.3 |
p14.1 |
p13.3 |
p13.2 |
p13.1 |
p12 |
q11.2 |
q12.1 |
q12.3 |
q13.2 |
q13.3 |
q14.1 |
q14.3 |
q15 |
q21.1 |
q21.3 |
q23.1 |
q23.2 |
q23.3 |
q31.1 |
q31.2 |
q31.3 |
q32 |
q33.1 q33.2 |
q33.3 |
q34 |
q35.1 |
q35.2 |
q35.3 |
Ген, кодирующий фермент, локализуется в сегменте 5q13-q14
Клинические проявления
Внешний вид. Грубые черты лица при нормальном интеллекте, тугоподвижность в суставах, помутнение роговицы.
Выделяют 3 формы заболевания: при тяжелой форме клинические симптомы появляются с 1–3 лет, при среднетяжелой — с 6 лет, при легкой — после 20 лет. Отставание в росте, диспропорциональное телосложение: карликовость с укорочением туловища.
Изменение формы лица: большой нос с запавшей переносицей, пухлые губы, маленькие зубы с широкими зубными промежутками, позднее прорезывание зубов, макроглоссия. Возможна глухота, пупочная и паховая грыжи, уплотнение и утолщение кожи, грубые волосы, умеренный гирсутизм.
188

Мукополисахаридозы
А |
Б |
В |
А–В. Внешний вид пациентки 17 лет с мукополисахаридозом тип VI
А |
Б |
А–Б. Контрактуры кистей рук у пациентки 17 лет с мукополисахаридозом тип VI
На поздних стадиях развивается глухота, слепота, деменция.
Костная система. Задержка роста: при быстром прогрессировании заболевания рост достигает 90–100 см. Максимальный рост 150 см. Множественные дизостозы. Умеренная тугоподвижность практически во всех суставах, сгибательные контрактуры пальцев и клешневидная деформация кисти. Особенно выражены изменения в тазобедренных суставах, что может привести к прогрессирующей инвалидизации (дисплазия головки бедренной кости, деформация эпифизов бедренных костей). Килевидная грудная клетка с широкими ребрами, дефект развития тел позвонков с передним переломом. Х-образное искривление ног. При рентгенографии могут наблюдаться точечные пястные кости.
Органы дыхания. Частые респираторные заболевания: риниты, отиты. В связи с гипертрофией миндалин и аденоидов, увеличением языка, утолщением надгортанника и голосовых связок могут развиться дыхательные нарушения разной степени тяжести. Особенности строения грудной клетки (жесткая грудная клетка в сочетании с кифосколиозом и усиленным поясничным лордозом) способствуют развитию рестриктивных заболеваний легких. Наблюдается обструктивное апноэ во время сна.
При проведении полисомнографии выявляются:
•синдром обструктивного апноэ сна тяжелой степени. Индекс апноэ–гипопноэ 24/час (норма < 1/час);
•структура сна нарушена;
•патологическая двигательная активность отсутствует;
•на ЭЭГ признаки дисфункции корково-подкорковых взаймодействий, раздражения срединных структур.
189