

3 курс / Патологическая физиология / Баранов_А_А_,_Намазова_Баранова_Л_С_ред_Атлас_редких_болезней_2016
.pdfАтлас редких болезней
•Биохимический анализ крови: снижение уровня холестерина, железа; повышение уровня ферритина.
•Пунктат костного мозга: инфильтрация клетками Гоше.
•Ультразвуковое исследование органов брюшной полости: гепатоспленомегалия, диффузная мелкоочаговая неоднородность, гиперэхогенность паренхимы печени и селезенки, очаги ишемии и фиброза как с повышенной, так и с пониженной интенсивностью сигнала, портальная гипертензия.
•Рентгенограммы костей: истончение надкостницы, деформация дистальных метафизов бедра в колбы Эрленмейера, переломы.
•Магнитно-резонансная томография костей скелета: инфильтрация костного мозга, остеонекроз.
•Денситометрия: системная остеопения, остеопороз.
•Электроэнцефалограмма (ЭЭГ): у больных с нейронопатическими типами БГ изменения неспецифичны и чаще проявляются дезорганизованным паттерном ЭЭГ сна, дисфункцией корково-подкорковых взаимодействий, дисфункцией и раздражением срединных и подкорковых структур, формированием вспышек полиморфной эпилептиформной активности и пароксизмами острых полифазных потенциалов.
Дифференциальный диагноз
БГ тип I:
•разнообразные экзогенные и наследственные болезни, сопровождающиеся висцеромегалией, острыми болями в костях, кровоточивостью (вирусный гепатит, остеомиелит, костный туберкулез, гемофилии, сфинголипидозы).
Мальчик, 3 года, БГ тип III |
Девочка, 7 лет, БГ тип III |
50

Болезнь Гоше
БГ типы II и III:
•всеинфантильныеформысфинголипидозовсгепатоспленомегалией(болезнь Нимана–Пика типы А, С);
•GM1-ганглиозидоз;
•галактосиалидоз;
•болезнь Вольмана;
•болезнь Фарбера (атипичные формы);
•врожденная окуломоторная апраксия.
Лечение
Единственный эффективный метод лечения БГ у детей — патогенетическая ферментная заместительная терапия (ФЗТ) рекомбинантной глюкоцереброзидазой. В РФ зарегистрировано 2 лекарственных препарата рекомбинантной глюкоцереброзидазы, которые показаны для применения в детском возрасте:
А |
Б |
А |
В |
Г |
Б |
А–Г. Пациент А., 11 лет, БГ тип I до начала ферментозаместительной |
А, Б. Пациентка Ш., 8 лет, |
|
терапии (А, Б) и на фоне лечения (В, Г) |
|
БГ тип I, до начала ферменто- |
|
|
заместительной терапии (А) |
|
|
и через год от начала лечения (Б) |
51

Атлас редких болезней
•имиглюцераза (все возрастные группы);
•велаглюцераза альфа (пациенты старше четырех лет).
Основные цели лечения пациентов с болезнью Гоше включают:
•устранение болевого синдрома, нормализацию самочувствия больных;
•регресс или уменьшение выраженности цитопенического синдрома;
•сокращение размеров селезенки и печени;
•предупреждение необратимого поражения костно-суставной системы и жизненно важных внутренних органов (печень, легкие, почки).
ФЗТ купирует основные клинические симптомы болезни, улучшает качество жизни больных и не оказывает выраженных побочных эффектов.
БГ тип II. Методы эффективной терапии для БГ тип II не разработаны.
Прогноз. Прогноз при БГ типы I и III зависит от выраженности клинических проявлений. Назначение патогенетической терапии на ранних стадиях заболевания определяет благоприятный прогноз и улучшает качество жизни детей с БГ, предотвращая их инвалидизацию. При БГ тип II прогноз крайне неблагоприятный (летальный исход на 1–2-м году жизни).
Клинический случай
Больная Сабина Д., 1993 года рождения, впервые поступила в гематологическое отделение Научного центра здоровья детей в феврале 2001 г.
При поступлении: печень до +6 см из-под края правой реберной дуги, селезенка — нижний полюс у входа в малый таз. В клиническом анализе крови: гемоглобин (Hb) 103 г/л, тромбоциты 74 109/л.
Проведена стернальная пункция. В миелограмме выявлены клетки Гоше.
ВМедико-генетическом научном центре проведено исследование активности лизосомных ферментов: снижение -D-глюкозидазы до 1,6 нМ/мг в час (норма 4,7–19); резкое повышение активности хитотриозидазы до 14575 нМ/мг в час (норма 4,5–198), что свидетельствовало о наличии у ребенка болезни Гоше.
В2001 г. девочка выехала на постоянное место жительства в Азербайджан, где в апреле 2002 г. в отделении гематологии РДКБ Азербайджана была проведена спленэктомия. ФЗТ не получала.
Впервые госпитализирована в отделение восстановительного лечения детей с болезнями органов пищеварительной системы в сентябре 2008 г. При поступлении: состояние ребенка средней тяжести по основному заболеванию. Жалоб активно не предъявляла. Вес 40 кг, рост 160 см. При осмотре: печень +5,0 +6,0 см из-под края правой реберной дуги, плотной консистенции. По данным лабо-
раторных исследований: в клиническом анализе крови Hb 124 г/л, лейкоциты 12,9 109/л, тромбоциты 446 109/л; в биохимическом анализе крови — без патологии. По результатам денситометрии выявлены снижение минеральной плотности костной ткани, остеопороз (Z-score в L2–L4 — 2,9, в области L2 — 4,0). Было рекомендовано проведение ФЗТ, однако по месту жительства указанная терапия не проводилась.
52
Болезнь Гоше
В течение 6 мес после выпи- |
|
|
ски появились жалобы на боли |
|
|
в бедренных костях, с апре- |
|
|
ля 2009 г. — повторные кост- |
|
|
ные кризы, боли и скованность |
|
|
в левом тазобедренном суставе, |
|
|
нарушение походки. ФЗТ (ими- |
Компьютерная томограмма тазобедренных суставов |
|
глюцеразу) девочка начала полу- |
||
с 3D-реконструкцией (август, 2010) |
||
чать только с июля 2009 г. При |
||
|
||
контрольной госпитализации в |
|
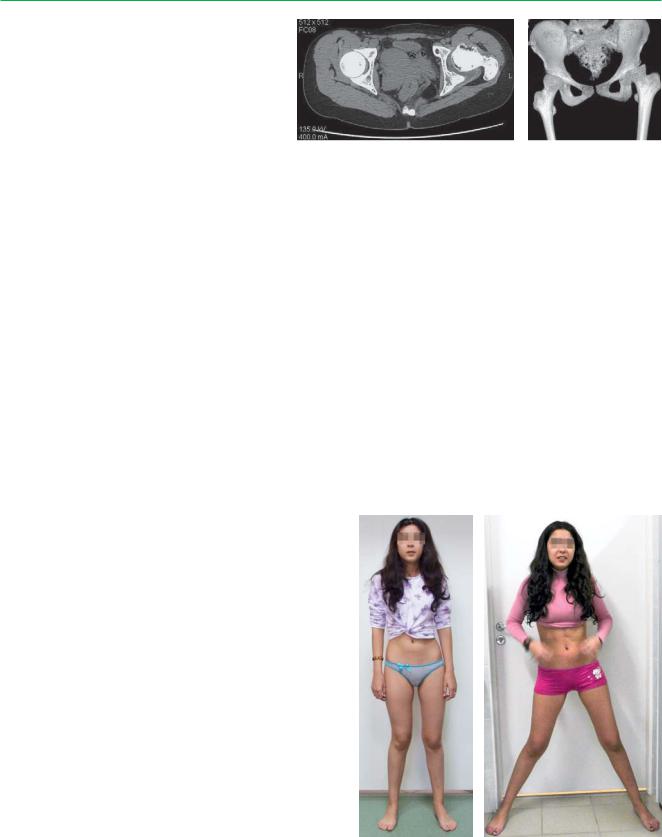
клинику в сентябре 2009 г. проведена компьютерная томография (КТ) левого тазобедренного сустава. Заключение: головка левой бедренной кости уплощена, визуализируется линия импрессионного перелома с деформацией контура по передним отделам. Заключение ортопеда: асептический некроз головки бедренной кости слева. Рекомендовано: динамическое наблюдение, контроль КТ тазобедренных суставов через 6 мес. По месту жительства продолжена ФЗТ имиглюцеразой в дозе 60 ЕД/кг.
При обследовании в клинике в августе 2010 г. отмечено, что на фоне проводимой ФЗТ девочка выросла на 5 см, прибавила в весе 6 кг, купировались костные кризы, улучшилась УЗ-картина (нормализовались размеры печени), увеличилась минеральная плотность костей в поясничном отделе позвоночника (Z-score в L2–L4 — 1,9), однако, сохранялись стойкое ограничение подвижности в левом тазобедренном суставе и нарушение походки. На серии компьютерных томограмм тазобедренных суставов по сравнению с сентябрем 2009 г. отмечается умень-
шение головки левой бедренной кости |
|
|
с уплощением, контуры ее деформирова- |
|
|
ны. Увеличились локальные участки раз- |
|
|
ряжения костной ткани вдоль передних |
|
|
и наружных отделов зоны роста с пора- |
|
|
жением аналогичных участков в проек- |
|
|
ции большого вертела и шейки бедрен- |
|
|
ной кости; расширение костномозгового |
|
|
канала левой бедренной кости и умерен- |
|
|
ное количество дополнительного жид- |
|
|
костного содержимого в полости сустава |
|
|
слева. |
|
|
Девочка консультирована в травмато- |
|
|
лого-ортопедическом отделении НЦЗД. |
|
|
Ортопедический статус: ребенок пра- |
|
|
вильного телосложения. Отмечается |
|
|
асимметрия стояния лопаток, надпле- |
|
|
чий, треугольников талии. Ось позво- |
Внешний вид |
Внешний вид больной через |
ночника отклонена влево в поясничном |
больной |
2 мес после операции. |
отделе. Верхние конечности сформи- |
до операции. |
Максимальное отведение |
рованы правильно, движение в суста- |
Максимальное |
нижних конечностей |
отведение |
|
|
вах рук в полном объеме. Больная ходит |
|
|
нижних |
|
|
без дополнительной опоры. Движения |
конечностей |
|
53

Атлас редких болезней
|
в голеностопных, коленных суставах |
|
в полном объеме. Укорочение левой |
|
нижней конечности на 2 см. Движения |
|
в тазобедренных суставах: с правой |
|
стороны сгибание 110°, разгибание 180°, |
|
внутренняя ротация 40°, наружная 60°, |
|
отведение 40°, приведение 20°; с левой |
|
стороны сгибание 30°, разгибание пол- |
|
ное, внутренняя ротация 10°, наруж- |
|
ная 10°, отведение 15°, приведение 5°. |
|
Нейроциркуляторных нарушений нет. |
|
Заключение. У ребенка с БГ деформи- |
|
рующий левосторонний коксартроз |
|
3-й степени. Ограничение сгибания, раз- |
|
гибания, наружной и внутренней рота- |
Рентгенография тазобедренных суставов |
ции, связанное с болевым синдромом. |
(июль, 2011) |
На компьютерной томографии тазобе- |
|
дренных суставов — вальгусная деформа- |
|
ция шейки правого бедра, асептический |
некроз и седловидная деформация головки левого бедра. Показано тотальное эндопротезирование левого тазобедренного сустава.
Учитывая нарушения структурно-анатомических соотношений в левом тазобедренном суставе, 6 октября 2010 г. в травматолого-ортопедическом отделении НЦЗД выполнена операция по тотальному эндопротезированию левого тазобедренного сустава (протез системы Bicontact/Plasmacup). Проведенное морфологическое исследование подтвердило клинический диагноз асептического некроза головки левой бедренной кости.
Период реабилитации прошел без осложнений. Двигательная активность начиналась с костылей (в течение 1 мес), затем ходьба с тростью. Девочка выполняла все необходимые физические упражнения, направленные на укрепление мышц обеих нижних конечностей. Она быстро освоила спуск и подъем по лестнице, у нее восстановился правильный стереотип ходьбы. Проведенная операция позволила устранить боль, значительно восстановить устойчивость в тазобедренном суставе благодаря натяжению мышц, выровнять осанку путем устранения контрактуры и удлинения ноги.
В июле 2011 г. девочка повторно проконсультирована ортопедом в травмато- лого-ортопедическом отделении НЦЗД. При осмотре: объем движений в левом тазобедренном суставе полный; движения безболезненные. Пациентка активно ходит, в том числе по лестнице, не хромает. На рентгенографии левого тазобедренного сустава в прямой проекции с захватом бедренного компонента: состояние после тотального эндопротезирования левого тазобедренного сустава; отмечается хорошее позиционирование и стабилизация компонентов эндопротеза.
Таким образом, у пациентки с БГ 1-го типа проведенная операция по эндопротезированию левого тазобедренного сустава позволила устранить боль и полностью восстановить функцию оперированной нижней конечности. Операция по тотальному эндопротезированию стала звеном в осуществлении главной зада-
54

Болезнь Гоше
чи — вернуть больного в социальную и бытовую среду. Хочется также отметить, что в настоящее время это пока единственная операция в России, проведенная ребенку с подобным диагнозом.
В настоящее время в НЦЗД наблюдается 85 детей с болезнью Гоше. Всем пациентам диагноз подтвержден по данным энзимодиагностики, практически всем (95%) в НЦЗД выполнено молекулярно-генетическое исследование с выявлением мутаций, определяющих развитие болезни.
Список рекомендованной литературы
1. Koprivica V., Stone D. L., Park J. K., Callahan M., Frisch A., Cohen I. J., Tayebi N., Sidransky E. Analysis and classification of 304 mutant alleles in patients with type 1 and type 3 Gaucher disease. Am J Hum Genet. 2000; 66 (6): 1777–1786.
2.Mankin H.J., Rosenthal D., Xavier R. Current concepts review Gaucher disease: new approaches to an ancient disease. J Bone Joint Surg Am. 2001; 83 A (5): 748–762.
3.Pastores G.M., Weinreb N.J., Aerts H., Andria G., Cox T.M., Giralt M., Grabowski G.A., Mistry P. K., Tylki-Szymanska A. Therapeutic goals in the treatment of Gaucher disease. Supp Semin Hematol. 2004; 41 (4): 4–14.
4.Wenstrup R.J., Roca-Espiau M., Weinreb N.J., Bembi B. Skeletal aspects of Gaucher disease: a review. Br J Radiol. 2002; 75 (Suppl. 1): A2–А12.
5.Vellodi A., Bembi B., de Villemeur T.B. et al. Management of neuronopathic Gaucher disease: a European consensus. J Inherit Metab Dis. 2001; 24: 319–327.
6.Baldellou A., Andria G., Campbell P.E., Charrow J., Cohen I.J., Grabowski G.A. et al. Paediatric non-neuronopathic Gaucher disease: recommendations for treatment and monitoring. Eur J Pediatr. 2004; 163 (2): 67–75.
7. Bembi B., Ciana G., Mengel E., Terk M. R., Martini C., Wenstrup R. J. Bone complications in children with Gaucher disease. Br J Radiol. 2002; 75 (Suppl. 1): A37–A43.
8.Andersson H., Kaplan P., Kacena K., Yee J. Eight-year clinical outcomes of longterm enzyme replacement therapy for 884 children with Gaucher disease type 1. Pediatrics. 2008; 122: 1182–1190.
9.Neal J. Weinreb & Jack Goldblatt & Jacobo Villalobos & Joel Charrow & J. Alexander Cole & Marcelo Kerstenetzky & Stephan vom Dahl & Carla Hollak. Long-term clinical outcomes in type 1 Gaucher disease following 10 years of imiglucerase treatment. J. Inherit Metab. Dis. 2012; 36 (3): 543–553.
55
БОЛЕЗНЬ НИМАННА–ПИКА
(NIEMANN–PICK DISEASE)
МКБ-10: Е 75.2
Определение. Болезнь Ниманна–Пика является лизосомной болезнью накопления, связана с дефицитом активности сфингомиелиназы и накоплением сфингомиелина в клетках ретикулоэндотелиальной системы. Болезнь описана в 1914 году, когда немецкий педиатр Albert Niemann представил характерные клинические проявления заболевания, а в 1927 г. немецкий патологоанатом Ludwig Pick описал патоморфологические признаки заболевания и на их основании отграничил связь с болезнью Гоше. В 1961 г. Crocker разделил болезнь Ниманна– Пика на 4 клинические формы: типы А, В, С, D.
Синонимы: отсутствуют.
Эпидемиология. Частота заболевания составляет 1 случай на 100000 новорожденных. Заболевание встречается во всех этнических группах, однако, частота типа А выше среди евреев-ашкенази и составляет 1:100.
Тип наследования: аутосомно-рецессивный.
Этиология, патогенез. Болезнь Ниманна–Пика объединяет группу сфингомиелолипидозов, характеризующихся накоплением сфингомиелина вследствие снижения активности фермента сфингомиелиназы, катализирующего гидролиз сфингомиелина с образованием фосфорилхолина и церамидных остатков, при котором происходит накопление сфингомиелина в лизосомах мозга, печени, ретикулоэндотелиальной системе.
Ген кислой сфингомиелиназы находится на хромосоме 11. Гены, ответственные за развитие болезни Ниманна-Пика А и В типов расположены на аллельных участках хромосомы 11, картированы на 11з15.4-15.1, описано большое количество мутаций. Ген, ответственный за развитие болезни Ниманна-Пика тип С, картирован на 18q11.2, кодирует белок, который переносит через цитоплазматическую мембрану холестерин и гликолипиды, поэтому при типе С накапливается не только сфингомиелин, но и холестерин. В норме сфингомиелин содержится в мозге, печени, почках и селезенке.
Клинические проявления
Раннее начало, гепатоспленомегалия, мышечная гипертония, переходящая в гипотонию, задержка психомоторного развития.
На сегодняшний день, когда понятна генетическая природа заболевания, расстройство классифицируется следующим образом:
•болезнь Ниманна–Пика, связанная с геном SMPD1, которая включает в себя типы А и В;
•болезнь Ниманна–Пика тип C, который включает в себя типы C1 и C2. Тип D возникает в результате мутации того же гена, что и тип C1.
56

Болезнь Ниманна–Пика
Болезнь Ниманна–Пика тип А (NIEMANN–PICK DISEASE TYPE A)
Тип А (классическая инфантильная форма, острая нейропатическая форма) наблюдается наиболее часто. Заболевание проявляется после рождения и характеризуется поражением внутренних органов и нервной системы. Уже в возрасте 3–5 месяцев отмечаются трудности вскармливания, гипотрофия, а в 5–6 месяцев выявляется гепатоспленомегалия.
Внешний вид больного: как правило, печень увеличивается раньше, чем селезенка. Дети истощены, характерны большой выступающий живот и тонкие конечности. Кожные покровы приобретают коричневатый оттенок. Иногда отмечаются небольшие или нодулярные ксантомы на коже. Увеличены лимфатические узлы. Дети отстают в весе (гипотрофия различной степени) вследствие снижения аппетита, вздутия и болей в животе (за счет гепатоспленомегалии).
На фоне болезни может усугубиться течение рахита с появлением рахитических изменений в костях и суставах (при рентгенографии суставов), появление контрактур, повышенная потливость (особенно ночью).
Кровь: следствием спленомегалии могут быть тромбоцитопения, гипохромная анемия.
В периферической крови, чаще в костном мозге, а также в печени, селезенке, почках, надпочечниках, лимфатических узлах и некоторых других органах обнаруживаются довольно крупные зернистые и вакуолизированные «пенистые» клетки (при микроскопии во всех органах обнаруживаются клетки, которые при фиксации спиртом выглядят «пенистыми» — клетки Ниманна–Пика). Они могут достигать значительной величины — 20–25 мкм, в некоторых случаях 90 мкм. Пенистость клеток — это артефакт, вызванный растворением жироподобных субстанций, содержащихся в клетках.
Костная система: рентгенологически в костях обнаруживаются признаки остеопороза и остеомаляции.
Со стороны органов дыхания: частые респираторные заболевания в виде ринитов, отитов. Достаточно быстро развиваются пневмонии различного генеза с выраженной картиной дыхательной недостаточности. Характерна инфильтрация легких, выявляемая рентгенологически.
Внешний вид пациента с болезнью Ниманна–Пика |
Симптом «вишневой косточки» у пациента 5 мес |
тип А (пунктиром показаны размеры печени) |
с болезнью Ниманна–Пика тип А |
57

Атлас редких болезней
Орган зрения: у больных снижается острота зрения, затем наступают слепота и глухота. Также описаны помутнение роговицы, коричневое прокрашивание передней капсулы хрусталика. У 20–30% детей при осмотре глазного дна обнаруживается симптом «вишневой косточки».
Центральная нервная система: неврологическая симптоматика включает остановку общего развития, затем утрату моторных навыков. Теряется интерес к окружающему. Выявляются также эпилептические припадки, уменьшение болевой чувствительности, снижение мышечного тонуса и угнетение сухожильных рефлексов, вплоть до их полного отсутствия.
Болезнь Ниманна–Пика тип В (NIEMANN–PICK DISEASE TYPE B)
Тип В — висцеральная форма без вовлечения нервной системы. Основные клинические проявления развиваются позже, чем при типе А. Спленомегалия появляется в возрасте 2–6 лет, позднее поражаются печень и легкие (больные подвержены частым инфекциям дыхательных путей). Симптоматика поражения ЦНС отсутствует: напротив, в ряде случаев отмечены высокие интеллектуальные способности. Продолжительность жизни не снижена.
Диагностика
•Гепатоспленомегалия;
•характерный внешний вид больного;
•«пенистые» клетки в костном мозге, печени и селезенке;
•накопление сфингомиелина в ретикулоэндотелиальных клетках и клетках других органов.
Дефицит сфингомиелиназы в лейкоцитах и фибробластах кожи, «вишневая
косточка» на глазном дне.
В пренатальной диагностике определяется уровень сфингомиелиназы в культуре амниотических фибробластов.
Дифференциальный диагноз:
•GM2-ганглиозидоз, тип I;
•GM1-ганглиозидоз, тип I;
•болезнь Волмана;
•болезнь Гоше;
•цирроз печени;
•болезнь Леттерера–Сиве;
•болезнь Ниманна–Пика тип С.
Лечение. Трансплантация костного мозга и печени оказалась неэффективной. Заместительная ферментотерапия также находится на стадии разработки. Несмотря на некоторый эффект симптоматической терапии, попытки лечения болезни Ниманна–Пика до сих пор заканчивались неудачей.
Прогноз. Прогноз неблагоприятный при болезни Ниманна-Пика тип А. Очень редко дети доживают до 3-летнего возраста. Несколько более благоприятен прогноз при типе В, который протекает без поражения центральной нервной системы.
58

Болезнь Ниманна–Пика
Болезнь Ниманна–Пика тип С (NIEMANN–PICK DISEASE TYPE C)
Определение. Болезнь Ниманна–Пика тип С (НП-С) является редким наследственным нейровисцеральным заболеванием. Это болезнь накопления липидов, которая характеризируется прогрессирующими, инвалидизирующими неврологическими расстройствами у большинства пациентов и преждевременной смертью.
Эпидемиология. Заболеваемость составляет 1 случай на 120000 живых новорожденных. Болезнь НП-С, как правило, возникает спорадически у представителей любой этнической группы, хотя были обнаружены генетические изоляты с эпидемиологией выше средних показателей.
Тип наследования: аутосомно-рецессивный.
Этиология, патогенез. Болезнь НП-С развивается в результате мутаций в какомлибо из двух генов: NPC1 локус 18q11-q12 (95%) или NPC2 локус 14q24 (4%). Мутации в одном из генов (NPC1 или NPC2) приводят к нарушению переработки и утилизации захваченного путем эндоцитоза холестерина с последующим внутриклеточным накоплением неэтерифицированного холестерина в органах и тканях, а также нарушению обмена гликосфинголипидов.
Клинические проявления
Клинические проявления болезни НП-С разнообразны, характеризуются сочетанием системных и неврологических признаков, которые возникают в любом возрасте и прогрессируют различными темпами. В результате гетерогенности клинических проявлений диагностика болезни НП-С затруднена: обычно выражается одним или несколькими неврологическими симптомами в детском возрасте, у пациентов с очень ранним началом — изолированными висцеральными проявлениями; кроме того, первые признаки могут развиться в подростковом или взрослом возрасте.
Выделяют следующие клинические формы болезни:
•неонатальная (до 3 месяцев): затяжная желтуха новорожденных, холестаз, гепатоспленомегалия, мышечная гипотония;
Изменения при магнитно-резонансной томографии и филиппиновом тесте у двух пацинтов с болезнью Ниманна–Пика тип С. Верхний ряд — МРТ пациентов на уровне гиппокампа, средний ряд — на уровне задней ямки, нижний ряд — тест с окрашиванием филиппином в культуре фибробластов (JIMD Short Report #180 (2009) Online DOI 10.1007/s10545-009-1173-1 Gender dimorphism in siblings with schizophrenia-like psychosis due to Niemann-Pick disease type C.
M. Walterfang, M. Fietz, L. Abel, E. Bowman, R. Mocellin, D. Velakoulis)
59