

- •Глава 1: Болезни женских половых органов и молочной железы
- •В патологической анатомии принято выделять три категории болезней женских половых органов и молочной железы: дисгормональные, воспалительные и опухолевые
- •Дисгормональные болезни
- •Дисгормональные болезни по своей природе связаны с нарушением выработки половых гормонов. Самый значимый этиологический фактор в данной категории - это эстроген, а точнее:
- •излишняя выработка эстрогена (беременность, половое созревание, аномалии развития половых органов, ожирение, перименопаузальный период, поликистоз яичников, наследственная предрасположенность)
- •неправильный прием некоторых противозачаточных средств (содержащих этинилэстрадиол и другие производные эстрогена)
- •Железистая гиперплазия слизистой оболочки шейки матки
- •Гиперэстрогения. Чем же она может быть обусловлена. Из опроса и анамнеза пациентки можно узнать следующие версии происходящего в ее организме:
- •Избыточная периферическая конверсия андрогенов в эстрогены при ожирении, особенно висцеральном, характеризующемся наибольшим ферментным потенциалом, обеспечивающим ароматизацию.
- •Наличием гормонпродуцирующих структур в яичнике (текоматоз, опухоли)
- •Патологией печени с нарушением инактивационной и белковосинтетической функций (снижение глобулина, связывающего половые стероиды (ГСПС), приводящее к увеличению биологически активной, так называемой «свободной» фракции стероидных гормонов)
- •Пролиферация клеток эндометрия
- •Дисгормональные нарушения. Прогестерон и эстрогены сочетанно стимулируют пролиферативный потенциал клеток путем индуцирования факторов роста (инсулиноподобного, трансформирующего и др.) и их рецепторов, а также регулируют процессы ангиогенеза.
- •Воспалительные вирусные заболевания, ингибирующие апоптоз, что позволяет вирусам закончить цикл репликации до гибели клетки и тем самым ускорить трансформацию поврежденных клеток.
- •Другие факторы (механические повреждения ткани, метаболические нарушения).
- •Классификация
- •Простая неатипическая гиперплазия эндометрия: увеличение количества как железистых элементов так и стромальных с незначительным преобладанием первых.
- •Сложная атипическая гиперплазия эндометрия характеризуется выраженной пролиферацией эпителиального компонента, которая сочетается с тканевой и клеточной атипией без инвазии базальной мембраны железистых структур.
- •На фоне железистой гиперплазии эндометрия развивается воспаление слизистой оболочки с переходом в склероз и рак тела матки, по этому ее рассматривают как предраковое состояние матки.
- •По старой классификации выделяют: железисто-кистозную и атипичную гиперплазии.
- •Псевдоэрозия шейки матки
- •Классификация
- •Морфологически различают поверхностную, железистую (фолликулярную), папиллярную, кистозную, заживающую (эпидермизирующуюся) псевдоэрозию.
- •Патогенез
- •Зоны трансформации шейки матки
- •В гинекологии принято выделять три типа «зон трансформации» благодаря которым можно судить о видимости и рисках патологического процесса.
- •Аденоматоз и полип шейки матки
- •Этиология
- •Классификация
- •Полипы цервикального канала бывают:
- •Железистый (покрытый однорядным циллиндрическим эпителием)
- •Эпидермизированный (покрытый метапластическим многослойным эпителием)
- •Доброкачественная дисплазия молочной железы
- •Нормальная физиология и гормональная регуляция молочных желез
- •Этиопатогенез
- •Доброкачественные изменения молочной железы
- •Классификация
- •1. Диффузная фиброзно-кистозная мастопатия (ФКМ):
- •2. Узловая ФКМ.
- •Кроме того, выделяют пролиферативные и непролиферативные формы мастопатии.
- •Клиника диффузной мастопатии
- •Диффузная ФКМ может проявляться в виде одного, двух или всех трех перечисленных признаков (боли, выделения из сосков и диффузные уплотнения в молочных железах).
- •Клиника узловой мастопатии
- •Фиброаденома
- •Непролиферативная мастопатия
- •Пролиферативная мастопатия
- •Характеризуется размножением эпителия, миоэпителия или содружественным разрастанием эпителия и соединительной ткани. Разновидности этой формы - аденоз и склерозирующий аденоз.
- •Аденоз (мазоплазия) - увеличение долек в связи с пролиферацией эпителия желез, а строение долек сохранено. Местами пролиферирует не только эпителий, но и миоэпителий; в концевых отделах долек появляются микроскопические кисты и участки склероза.
- •В зависимости от количества новообразований в груди и их типа выделяют следующие формы заболевания:
- •Воспалительные болезни женских половых органов и молочных желез
- •Каждое из этих заболеваний, в связи со своей инфекционной природой, может быть острым и переходить в хроническую форму.
- •Развитие заболевания происходит в родах путем восходящего инфицирования при лечебно-диагностических процедурах или в послеродовом периоде через раневую поверхность. А для постановки диагноза достаточно всего двух симптомов из четырех имеющихся:
- •повышение температуры тела (38 и выше)
- •болезненная матка при пальпации
- •выделения из половых путей с неприятным запахом
- •возможно умеренное кровотечение из половых путей
- •Эндометрит
- •Патогенез
- •Пути распространения инфекции:
- •Хронический эндометрит
- •Клиническая классификация:
- •Этиология
- •Клиническая картина
- •Цервицит
- •Цервицитом называют инфекционное и реже неинфекционное воспаление шейки матки. Кроме наиболее распространенной причины цервицита – венерических болезней, выделяют ряд предрасполагающих факторов, способствующих развитию патологии:
- •Раздражение шейки матки механическими или химическими противозачаточными средствами;
- •Ослабление иммунитета, например, при других соматических заболеваниях;
- •Травма при аборте;
- •Не ушитые разрывы шейки и промежности после родов;
- •Опущение половых органов;
- •Климактерический период.
- •Классификация
- •И уже затем можно уточнить конкретный патоген - например «Хламидийный цервицит».
- •По локализации различают: эндоцервицит – воспаление слизистой оболочки цервикального канала; экзоцервицит – поражение влагалищной части шейки матки.
- •Клиническая картина заболевания
- •Цервицит у большинства женщин, как уже говорилось выше, протекает бессимптомно. Однако иногда могут присутствовать следующие косвенные признаки заболевания:
- •Наботовые и ретенционные кисты шейки матки как осложнение воспалительных заболеваний
- •Мастит
- •Этиология
- •Патогенез
- •В возникновении процесса принимают участие различные неблагоприятные факторы, носящие название предрасполагающих:
- •Причины снижения резистентности организма:
- •простудные заболевания.
- •Причины лактостаза:
- •Пути контаминации:
- •Патологическая анатомия
- •Выделяют клинические формы мастита:
- •4. Ретромаммарные.
- •5. Панмастит.
- •Клиническая картина
- •Заболевание начинается спустя 2-4 недели после родов.
- •Опухоли женских половых органов и молочной железы
- •В данном разделе будут рассмотрены самые распространенные опухолевые заболевания: Рак тела матки, рак шейки матки, рак яичников, рак молочной железы.
- •Этиология и патогенез
- •Выделяют два патогенетических типа РТМ.
- •Морфология
- •Гистологически рак эндометрия чаще всего представлен:
- •Клиника
- •Метастазирование
- •Прежде всего метастазы наблюдаются в лимфатических узлах малого таза и поясницы. Гематогенные метастазы встречаются редко, в легких. Имплантационные в прямой кишке, яичниках, большом сальнике, мочевом пузыре.
- •Неэнодометриоидные виды аденокарциномы
- •Поражение более глубокого мышечного слоя происходит реже, а его выявление представляет сложности в связи с затруднением взятия образцов тканей. В случае поражения мышечного и других слоев тела матки различают неэндометриоидные виды аденокарциномы:
- •Рак шейки матки
- •Этиология
- •Раннее начало половой жизни (до 16 лет)
- •Курение
- •Инфицирование вирусом папилломы человека
- •Патогенез
- •Клиническая картина
- •Симптоматика на ранних стадиях может отсутствовать или проявляться в виде трудно дифференцируемого дискомфорта. На более поздних стадиях развития заболевания могут возникать следующие клинические признаки.
- •Патологические вагинальные кровотечения.
- •Кровотечения после полового акта, спринцевания или вагинального осмотра гинекологом.
- •Изменения характера и длительности менструации.
- •Боль в области малого таза.
- •Боль во время полового акта.
- •Все приведённые выше клинические признаки неспецифичны. Также развитие рака шейки матки может сопровождаться системными эффектами, например:
- •Нарастающая слабость, утомляемость.
- •Быстрая потеря веса.
- •Метастазирование
- •Метастазирует рак шейки матки рано и прежде всего по лимфатическим путям в лимфатические узлы малого таза, паховые и забрюшинные; позже наблюдается гематогенное метастазирование в легкие, кости и печень.
- •Рак яичников
- •В 2018 году по уровню заболеваемости раком яичников на первом месте находилась Сербия, на втором Бруней и на третьем Белоруссия.
- •Этиология
- •Клиника
- •Опухоль Бреннера
- •Метастазирование
- •Лимфо- и гематогенное: метастазы встречаются в лимфатических узлах, брюшине и во внутренних органах.
- •Рак молочной железы
- •Этиология
- •Морфология
- •Гистологические типы:
- •неинфильтрирующий (неинвазивный) рак (внутридольковый и внутрипротоковый) инфильтрирующий (инвазивный) рак (протоковый и дольковый; обычно имеет строение скирра), а так же болезнь Педжета.
- •Болезнь Педжета
- •Метастазирование
- •Распространение рака молочной железы связано с прорастанием в мягкие ткани. Лимфогенные метастазы появляются в регионарных лимфатических узлах: подмышечных, передних грудных, подключичных, надключичных и парастернальных.
- •Гематогенные метастазы обнаруживают часто в легких, костях, печени и почках.
- •Глава 2: Болезни беременности и послеродового периода
- •Глава 3: Болезни желез внутренней секреции
- •Общие понятия о нейроэндокринно-иммунной регуляторной системе
- •Гипофиз
- •Гипофизарные расстройства
- •Гиперпитуитаризм
- •Лактотропные аденомы гипофиза
- •Соматотропные аденомы гипофиза
- •Другие аденомы гипофиза
- •Гипопитуитаризм
- •Клиника
- •Гипофизарный нанизм
- •Классификация
- •Церебрально-гипофизарная кахексия (Болезнь Симмондса)
- •Клиника и патогенез
- •аменорею;
- •исчезновение полового влечения.
- •спутанность сознания;
- •головокружения;
- •потеря психической живости.
- •Адипозо-генитальная дистрофия
- •Этиология
- •внутриутробная инфекция (токсоплазмоз),
- •родовая травма,
- •хроническая инфекция (туберкулёз, сифилис),
- •травматическое поражение мозга в раннем детском возрасте,
- •водянка третьего желудочка головного мозга,
- •тромбозы, эмболии сосудов головного мозга,
- •кровоизлияния (в детском возрасте).
- •Патогенез
- •Клиника
- •Адипозогенитальная дистрофия у пациентов с новообразованием гипоталамической области:
- •ожирение (жировая клетчатка откладывается неравномерно - на бёдрах, животе, ягодицах, голенях и в области грудных желёз);
- •гипогенитализм;
- •мозговые симптомы (головная боль, эпилептиформные припадки, гетеронимная гемианопсия);
- •малый рост;
- •часто сочетается с клинической картиной несахарного диабета, тенденцией к пониженной температуре тела.
- •Центральный несахарный диабет
- •Этиология
- •Патогенез
- •Клиника
- •Основные симптомы несахарного диабета у взрослых следующие:
- •Надпочечники
- •Гиперальдостеронизм (Синдром Конна)
- •Этиология
- •Причиной первичного гиперальдостеронизма обычно является гормонально-активная опухоль (альдостерома), происходящая из клубочковой зоны.
- •Болезнь Аддиссона (хроническая надпочечниковая недостаточность)
- •Этиология
- •Патогенез
- •Клиника
- •Вторичная недостаточность коры надпочечников
- •Морфология
- •Адреногенитальный синдром
- •Морфология
- •Клиника
- •Феохромоцитома или хромаффинома
- •Хромаффинома (феохромоцитома) - опухоль, развивающаяся из клеток мозгового слоя надпочечников, симпатических ганглиев, параганглиев.
- •Патогенез
- •- Со стороны сердцебиения: тахиаритмия, фибрилляция.
- •Тахикардия ведет к повышенному потреблению кислорода миокардом, что может привести к ишемии миокарда и стенокардии.
- •В крови: повышение уровня глюкозы, жирных кислот, калия и лактата (ацидоз). Повышение свертываемости крови.
- •В конце криза - усилены эффекты вагуса. Резкое падение артериального давления, снижение ЧСС (брадикардия). Гиперемия лица, гиперсаливация, рвота.
- •Со стороны мочевыделительной системы : полиурия, повышение клубочковой фильтрации, глюкозурия.
- •Клиника
- •Осложнения
- •- Фибрилляция сердца может привести к острой сердечной недостаточности и кардиогенному отеку легких (или инфаркту миокарда).
- •При длительном течении может развиться сахарный диабет II типа (связано с повреждением поджелудочной железы во время приступов гипертонического криза и гипергликемией (инсулинорезистентностью).
- •Патогенез
- •Клиника болезни ИК
- •Болезни щитовидной железы - зоб, тиреоидиты и опухоли - сопровождаются гипертиреозом или гипотиреозом.
- •Гипертиреоз
- •Клиническое значение: все другие причины тиреотоксикоза сопровождаются снижением ТТГ по механизму отрицательной обратной связи.
- •Клиника
- •Болезнь Грейвса
- •Этиология
- •Так называемая «тиреоидная болезнь глаз», характерная для болезни Базедова, включает в себя следующие симптомы:
- •симптом Грефе — отставание верхнего века от края радужки при взгляде вниз
- •симптом Кохера — отставание глазного яблока от движения верхнего века при взгляде вверх
- •симптом Елинека — гиперпигментация вокруг глаз
- •симптом Дальримпля — расширение глазной щели
- •симптом Розенбаха — тремор век при неплотно закрытых глазах
- •симптом Мебиуса — нарушение конвергенции
- •симптом Жоффруа — отсутствие наморщивания при взгляде вверх, а также подъём верхнего века, опущение (зияние) нижнего века, периорбитальный отек и разрастание периорбитальных тканей.
- •Гипотиреоз
- •Кретинизм
- •Микседема
- •Микседема — синдром приобретенного гипотиреоза у взрослых, который проявляется характерным «слизистым отеком» и энцефалопатией (сонливость, заторможенность, апатия, снижение памяти, ослабление рефлексов).
- •Патогенез
- •Из-за дефицита гормонов щитовидной железы:
- •снижается активность окислительных ферментов, снижается количество АТФ
- •снижается утилизация кислорода в тканях
- •снижается основной обмен. Снижается теплопродукция, что приводит к гипотермии
- •повышается масса тела на фоне сниженного аппетита, что приводит к ожирению и гипергидратации.
- •угнетены все виды обмена веществ: склонность к гипогликемии( снижена всасываемость глюкозы и др.), развивается азотемия (снижены утилизация и выведение продуктов липолиза, что приводит к повышению риска атеросклероза и его осложнениям).
- •«Слизистый отек» Патогенез
- •У больных снижен ПНФ (ПНУП), что ведет к снижению выведения натрия с мочой, повышению его концентрации в плазме крови и как следствие, усилению выработки АДГ. В конечном итоге это приводит к задержке воды, гипергидратации и отекам.
- •Повышается проницаемость сосудов для белка, повышается онкотическое давление в интерстиции, а в плазме крови снижается (гипоальбуминемия).
- •Повышается гидрофильность соединительной ткани ( повышение концентрации кислых ГАГ глюкуроновой и хондроитинсерной кислот), как следствие вода связывается с мукополисахаридами.
- •К общим симптомам относятся:
- •Этиология
- •Классификация
- •Выделяют несколько форм аутоиммунного тиреоидита:
- •Патогенез
- •Этиология
- •с отсутствием компрессионного синдрома;
- •с наличием компрессионного синдрома;
- •Патогенез
- •Классификация зобов
- •Классификация зоба учитывает морфологические признаки, эпидемиологию, причины развития, функциональные и клинические особенности.
- •способствует превращению витамина D в почках в активную форму;
- •Клиника
- •Патогенез
- •Клиника
- •Гипопаратиреоз
- •Клиника
- •Поджелудочная железа
- •Сахарный диабет
- •Классификация
- •Этиология
- •В развитии СД1 можно выделить несколько периодов.
- •II – в этом периоде происходит аутоиммунное разрушение β-клеток, но продукция инсулина оставшимися клетками вполне достаточна.
- •Патогенез сахарного диабета I типа
- •Аутоиммунная форма СД1 ассоциируется с внутренними (генетическими) и внешними (провоцирующими) факторами, которые в комбинации друг с другом «запускают» иммунные реакции повреждения островкового аппарата.
- •Патогенез инсулиновой недостаточности при СД2 типа
- •Острые осложнения сахарного диабета
- •К острым осложнениям СД относят кетоацидотическую, гиперосмолярную (гипергликемическую) и лактацидотическую комы. Отдельно рассматривается гипогликемическая кома, которая может осложнять сахароснижающую терапию СД.
- •Предрасполагающими факторами являются:
- •назначение слишком малых доз инсулина при лечении;
- •тяжелые поражения печени и почек, т.е. органов, в которых метаболизируется молочная кислота;
- •диабетическая катаракта.
- •Глава 4: Болезни сердечно-сосудистой системы
- •Инфекционный эндокардит
- •Этиология
- •Патогенез
- •Бактериемия — это циркуляция тех или иных инфекционных агентов в кровяном русле. Источником бактериемии могут служить:
- •высокие титры аутоантител (криоглобулинов, ревматоидного фактора, антимиокардиальных антител и др.);
- •снижение содержания комплемента;
- •образование циркулирующих иммунных комплексов (ЦИК).
- •Гистологическая классификация активности инфекционного эндокардита
- •По анатомическому субстрату:
- •По клиническим проявлениям и гистологии удаленного материала:
- •По течению:
- •Клиника
- •Возможные проявления:
- •Физикальное обследование
- •При типичном классическом течении инфекционного эндокардита рекомендуется проведение общего осмотра, что позволяет выявить многочисленные неспецифические симптомы:
- •2. Похудание весьма характерно для больных инфекционным эндокардитом. Иногда оно развивается очень быстро, в течение нескольких недель.
- •Пальпация и перкуссия сердца:
- •Аускультация сердца:
- •Рекомендуется выполнение аускультации для выявления аускультативных признаков формирующегося порока сердца; обычно начинают проявляться через 2–3 месяца после лихорадочного периода.
- •Миокардит
- •Этиология
- •Классификация
- •Патогенез
- •Микроскопия
- •Существует мнение, что в некоторых случаях хронические миокардиты тяжелого течения со временем трансформируются в ДКМП.
- •Клиника
- •Клинические проявления:
- •Острая боль в груди, в т.ч. по типу перикардита, или псевдоишемическая
- •Этиология
- •Основные причины заболевания
- •Общепринятая терминология и классификация перикардитов отсутствует.
- •По этиологическому принципу выделяют: инфекционные (вирусные, бактериальные, туберкулезные); аллергические; аутоиммунные; асептические.
- •По течению выделяют: острые (менее 1 недели); подострые (от 1 недели до 3 месяцев); хронические (более 3 месяцев).
- •При туберкулезном перикардите различают две формы поражения:
- •Клиника
- •Фазы Сподика для острого перикардита: (ЭКГ)
- •Фаза 1. Диффузная элевация сегмента ST и депрессия сегмента PR. Изменения сохраняются от нескольких часов до нескольких дней.
- •Фаза 2. Элевация сегмента ST возвращается к изолинии
- •Фаза 3. Инверсия зубцов Т. Сохраняется от нескольких дней до нескольких недель и месяцев.
- •Фаза 4. Нормализация ЭКГ
- •Симптомами гемодинамически значимого выпота являются:
- •1. Глухие сердечные тоны
- •2. Исчезновение шума трения перикарда
- •Осложнения
- •Приобретенные пороки сердца
- •Недостаточность митрального клапана
- •Этиология
- •Изменения гемодинамики
- •Митральный стеноз
- •Этиология
- •Изменения гемодинамики
- •Стеноз устья аорты
- •Этиология
- •Изменения гемодинамики
- •Недостаточность аортального клапана
- •Недостаточность аортального клапана (аортальная недостаточность) характеризуется неполным смыканием створок клапана во время диастолы, что приводит к возникновению обратного диастолического тока крови из аорты в ЛЖ
- •Этиология
- •Ревматическая лихорадка и ревматическая болезнь сердца
- •Морфология
- •Патогенез
- •Клиника
- •Осложнения ревматизма:
- •1. Приобретенные пороки сердца
- •3. Шаровидный тромб
- •4. Спаечные процессы в полости перикарда
- •Другие формы ревматизма:
- •Ишемическая болезнь сердца (острые формы)
- •Этиология
- •Нарушение баланса между реальным коронарным кровотоком и потребностями миокарда в кислороде может быть обусловлено:
- •Патогенез атеросклероза
- •В современных научных работах большое количество внимания посвящено воспалению - как главному фактору атеросклероза сосудов.
- •Классификация
- •Внезапная сердечная смерть
- •Морфология
- •Стенокардия
- •Стенокардия напряжения
- •Стенокардия напряжения подразделяется на следующие формы:
- •Для вариантной стенокардии характерно:
- •Инфаркт миокарда
- •Этиология
- •Основными патогенетическими факторами ИМ являются:
- •Патогенез
- •Стадии патогенеза ИМ следующие:
- •4) рубцевание.
- •Рассмотрим механизмы развития и по следствия ишемии миокарда.
- •3. Медиаторы, высвобождаемые тромбоцитами, стимулируют спазм сосудов.
- •Классификация
- •Реакция миокарда
- •продолжительность окклюзии;
- •метаболические потребности миокарда и его потребность в кислороде в зоне повышенного риска;
- •степень развития коллатеральных кровеносных сосудов;
- •наличие, локализация и тяжесть коронарного спазма;
- •Морфология
- •Модификация инфаркта путем реперфузии
- •Потенциальная польза реперфузии зависит от двух факторов:
- •Клиника
- •Патологическая физиология: влияние токов повреждения на комплекс QRS
- •Последствия и осложнения инфаркта миокарда
- •Аневризма аорты
- •Классификация
- •Морфология
- •Этиопатогенез
- •Расслоение аорты
- •Классификация расслаивающей аневризмы аорты
- •Согласно классификации ДеБейки, определяют 3 типа расслоения:
- •I – надрыв интимы в восходящем сегменте аорты, расслоение распространяется до грудного и брюшного отделов;
- •II – место надрыва и расслоение ограничено восходящим отделом аорты,
- •Клиника
- •Патология мозгового кровообращения
- •Со стороны физиологии патологических процессов
- •Возможность развития коллатерального кровообращения обусловливается:
- •Нарушения саморегуляции мозгового кровообращения возникают в следующих случаях.
- •4. При значительном снижении интенсивности насыщения крови кислородом или увеличении напряжения углекислоты в мозге. При этом активность мозгового кровотока также меняется вслед за изменением системного АД.
- •Она может возникнуть:
- •Патологические изменения мозгового кровотока
- •Патологическое снижение интенсивности мозгового кровотока возможно в следующих случаях:
- •Патологическое усиление интенсивности мозгового кровотока возникает:
- •Клиника
- •Общемозговая симптоматика:
- •1. Нарушение и расстройство сознания: количественное (содержание сознания) и качественное (степень бодрствования).
- •2. Головная боль: диффузная, интегрально нарастающая
- •3. Головокружение: системное (кружатся предметы, болезнь Меньера) и несистемное (кружится больной.
- •4. Тошнота и рвота. (Мозговая рвота может приносить облегчение вопреки классической дифференциальной диагностике).
- •Менингеальные симптомы:
- •1. Общая гиперестезия: фотофобия, усиление ощущения звуков, запахов, прикосновения вплоть до ощущения боли от привычных внешних раздражителей, в норме не вызывающих боль.
- •2. Ригидность затылочных мышц (невозможность приведения подбородка к груди). Важно запомнить, что если имела место травма головы и шеи, то перед тем как проверять данный симптом, необходимо убедиться в отсутствии переломов шейных позвонков.
- •3. Симптом Кернига положительный. (Больной поднимает ногу, согнутую в коленном суставе, затем врач просит больного разогнуть ногу в коленном суставе. Если больной не может это выполнить - симптом положительный).
- •4. Скуловой прием Бехтерева. Постукивание по скуловой дуге вызывает «гримасы» у больного.
- •5. Симптом Брудзинского. Непроизвольное сгибание ног и подтягивание их к животу при попытке пассивного сгибания головы ( перед этим следует убедиться что шея цела).
- •Очаговая симптоматика
- •Очаговая симптоматика соответствует очагу локализации гематомы или ишемизированному участку коры головного мозга. При субарахноидальном кровоизлиянии очаговая симптоматика отсутствует.
- •Лобная симптоматика
- •Центральный парез, инертность, пассивность, гипокинезия.
- •Теменная симптоматика
- •Височная доля
- •Затылочная доля
- •Нарушение зрительного ощущения и восприятия. Полная или частичная гомонимная гемианопсия на противоположной очагу стороне. Нарушение зрительной памяти, фотопсии, выраженные головные боли.
- •Эссенциальная артериальная гипертензия
- •Этиология
- •Патогенез
- •Уровень АД, как известно, определяется тремя основными гемодинамическими показателями:
- •Классификация
- •Классификация эссенциальной АГ (гипертонической болезни) (ВОЗ, 1996)
- •Гемодинамические последствия АГ и поражение органов мишеней
- •Сосуды
- •Сердце
- •Почки
- •Таким образом, характеристика ГБ складывается из оценки трех основных параметров:
- •Системные васкулиты
- •Классификация
- •Определения основных нозологических форм системных васкулитов
- •Неинфекционный васкулит
- •отложение иммунных комплексов;
- •Гигантоклеточный артериит
- •Патогенез
- •Морфология
- •Клиника
- •Артериит Такаясу
- •Морфология
- •Клиника
- •Узелковый полиартериит
- •Морфология
- •Клиника
- •Болезнь Кавасаки
- •Морфология
- •Клиника
- •Патогенез
- •Морфология
- •Клиника
- •Синдром Черджа-Стросс
- •Гранулематоз Вегенера
- •Морфология
- •Клиника
- •Патогенез
- •Морфология
- •Клиника
- •Феномен Рейно
- •Болезни вен
- •Острая венозная недостаточность
- •Этиопатогенез
- •Классификация венозных тромбозов:
- •Клиническая картина венозного тромбоза
- •Симптомы тромбоза глубоких вен голени (периферического флеботромбоза):
- •Известны следующие специфические симптомы тромбоза глубоких вен (определяются только в течение первых суток от начала заболевания):
- •Хроническая венозная недостаточность. Варикозная болезнь нижних конечностей.
- •Этиология
- •Патогенез
- •Классификация
- •Клиническая классификация (С):
- •2. Стадия 1. Телеангиэктазии или ретикулярные вены.
- •3. Стадия 2. Варикозно расширенные вены.
- •Клиника
- •Глава 5: Ревматические болезни
- •В основной своей массе системные заболевания соединительной ткани - это аутоиммунные болезни и болезни обусловленные врожденными генетическими мутациями.
- •Далее будут рассмотрены реакции гиперчувствительности II, III, IV типов - так как они лежат в основе патогенеза многих аутоиммунных заболеваний и соответственно системных заболеваний соединительной ткани.
- •Реакция гиперчувствительности II типа
- •Примеры заболеваний, опосредованных реакцией гиперчувствительности II типа.
- •Гиперчувствительность типа III
- •Системные болезни иммунных комплексов
- •Морфология
- •Примеры заболеваний с участием реакции гиперчувствительности III типа
- •Гиперчувствительность IV типа
- •Реакции Т-клеток CD8+: клеточно-опосредованная цитотоксичность
- •Примеры заболеваний, опосредованных реакцией гиперчувствительности IV типа.
- •Системная красная волчанка
- •Этиология
- •Спектр аутоантител
- •Этиология
- •Ряд данных подтверждает генетическую предрасположенность СКВ:
- •Иммунологические факторы
- •Патогенез
- •Морфология
- •гемотоксилиновые тельца — очаги внеклеточно расположенного базофильного вещества, являющегося продуктом деградации ядер;
- •Клиника
- •Диагностические критерии системной красной волчанки (1997)
- •Анкилозирующий спондилит (Болезнь Бехтерева)
- •Этиология и патогенез
- •Клиника
- •Основные приципы лежащие в основе современных клинических рекомендаций:
- •Оптимальное ведение пациента АС требует комбинации нефармакологических и фармакологических методов лечения.
- •Воспалительные поражения периферических суставов (артриты) и энтезов (энтезиты) часто встречаются и характерны для АС;
- •Для АС нет специфических диагностических лабораторных тестов;
- •Патологическая анатомия
- •Системный склероз
- •Этиология
- •Патогенез
- •Морфология
- •Классификация
- •1. Диффузная форма:
- •2. Лимитирующая форма:
- •3. Склеродермия без склеродермы:
- •5. Ювенильная склеродермия — до 16 лет.
- •6. Пресклеродермия.
- •Течение
- •1. Острое, быстропрогрессирующее с генерализованным фиброзом кожи и внутренних органов.
- •2. Подострое, умеренно прогрессирующее с отеками кожи, артритом, миозитом.
- •Стадии:
- •Клиника
- •Дерматомиозит
- •Этиология
- •Патогенез
- •Морфология
- •Классификация
- •По течению выделяют 3 основные формы: острую, подострую и хроническую.
- •Подострое течение характеризуется постепенным нарастанием симптомов, цикличностью, через 1–2 года от начала ДМ отмечается развернутая клиническая картина.
- •Ревматоидный артрит
- •Этиология
- •Патогенез
- •Классификация
- •1. Основной диагноз:
- •- Болезнь Стилла взрослых
- •q. Ревматоидный артрит вероятный
- •2. Клиническая стадия:
- •3. Активность болезни [4]:
- •4. Внесуставные (системные) проявления:
- •1. ревматоидные узелки
- •3. васкулиты других органов
- •4. нейропатия (мононеврит, полинейропатия)
- •5. плеврит (сухой, выпотной), перикардит (сухой, выпотной)
- •6. синдром Шегрена
- •7. поражение глаз (склерит, эписклерит)
- •8. интерстициальное заболевание легких
- •7. Функциональный класс:
- •I - полностью сохранены: самообслуживание, непрофессиональная и профессиональная деятельность
- •II - сохранены: самообслуживание, профессиональная деятельность, ограничена: непрофессиональная деятельность
- •III - сохранено: самообслуживание, ограничены: непрофессиональная и профессиональная деятельность
- •IV - ограничены: самообслуживание, непрофессиональная и профессиональная деятельность
- •Клиника
- •Генерализованная миалгия: скованность, депрессия, двухсторонний синдром запястного канала, похудание (обычно развивается в пожилом возрасте и напоминает ревматическую полимиалгию); характерные клинические признаки РА развиваются позднее.
- •В практике чаще всего встречаются следующие клинические варианты НДА:
- •Терапевтические подходы при НДА близки к таковым при РА.
- •- У всех пациентов с РА или подозрением на это заболевание рекомендуется проводить общетерапевтический осмотр, для выявления патологии кожи, мышц, желудочно-кишечного тракта, легких, сердечно-сосудистой, мочевыделительной и эндокринной систем.
- •Глава 6: Болезни органов дыхания
- •Острая дыхательная недостаточность
- •ОДН является патологическим cостоянием, при котором аппарат внешнего дыхания не может обеспечить организм достаточным количеством кислорода и осуществить элиминацию углекислого газа при нормальных затратах энергии.
- •Классификация
- •Нам представляется целесообразным выделить два типа ДН:
- •Этиопатогенез
- •Острое повреждение легких и острый респираторный дистресс синдром (ОРДС)
- •Состояния, ассоциированные с острым респираторным дистресс синдромом
- •Морфология
- •Патогенез
- •Клиника
- •Отек легких
- •Классификация и причины отека легких
- •Пневмонии
- •застоем и отеком легких.
- •Классификация
- •Этиология
- •Морфология
- •Классическая долевая пневмония имеет 4 стадии:
- •Осложнения пневмонии:
- •На рисунке представлены: бронхопенвмония слева и долевая пневмония справа.
- •Патогенез
- •Формирование внебольничной или госпитальной пневмонии происходит в результате реализации нескольких патогенетических механизмов, важнейшими из которых являются:
- •нарушение сложной многоступенчатой системы защиты органов дыхания от проникновения микроорганизмов в респираторные отделы легких
- •механизмы развития локального воспаления легочной ткани
- •формирование системных проявлений заболевания
- •формирования осложнений
- •Клиника
- •Внебольничная атипичная пневмония
- •Этиология
- •Морфология
- •Клиника
- •Внутрибольничная пневмония
- •Аспирационная пневмония
- •Нагноительные заболевания легких
- •К нагноительным заболеваниям легких относятся: абсцесс легкого, гангрена легкого и бронхоэктатическая болезнь.
- •Абсцесс легкого
- •Классификация
- •3. По этиологии:
- •4. По механизму инфицирования: – бронхогенные (аспирационные, ингаляционные, постпневмонические, обтурационные), гематогенные (тромбоэмболические постинфарктные, септикопиемические) и травматические;
- •Типы абсцедирования (по И. С. Колесникову, 1988):
- •Морфология
- •Полостная — полость абсцесса опорожняется, образуется фиброзная капсула, появляются участки ателектаза, в процесс вовлекается плевра.
- •Клиника
- •Особенности течения гематогенных абсцессов легкого
- •Хронический абсцесс легкого
- •Осложнения
- •Гангрена легкого
- •Бронхоэктатическая болезнь
- •Этиология и патогенез
- •Морфология
- •Клиника
- •Обструктивные болезни легких
- •Эмфизема
- •Классификация
- •Патогенез
- •Установлена следующая патогенетическая последовательность событий:
- •Морфология
- •Клиника
- •Эмфизема и хронический бронхит (Pink Puffer & Blue Bloater)
- •Другие типы эмфиземы
- •Хронический необструктивный бронхит
- •Этиология
- •Патогенез
- •Морфология
- •Клиника
- •Клиническое течение ХНБ в большинстве случаев характеризуется длительными периодами стойкой клинической ремиссии и сравнительно редко возникающими обострениями заболевания (не чаще 1-2 раза в год).
- •При расспросе больного с обострением ХНБ выявляют три клинических признака:
- •Бронхиальная астма
- •Этиология
- •Патогенез
- •Морфология
- •Ремоделирование дыхательных путей
- •Клиника
- •Основными клиническими симптомами БА являются:
- •Оценка одышки по шкале MRS:
- •Визуальная аналоговая шкала (модифицированная по Borg)
- •Удушье. Является наиболее ярким клиническим проявлением БА. В развитии приступа удушья выделяют три периода :
- •период предвестников (продромальный период)
- •период удушья
- •период обратного развития приступа;
- •Тромбоэмболия легочной артерии (ТЭЛА)
- •Этиология
- •Патогенез
- •Патофизиология
- •Морфология
- •Клиника
- •Туберкулез органов дыхания
- •Этиология
- •В патогенезе туберкулеза имеется два этапа — инфицирование МБТ (затем иногда очень длительное — до нескольких десятков лет персистирование в организме человека измененных форм МБТ) и развитие собственно заболевания.
- •Патогенез
- •пиема). ○ Туберкулез бронхов, трахеи, верхних дыха-
- •Эти клинические формы наиболее часто встречаются у взрослых и характеризуют течение вторичного туберкулеза.
- •III. Туберкулез других органов и систем.
- •Морфология
- •Клинические формы туберкулеза могут переходить одна в другую по мере прогрессирования или, наоборот, регрессии специфического процесса.
- •Глава 7: Болезни органов желудочно-кишечного тракта
- •Диспепсия
- •Классификация:
- •Формы диспепсии по патогенезу: функциональная и органическая, а по локализации желудочная и кишечная.
- •Этиология
- •Причины развития функциональной диспепсии:
- •Нарушения в питании, преобладание тех или иных групп питательных веществ (белков, жиров или углеводов)
- •Гиперсекреция соляной кислоты в желудке
- •Прием некоторых лекарственных препаратов (антибиотиков, гормональных препаратов, противотуберкулезных и противоопухолевых препаратов).
- •Интоксикация организма при вирусных инфекциях, гнойных заболеваниях, профессиональных и бытовых отравлениях.
- •Нарушения моторики желудка и кишечника.
- •Симптомы диспепсии
- •Желудочная диспепсия
- •Варианты течения желудочного диспептического синдрома: язвенноподобный, дискинетический и неспецифический. Ниже они будут описаны в патогенезе как нарушения моторики, чувствительности и секреции.
- •Патогенез
- •Повышение секреции соляной кислоты. Высокая продукция соляной кислоты и нарушение ощелачивания в антральном отделе желудка являются причиной болей в эпигастральной области.
- •Так же при разных заболеваниях ЖКТ могут появляться желудочные дисритмии: тахигастрия, брадигастрия, антральная фибрилляция, дуоденогастральный рефлюкс.
- •Изменение висцеральной чувствительности (гиперчувствительность). Висцеральная гиперчувствительность - это повышенная чувствительность рецепторного аппарата стенки желудка и двенадцатиперстной кишки к растяжению.
- •Кишечная диспепсия
- •Варианты функциональной кишечной диспепсии:
- •Бродильная диспепсия. Возникает при преобладании в пище углеводных продуктов и бродильных напитков. Симптомы: метеоризм, урчание в животе, отхождение большого количества кишечных газов, частый, слабо окрашенный жидкий пенистый кал с кислым запахом.
- •Гнилостная диспепсия. Возникает при преобладании в пище белковых, а также не свежих мясных продуктов. Симптомы: понос. Цвет испражнений насыщенно темный, запах - гнилостный.
- •Жировая (мыльная) диспепсия. Возникает при чрезмерном поступлении в пищей жиров, особенно тугоплавких. Симптомы: испражнения светлые, обильные, с жирным блеском.
- •Симптомы кишечной диспепсии
- •Метеоризм, учрание в кишечнике, выделение большого количества газов, частый, слабо окрашенный жидкий пенистый стул с кислым запахом.
- •Органическая диспепсия
- •Заболевания и повреждения пищевода
- •Кардиоспазм пищевода
- •Этиология
- •Патогенез
- •Клиника
- •Эзофагоспазм
- •Этиология и патогенез
- •Клиника
- •Халазия пищевода
- •Этиология и патогенез
- •Клиника
- •Дивертикулы пищевода
- •Дивертикул пищевода — это мешковидное выпячивание его стенки.
- •Классификация
- •По патогенному происхождению дивертикулы бывают пульсионные — выпячивающиеся, тракционные — вытянутые и смешанные — пульсионно-тракционные.
- •По локализации они бывают глоточно-пищеводные (3–5 %), бифуркационные (70–80 %) и эпифренальные (10–15 %). Размеры дивертикула колеблются в больших проделах, вплоть до огромного мешка.
- •Патогенез
- •Клиника
- •Желудок
- •Гастриты
- •Этиология
- •Острый гастрит
- •Патогенез
- •Морфология
- •Клиника
- •Хронический гастрит
- •Классификация
- •Хронический гастрит типа А
- •Морфология
- •Клиника
- •Хронический гастрит типа В
- •Патогенез
- •Морфология
- •Клиника
- •Патогенез
- •Клиника
- •Другие виды гастрита
- •Язвенная болезнь желудка и двенадцатиперстной кишки
- •Этиология и патогенез
- •Морфология
- •Классификация
- •Клиника
- •Осложнения
- •Кишечная непроходимость
- •Классификация
- •По клиническому течению непроходимость делят на острую, подострую и хроническую (проявляется замедлением пассажа по кишечнику), по степени — на полную и неполную, по уровню — на тонкокишечную (высокую) и толстокишечную (низкую).
- •Первая стадия — нервно-рефлекторная (болевая, острого нарушения пассажа).
- •Третья стадия — терминальная (стадия перитонита).
- •Этиология
- •Предрасполагающие факторы:
- •Патогенез
- •Патологическая анатомия
- •Клиника
- •Жажда, сухость во рту появляются и нарастают по мере прогрессирования секвестрации жидкости.
- •Синдром Валя дополняется другими проявлениями и включает:
- •Симптом Руша — усиление боли в животе при пальпации инвагината.
- •«Стоп-симптом» — при изучении пассажа бария снимки мало отличаются друг от друга, расположение чаш в целом не меняется, появляются новые.
- •Воспалительные заболевания кишечника
- •Этиология и патогенез
- •Генетические факторы
- •Иммунологическая концепция патогенеза
- •Морфология язвенного колита
- •Морфология болезни Крона
- •Клиника
- •Кишечные симптомы
- •Предложено 4 стадии развития хронического гепатита, ассоциированного с ПСХ, по которым можно оценить клиническое течение и исход заболевания:
- •Клинические проявления коллагенового и лимфоцитарного колита
- •Острый аппендицит
- •Этиология
- •На сегодняшний день известны следующие теории возникновения воспаления червеобразного отростка:
- •Теория Делафуа — рассматривает развитие аппендицита вследствие нарушения опорожнения и последующего застоя содержимого из-за деформации, сужения просвета, перегибов и других причин.
- •Теория Риккера — развитие аппендицита вследствие ангионевроза, что приводит к нарушению кровообращения и питания, снижению резистентности к инфекции.
- •Патогенез
- •Классификация
- •Морфология
- •Клиника
- •Аускультация. При аускультации живота наблюдается ослабление перистальтических шумов кишечника.
- •На клинические проявления ОА могут влиять: расположение ЧО, длительность заболевания, возраст, беременность, сопутствующая патология и другие факторы.
- •Осложнения
- •Хронический аппендицит
- •Острый перитонит
- •Перитонит – воспаление брюшины в результате интраабдоминального инфицирования.
- •Этиология
- •Патогенез
- •Классификация
- •2. Характер развития:
- •Морфология
- •Геморрой
- •Синонимы: геморроидальная болезнь.
- •Этиология и патогенез
- •Классификация
- •По форме выделяют: внутренний, наружный и комбинированный. По течению: острое и хроническое. Классификация хронического геморроя
- •Классификация острого тромбированного геморроя
- •Морфология
- •Клиника
- •Глава 8: Болезни системы крови
- •Болезни эритроцитарной системы
- •Анемии
- •Классификация
- •микроцитарные: СОК< 75 мкм , а СД< 7 мкм;
- •P.S. Для расчета цветового показателя можно использовать следующее мнемоническое правило:
- •Регенераторные (и гиперрегенераторные) - ИР>2%;
- •Индекс ретикулоцитов рассчитывается по формуле:
- •Анемии вследствие нарушенного кровобразования
- •А.1. Железо-дефицитные анемии (ЖДА)
- •Этиология
- •Патогенез
- •Морфология
- •Клиника
- •Метаболическая роль витамина В12 и фолатов
- •А.2. В12-дефицитные анемии и фолиево-дефицитные анемии
- •dUMP
- •Этиология
- •IV. Быстро растущие опухоли
- •V. Повышенная потребность в витамине В12 (беременность, лактация)
- •VI. Длительное жесткое вегетарианство
- •Патогенез
- •Морфология
- •Клиника
- •1. мегалобластическая анемия, как результат нарушения синтеза ДНК в эритроидных клетках костного мозга;
- •А.3. Сидеробластные анемии
- •Клиника
- •Лабораторные показатели железодефицитных и сидеробластных анемий
- •Б. Гипо-апластические анемии
- •Классификация
- •Патогенез
- •Морфология
- •На микропрепарате слева представлен препарат костного мозга больного с апластической анемией, справа микропрепарат «в норме».
- •Клиника
- •легкая утомляемость и бледность кожных покровов
- •инфекция
- •II. Гемолитические анемии
- •Морфология
- •Клиника
- •А.2. Гемоглобинопатии (гемоглобинозы)
- •β-талассемия
- •Патогенез
- •Клиника
- •α-талассемия
- •Серповидноклеточная анемия
- •Патогенез
- •хронического гемолиза;
- •На скорость развития и тяжесть заболевания влияют различные факторы:
- •Морфология
- •Глюкозо-6-фосфатдегидрогеназная недостаточность
- •Гемолиз инфекционного происхождения
- •Иммунные гемолитические анемии
- •Наиболее частыми причинами иммунного разрушения эритроцитов являются:
- •идиопатические формы, этиология которых неизвестна.
- •Гемолитическая болезнь новорожденных
- •Описание микропрепаратов:
- •1. Эритробластоз печени. Обилие эритробластов и нормобластов (тёмные клетки) в синусоидах печени при отёчной форме гемолитической болезни новорожденных (окраска гематоксилин-эозином; × 400).
- •2. Накопление гемосидерина (синего цвета) в макрофагах красной пульпы селезёнки при желтушной форме гемолитической болезни новорожденных (окраска по Перльсу; × 400).
- •3. Острое набухание, ишемические изменения нейронов (указано стрелками) в аммоновом роге головного мозга при желтушной форме гемолитической болезни новорожденных (окраска по Нисслю; × 400).
- •Механизм лекарственно-опосредованного гемолиза
- •Аутоиммунные гемолитические анемии
- •Морфология
- •Анемии вследствие кровопотери
- •Эритроцитозы и эритремии
- •Автономная продукция эритропоэтинов
- •Эритремия или истинная полицитемия (болезнь Вакеза)
- •Клиника
- •Патология белой крови
- •Этиология
- •Этиология
- •Клиника
- •Лимфоцитозы
- •Лимфоцитопении
- •Приобретенные лимфоцитопении – вторичные иммунодефициты:
- •Существуют 4 основные теории канцерогенеза: вирусная; химическая; лучевая; наследственная.
- •В своем большинстве теории канцерогенеза опираются на данные, полученных в экспериментах на животных и данные клинического наблюдения.
- •Клиника
- •Влияние ОЛЛ на организм больного
- •Принципы лечения ОЛЛ
- •Лабораторная диагностика ХЛЛ
- •Влияние ХЛЛ на организм больного
- •Иммунодефицит и аутоиммунные реакции при ХЛЛ
- •Лимфома Беркитта
- •Классификация
- •Клиника
- •Морфология
- •одна экстранодальная опухоль с поражением региональных лимфоузлов
- •две и более групп лимфоузлов по одну сторону диафрагмы
- •две одиночные экстранодальные опухоли с/без поражения региональных лимфоузлов по одну сторону диафрагмы
- •две одиночные экстранодальные опухоли по обе стороны диафрагмы
- •все первичные внутригрудные опухоли (медиастинальные, плевральные)
- •все обширные внутрибрюшные опухоли (нерезектабельные)
- •Лимфома Ходжкина
- •Морфология
- •Гистологическая классификация
- •Физикальное обследование
- •2. Экстранодулярные очаги
- •Глава 9: Неотложные состояния
- •ДВС-синдром
- •Механизмы активации и ингибиции гемостаза
- •Каскадно-матричная теория коагуляции
- •Необходимость создания новой каскадно-матричной (клеточной) теории свертывания крови назрела в результате нестыковок классической теории с данными клинических наблюдений.
- •Примечание:
- •по классической версии отсутствие факторов VIII и IX у больных гемофилиями А и В не должно приводить к кровотечениям, т.к. существует внешний путь активации тромбина. Однако на деле внешнего пути оказывается недостаточно.
- •отсутствие фактора Хагемана, инициатора внутреннего пути свертывания в классической теории, приводит к тромбофилиям, но не к кровотечениям,
- •влияние прекалликреина и высокомолекулярного кининогена (активаторов ф.XII) на свертывание крови проявляется только в исследованиях in vitro, и их недостаток у пациента выявляется случайно, т.к. клинически никак не проявляется.
- •Взаимодействие факторов свертывания плазмы и образование протромбиназы укладывается в 3 стадии:
- •Инициация.
- •Амплификация (лат. amplificatio - расширение, усиление).
- •Пропагация (лат. propagatio - разведение, распространение).
- •1 стадия – инициация
- •В каскаде коагуляции комплекс VIIа-TF-Ca2+ играет главную, пусковую роль.
- •по положительной обратной связи стимулирует образование новых порций фактора VIIa,
- •под влиянием фактора Ха начинает появляться тромбин, пока еще в небольшом количестве.
- •2 стадия – амплификация
- •3 стадия – пропагация
- •Этиология
- •*массивные гемотрансфузии и реинфузии крови; введение гемопрепаратов, содержащих активаторы факторов свертывания;
- •Первое место среди причин, вызывающих ДВС синдром, занимают генерализованные инфекции (бактериальные и вирусные), септицемии, иммунокомплексные процессы, патология беременных.
- •Патогенез
- •В рамках острого, подострого и хронического течения ДВС – синдрома согласно классификации М.С.Мачабели можно выделить четыре стадии:
- •Вторая стадия – коагулопатия потребления с подстадиями 2а и 2b.
- •Морфология
- •Клиника
- •Шоковые состояния
- •Этиология
- •Патогенез
- •Течение травматического шока различают как по времени, так и по фазе.
- •По фазе различаются:
- •В зависимости от тяжести состояния пострадавших клинически принято различать 3 степени торпидного шока, что весьма существенно для проведения сортировки пострадавших.
- •При травме таза на течение шока существенное влияние оказывают часто встречающаяся массивная кровопотеря и резкая интоксикация (повреждение подвздошных, ягодичных сосудов), а также повреждение органов малого таза.
- •Шокогенные травмы
- •Сепсис. Септический шок
- •Необходимо наличие двух и более обязательных факторов:
- •Этиология
- •Патогенез
- •В патогенезе шока при сепсисе имеют значение три основных механизма:
- •снижение периферического сосудистого тонуса
- •прогрессирующая миокардиальная дисфункция
- •Классификация
- •По типу течения сепсис может быть:
- •Анафилаксия – это серьезная, жизнеугрожающая, генерализованная или системная реакция гиперчувствительности.
- •Этиология
- •Вид триггера, наиболее часто вызывающего анафилаксию, зависит от возраста пациента. Так, в детском возрасте наиболее частая причина — пищевые продукты, у взрослых – ЛС и яд перепончатокрылых.
- •Патогенез
- •Классификация
- •III. В зависимости от характера течения АШ.
- •Клиническая картина
- •Согласно международным рекомендациям, врач должен подумать об анафилаксии:
- •б) Респираторные проявления (затруднение дыхания, одышка, кашель, заложенность носа, чихание, хрипы в груди, стридор, гипоксемия).
- •а) Взрослые: систолическое давление ниже 90 мм.рт.ст. или снижение более, чем на 30% от исходного систолического АД.
- •Эмболия
- •Легочная эмболия
- •Системная тромбоэмолия
- •Жировая и костномозговая эмболия
- •Патогенез
- •Воздушная эмболия
- •Эмболия амниотической жидкостью
- •Синдром Уотерхауза-Фридериксена (Острая надпочечниковая недостаточность)
- •Этиология
- •Причинами возникновения данного синдрома могут быть:
- •3. Тромбозы сосудов надпочечников.
- •5. Внутриутробное кровоизлияние в надпочечники во время тяжелых или осложненных родов.
- •7. Адреналэктомия.
- •Патогенез
- •Морфология
- •Клиника
- •Острое повреждение почек
- •Этиология
- •Происходящие изменения могут быть обусловлены следующими причинами:
- •Патогенез
- •Основные компоненты ишемического и нефротоксического ОПП:
- •Морфология
- •Клиника
- •Глава 10: Болезни почек
- •Почечная недостаточность
- •Острая почечная недостаточность
- •Острая почечная недостаточность возникает «внезапно» и быстро прогрессирует. Это состояние потенциально обратимо. Однако, нередко острая почечная недостаточность приводит к смерти пациентов.
- •Этиология
- •Различают преренальные, ренальные и постренальные причины острой почечной недостаточности.
- •Ренальные. Факторы этого рода оказывают прямое повреждение действия на ткань почек.
- •Постренальные. Обусловливают нарушение (вплоть до прекращения) оттока мочи по мочевыводящим путям.
- •- Обтурация мочевыводящих путей (почечными камнями, опухолью, инородными телами [например, длительно находящимися в мочеточниках катетерами], сгустком крови, воспалительным отёком).
- •Перегиб мочеточника (например, при мигрирующих почках, избыточной длине).
- •Патогенез
- •Значительное и быстро нарастающее снижение объёма клубочковой фильтрации.
- •Сужение или обтурация большого числа канальцев почек.
- •Хроническая почечная недостаточность
- •Этиология
- •Как и при острой почечной недостаточности, различают преренальные, ренальные и постренальные причины.
- •Преренальные: хронические артериальные гипертензии, медленно прогрессирующий стеноз почечных артерий, двусторонняя эмболия артерий почек.
- •Постренальные. Факторы, вызывающие длительное нарушение оттока мочи (закрывающие изнутри или сдавливающие снаружи мочевыводящие пути).
- •Патогенез
- •Уремия
- •Уремия — синдром, заключающийся в аутоинтоксикации организма продуктами метаболизма (нормального и нарушенного), «уремическими токсинами» и экзогенными соединениями, в норме выводящимися почками.
- •Этиология
- •Непосредственной причиной развития уремии является почечная недостаточность (острая или хроническая).
- •К основным факторам повреждения тканей и органов при уремии и почечной коме относятся:
- •«Уремические токсины».
- •Патогенез
- •В механизме развития наблюдаемых при уремии нарушений большое значение имеют расстройства метаболизма, отравление эндогенными токсинами, гормональные расстройства.
- •Клиника
- •Выделяют 3 формы клинического течения уремии:
- •Для второй-Б-формы характерны те же проявления, что и для второй-А-формы, но с более выраженными внутриорганными нарушениями.
- •Морфология
- •При уремии функционально страдают многие органы и системы. Различные аспекты поражений органов выглядят следующим образом:
- •Патологическая анатомия кратко
- •Проявления почечной патологии
- •Расстройства функций почек проявляются изменением параметров крови и мочи и развитием общих нефрогенных синдромов.
- •Изменения диуреза (количества выделяемой мочи).
- •Полиурия — выделение за сутки более 2000–2500 мл мочи. Причины: увеличение клубочковой фильтрации и/или уменьшение канальцевой реабсорбции.
- •Олигурия — выделение в течение суток менее 500–300 мл мочи. Обычно является следствием уменьшения фильтрации и/или увеличения реабсорбции.
- •Анурия — прекращение поступления мочи в мочевой пузырь. Как правило, это результат значительного снижения фильтрации, что может сочетаться с увеличением реабсорбции.
- •Изменения относительной плотности и состава мочи.
- •Гиперстенурия — увеличение плотности мочи выше нормы (более 1,029– 1,030). Как правило, является следствием увеличения реабсорбции.
- •Гипостенурия — снижение плотности мочи ниже нормы (менее 1,009). Наблюдается при нарушении концентрационной функции почек.
- •Изостенурия — мало меняющаяся в течение суток относительная плотность мочи. Свидетельствует об уменьшении эффективности канальцевой реабсорбции и снижения концентрационной способности почек.
- •Колебания (за пределы нормы) содержания нормальных её компонентов: глюкозы, ионов, воды, азотистых соединений.
- •Изменения ритма мочеиспускания
- •Поллакиурия — частое мочеиспускание. Причины: полиурия и/или раздражение мочевыводящих путей (при воспалении, прохождении мелких конкрементов — «песка» и др.).
- •Никтурия — преимущественное мочеиспускание ночью. Причины: нарушение кровоснабжения почек, развитие аденомы простаты, поражения почек (амилоидоз) и/или мочевыводящих путей (уретрит, цистит).
- •Показатели изменения объема и состава крови
- ••Гиперволемия (почечного генеза). Причины: снижение клубочковой фильтрации и/или увеличение канальцевой реабсорбции.
- ••Гиповолемия (почечного происхождения). Причины: как правило, это результат увеличения фильтрации и/или уменьшения реабсорбции.
- ••Азотемия (повышение уровня небелкового азота в крови). Причина:
- ••При различных заболеваниях почек могут развиваться также гипер(гипо)фосфатемия, гипер(гипо)калиемия, гипер(гипо)натриемия, гипер(гипо)кальциемия, гипер(гипо)магниемия и другие изменения содержания компонентов крови.
- •Характер отклонений определяется конкретным заболевание почек и нарушением процессов фильтрации, реабсорбции, и секреции.
- •Гломерулярные болезни
- •Острый пролиферативный (постстрептококковый) гломерулонефрит
- •Этиология и патогенез
- •Клиническая картина
- •При ОПСГН наблюдаются следующие клинические и лабораторные проявления:
- •Постинфекционный нестрептококковый гломерулонефрит
- •Быстропрогрессирующий гломерулонефрит
- •Этиология
- •Классификация
- •Патогенез
- •Морфология
- •(1–2) недель, что соответствует критериям ОПН.
- •Дифференциальная диагностика
- •Мембранозная нефропатия
- •Этиология
- •Патогенез
- •Морфология
- •Клиника
- •Нефротический синдром
- •Патофизиология
- •Причины
- •Болезнь минимальных изменений
- •Морфология
- •Клиника
- •Осложнения
- •Фокально-сегментарный гломерулосклероз
- •Этиология и классификация
- •Различают первичную (идиопатическую) и вторичную формы ФСГС
- •Патогенез
- •Морфология
- •Клиника
- •Мембранопролиферативный гломерулонефрит
- •Первичный МПГН
- •Патогенез
- •Морфология
- •Клиника
- •Морфология
- •Осложнения
- •Осложнением гломерулонефрита при его хроническом течении характерна хроническая почечная недостаточность с проявлениями уремии. Возможны сердечно-сосудистая недостаточность, кровоизлияние в мозг, которые становятся причиной смерти.
а также нарушение распределения альбумина между внутри- и внесосудистым пространствами. Снижение синтеза альбумина связывают с наличием воспаления, а также с повышенным уровнем цитокинов, таких, как тумор некротизирующий фактор альфа (ТНФα).
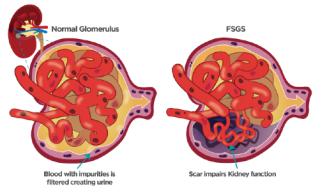
Фокально-сегментарный гломерулосклероз
Фокально-сегментарный гломерулосклероз (ФСГС) — форма гломерулопатии, для которой характерно склерозирование отдельных сегментов (cегментарные изменения) в части клубочков (фокальные изменения); остальные клубочки в начале болезни интактны. Этот морфологический тип изменений трудно отличим от болезни минимальных изменений (БМИ). Полагают, что это разной тяжести варианты или разные стадии одного и того же заболевания, объединяемые термином «идиопатический нефротический синдром»
Этиология и классификация
Различают первичную (идиопатическую) и вторичную формы ФСГС
Первичный (идиопатический) ФСГС – причины неизвестны. Вторичный ФСГС 1. Генетически обусловленный
-Мутации ACTN4, NPHS1, NPHS2, CD2AP, TRPC6, WT-1 и др.
-Связанный с другими наследственными заболеваниями (с-м Альпорта, Дауна, митохондриальные цитопатии, болезнь Шарко–Мари–Туф)
2.Ассоциированный с вирусами (HIV, парвовирус В19, цитомегаловирусы, вирус Эпштейн–Барр, вирус Коксаки и др.)
3.Индуцированные лекарствами (героин, интерферон-α, адриамицин, доксорубицин, литий, анаболические стероиды, памидронат и др.)
4.Адаптивные структурно-функциональные изменения, вызванные гипертрофией клубочков или гиперфильтрацией. При уменьшении массы почечной ткани (олигомеганефрония, односторонняя агенезия, дисплазия почечной ткани, кортикальный некроз, рефлюкс-нефропатия, нефрэктомия, хроническая трансплантационная нефропатия, низкая масса тела при рождении, поздняя стадия любого заболевания почек со снижением массы действующих нефронов и др.). При изначально нормальном числе нефронов (артериальная гипертензия, сахарный диабет, ожирение, врожденные «синие» пороки сердца, серповидно-клеточная анемия)
5.Злокачественные новообразования (лимфома и др.)
6.Неспецифические ФСГС-подобные изменения, вызванные сморщиванием почек при гломерулярных заболеваниях (очаговый пролиферативный ГН, пурпура Шенлейна–Геноха, мембранозная нефропатия, волчаночный нефрит, тромботические микроангиопатии)
Патогенез
При ФСГС выявляется поражение подоцитов различной этиологии, в связи с чем это заболевание относят к группе «подоцитопатий».
Основную роль в развитии ФСГС отводят повреждению подоцитов вследствие молекулярного генетического дефекта, воздействия циркулирующих факторов проницаемости или внешних повреждающих агентов. - Структурные нарушения подоцитов, дезор-
429
ганизация их актинового цитоскелета, сглаживание ножек, слияние фильтрационных щелей приводят к развитию протеинурии (ПУ). При длительном и/или выраженном воздействии повреждающего фактора активируются механизмы апоптоза, подоциты погибают, теряют связь с гломерулярной базальной мембраной (ГБМ), слущиваются в мочевое пространство, оголяя в этих местах участки ГБМ. Имея высокие адгезивные свойства, оголенная ГБМ формирует синехии с капсулой Боумена. В местах «сращивания» с ГБМ и мезангиуме образуются очаги фиброза. В зонах фокально-сегментарного склероза фильтрация меняет свое направление в сторону интерстиция, окружающего клубочек. В итоге образуются глобальный гломерулосклероз и интерстициальный фиброз. Кроме того, подоциты в ходе повреждения подвергаются трансдифференциации, приобретают свойства фибробластов и участвуют в синтезе экстрацеллюлярного матрикса, ускоряя формирование очагов фиброза
-В качестве факторов проницаемости рассматривают кардиотропиноподобный цитокин-1 (из семейства интерлейкина-6), растворимый рецептор к урокиназе, гемопексин и др. При ФСГС и БМИ активность циркулирующих факторов проницаемости зависит от баланса между продукцией этих факторов (в результате Т-клеточной дисрегуляции) и потерей с мочой их ингибиторов (предположительно липопротеидов высокой плотности). Мишенью факторов проницаемости могут являться белки щелевидной диафрагмы подоцитов (подоцин, нефрин, СD2AP и др.), участвующие в поддержании структуры и селективности гломерулярного фильтра . При вирус-индуци- рованном ФСГС допускают прямое повреждающее действие вируса на подоциты или через освобождение воспалительных цитокинов, взаимодействующих с подоцитарными рецепторами.
-В повреждении подоцитов при вторичном ФСГС, связанном с уменьшением массы почек, ре- флюкс-нефропатией, ожирением, важную роль играют гемодинамические механизмы – адаптивная внутриклубочковая гипертензия и гиперфильтрация с увеличением объема клубочков, ведущие к повышению механической нагрузки на подоциты. Гиперпродукция ангиотензина II и усиление синтеза TGF-β вызывают активацию апоптоза, реорганизацию цитоскелета и дедифференциацию подоцитов.
Изучение генетических основ некоторых вариантов ФСГС и других причин нефротического синдрома по зволило расширить представления о патогенезе протеинурии и разработать новые методы диагностики и прогнозирования исхода у пациентов с ФСГС. Первым был идентифицирован ген NPHS1, который расположен на хромосоме 19ql3 и кодирует белок нефрин. Некоторые мутации гена NPHS1 приводят к развитию врожденного нефротического синдрома финского типа, проявляющегося гломерулопатией, подобной болезни минимальных изменений, с выраженным сглаживанием отростков подоцитов. Анализ гена NPHS1 позволяет пренатально диагностировать врожденный нефротический синдром.
В результате мутаций гена NPHS2, который расположен на хромосоме Iq25-q31 и кодирует синтез белка подоцита, локализующегося в щелевой диафрагме, развивается аутосомно-рецессив- ный ФСГС. Мутации NPHS2 в ~ 30% случаев обусловливают развитие у детей резистентного к кортикостероидной терапии нефротического синдрома. У больных детей обычно наблюдаются признаки ФСГС. Мутации гена, кодирующего актинсвязывающий белок а-актинин 4, обусловливают развитие аутосомно-доминантного ФСГС, протекающего вначале без выраженных клинических симптомов, но способного быстро приводить к почечной недостаточности.
Мутации гена, кодирующего TRPC6, были обнаружены у некоторых взрослых единокровных родственников, заболевших ФСГС. TRPC6 секретируется многими клетками, в т.ч. подоцитами. Патогенные мутации нарушают функцию подоцитов из-за повышения поступления кальция в эти клетки. Объединяет все перечисленные белки то, что все они расположены в щелевой диафрагме и цитоскелете соседних подоцитов. Их специфические функции и механизмы взаимодействия изучены не полностью, но ясно, что целостность каждого из них необходима для обеспечения нормального функционирования фильтрационного барьера клубочка.
430
Данная книга в списке рекомендаций к покупке и прочтению форума сайта https://meduniver.com/
Исследования на мышиной модели позволили установить роль еще одного белка —СD2-ассоци- ированного белка (CD2AP), который редко обнаруживается у человека и может быть связан с развитием протеинурии. До момента идентификации этих генетических нарушений патогенез некоторых случаев идиопатического нефротического синдрома объясняли другими факторами, приводящими к нарушению проницаемости: взаимодействиями между клетками и между клетками и матриксом, особенно теми, которые опосредованы осфгинтегринами, дистрогликанами и лиминами. Нарушение этих взаимодействий может привести к потере адгезии подоцита к БМК. Аблациониая нефропатия —вторичный ФСГС, развивающийся как осложнение гломерулярных и негломерулярных заболеваний, которые характеризуются снижением массы функционирующей почечной ткани (особенно при рефлюксной нефропатии и односторонней агенезии почки) и могут приводить к гломерулосклерозу и почечной недостаточности.
Морфология
При световой микроскопии выявляют фокальные сегментарные поражения, затрагивающие лишь небольшое количество клубочков. При недостаточном объеме биоптата такие поражения диагностировать трудно. Сначала поражаются клубочки юкстамедуллярной зоны, затем изменения приобретают генерализованный характер. В склерозированных сегментах происходят коллапс петель капилляров, накопление матрикса и сегментарное отложение белков плазмы вдоль стенки капилляра (гиалиноз). Гиалиноз может быть настолько выраженным, что происходит окклюзия просвета капилляра. Часто обнаруживают жировые капли и пенистые клетки. Клубочки, которые не имеют сегментарных поражений, при световой микроскопии обычно выглядят неизмененными, однако возможно накопление мезангиального матрикса.
При электронной микроскопии выявляют диффузное сглаживание малых отростков подоцитов (в склерозированных и в несклерозированных областях клубочка). При этом происходит очаговое отслоение эпителиальных клеток и оголение базальной мембраны. При иммунофлуоресцентном исследовании в очагах склероза и/или мезангии можно выявить IgМ и компонент системы комплемента СЗ. Помимо фокального сегментарного гломерулосклероза возможны выраженный гиалиноз и утолщение стенок приносящих артериол. При прогрессировании заболевания увеличиваются количество склерозированных клубочков и площадь повреждения. Со временем это приводит к тотальному склерозу клубочков с выраженной атрофией канальцев и интерстициальным фиброзом.
Коллаптоидная гломерулопатия (один из морфологических вариантов ФСГС) характеризуется сморщиванием всего клубочка в сочетании с различными типами ФСГС или без них. Типичными для коллаптоидной гломерулопатии являются пролиферация и гипертрофия висцеральных эпителиальных клеток клубочка. Повреждение может быть идиопатическим, однако наиболее ха-
431
рактерно для ВИЧ-ассоциированной нефропатии. В обоих случаях гломерулопатия сопровождается выраженными повреждениями канальцев с формированием микрокист. Прогноз при таком ФСГС чрезвычайно неблагоприятный
Клиника
•НС развивается более чем у 70% больных, персистирующую ПУ без НС имеют <30% пациентов
•У большинства больных НС и ПУ сочетаются с микрогематурией, макрогематурия редка;
•Более чем у 50% больных наблюдается артериальная гипертензия (АГ);
•У 25–50% больных уже в дебюте заболевания отмечается ПН;
•При генетических вариантах ФСГС в начале проявлений болезни ПУ часто носит субнефротический характер;
•Развитие вторичной стероидрезистентности у детей в большинстве случаев может быть связано с трансформацией минимальных изменений в ФСГС;
•При ряде вторичных форм ФСГС отмечают клинические и лабораторные признаки заболевания, вызвавшего поражение почек;
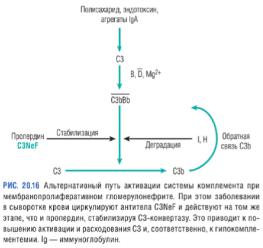
Мембранопролиферативный гломерулонефрит
Мембранопролиферативный (мезангиокапиллярный) гломерулонефрит характеризуется пролиферацией мезангиальных клеток и диффузным утолщением и удвоением базальной мембраны капилляров клубочка. Морфологическая картина при МПГН характеризуется повреждением базальной мембраны, пролиферацией клеток клубочка и лейкоцитарной инфильтрацией. Поскольку пролиферирует преимущественно мезангий и вовлекаются в патологический процесс петли капилляров, данное заболевание часто называют мезаигиокапиллярным гломерулонефритом. МПГН обусловливает 10-20% случаев нефротического синдрома у детей и взрослых. У некоторых пациентов заболевание проявляется только гематурией или незначительной протеинурией, у других наблюдается смешанная картина: нефротический и нефритический синдромы. Как и многие другие гломерулонефриты, МПГН может быть первичным (идиопатическим) или вторичным, обусловленным системными заболеваниями либо другими известными этиологическими агентами.
Первичный МПГН
Первичный МПГН подразделяют на 2 типа в зависимости от патоморфологических и ультраструктурных особенностей и характеристик при иммунофлуоресцентном исследовании: МПГН типа I и МПГН типа II (болезнь плотного осадка).
Патогенез
В большинстве наблюдений МПГН типа I выявляют иммунные комплексы в клубочке и классический и альтернативный пути активации системы комплемента. Антигены, участвующие в патогенезе первичного МПГН, неизвестны. Во многих случаях это могут быть белковые дериваты вируса гепатита В или С. Белки, вероятно, либо осаждаются после контакта с антигенами при первой контаминации, либо прямо захватываются клубочковыми структурами, либо содержатся в осаждаемых циркулирующих иммунных комплексах. Большинство пациентов с МПГН типа II имеют изменения, которые свидетельствуют об альтернативном
432
Данная книга в списке рекомендаций к покупке и прочтению форума сайта https://meduniver.com/