

МИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ УКРАИНЫ
ТАВРИЧЕСКИЙ НАЦИОНАЛЬНЫЙ УНИВЕРСИТЕТ
им. В.И.ВЕРНАДСКОГО
Кафедра физической и аналитической химии
МЕТОДИЧЕСКИЕ УКАЗАНИЯ
ПО ТИТРИМЕТРИИ / окислительно-восстановительное и комплексонометрическое титрование /
для студентов 2 курса дневной формы обучения 6.07.0300 «химия»
образовательно-квалификационного уровня « бакалавр »
СИМФЕРОПОЛЬ 2003
Печатается по решению научно-методического совета
Таврического Национального Университета от
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ ТИТРОВАНИЕ.
Методы окислительно-восстановительного титрования основаны на использовании реакций, связанных с переносом электронов. Название методам даётся по используемому титрованному раствору реагента, например: перманганатометрия, дихроматометрия, Йодометрия и т.д. Реагенты: KMnO4, K2Cr2O7, I2 и т.д.
ПЕРМАНГАНАТОМЕТРИЯ.
Перманганат-ион является одним из сильнейших окислителей. В зависимости от среды, восстановление MnO4--- протекает по-разному.
В сильнокислой среде протекает полуреакция:
MnO4--- + 8H+ + 5e- ↔ Mn2+ + 4H2O; EoMnO4--- / Mn2+ =1,51В (fэ=1/5)
В нейтральной и близких к ней средах восстановление идет до MnO2:
MnO4--- + 2H2O + 3e- ↔ MnO2 + 4OH---; EoMnO4--- /MnO2 =0,588В (fэ=1/3)
В анализе некоторых органических соединений используется восстановление в сильнощелочной среде по уравнению:
MnO4--- + e- ↔ MnO42--; EoMnO4--- / MnO42--- =0,56В.
В титриметрическом анализе определение восстановителей проводится в кислых средах, при [H3O+] = 1-2 моль/л. Индикаторы, как правило, не применяются, т.к. реагент сам является весьма чувствительным индикатором, и лишняя капля титранта окрашивает раствор в бледно-розовый цвет. Окраска раствора в к.т.т. неустойчива, раствор постепенно обесцвечивается вследствие реакции:
2MnO4--- + 3Mn2+ + 2H2O ↔ 5MnO2 + 4H+
Это надо иметь в виду во избежание перетитрования раствора.
KMnO4 легко восстанавливается органическими веществами (бумага, пыль, резиновые пробки) до MnO2, поэтому твёрдый KMnO4 и его растворы содержат примесь MnO2, каталитически ускоряющего процесс разложения:
4KMnO4 + 2H2O 4MnO2 + 4КOH + 3О2
Такое же взаимодействие оказывает прямой солнечный свет. Поэтому готовят раствор примерной концентрации, выдерживают в тёмной склянке в течение 7-10 дней, затем отфильтровывают через стеклянный фильтр от образовавшегося MnO2 и устанавливают титр KMnO4 по Н2С2O4∙2H2O или по Na2C2O4; металлическому Fe; As2O3, (NH4)2Fe(SO4)2∙6H2O методом пипетирования или отдельных навесок. Для создания среды используют раствор H2SO4 , растворы HCl и HNO3 не используют, т.к. Cl—- ион окисляется перманганатом, а HNO3 сама является сильным окислителем.
Практическое применение перманганатометрии многообразно: определение восстановителей (Fe(II), нитриты, пероксид водорода и другие соединения); окислителей – методом обратного титрования или титрования по остатку (MnO2, PbO2) и других веществ, в том числе органических соединений.
К недостаткам этого метода относятся: невозможность приготовления стандартного раствора титранта по точной навеске, нестабильность при хранении, необходимость строгого соблюдения условий проведения титрования.
К достоинствам относятся: титрование без постороннего индикатора, в широком диапазоне pH; высокое значение EoMnO4---, H+ /Mn2+; перманганат калия доступен и сравнительно не дорог.
Работа 1. Стандартизация раствора перманганата калия по щавелевой кислоте методом отдельных навесок
Взаимодействие протекает по схеме:
2MnO4--- + 5H2C2O4 + 6H+ → 2Mn2+ + 10CO2 + 8H2O
Данная реакция протекает сложно, в несколько стадий; для того чтобы она началась, необходимо присутствие в растворе хотя бы следов Mn2+:
MnO4--- + MnC2O4 → MnO42-- + MnC2O4+
Далее происходят процессы:
Mn (VI) + Mn (II) → Mn (IV)
Mn (IV) + Mn (II) → Mn (III)
Марганец(III) образует оксалатные комплексы состава Mn(C2O4)(3-2n)+, где n = 1,2,3, которые медленно распадаются с образованием Mn(II) и CO2. Таким образом, реакция катализируется ионами Mn(II). Первые капли KMnO4 даже в горячем растворе обесцвечиваются очень медленно. По мере возрастания концентрации Mn(II) скорость реакции увеличивается.
Описание работы в лабораторном журнале сделать по следующему плану:
1.Уравнение реакции, обоснование направленности (с указанием промежуточных стадий).
2.Обоснование условий титрования (pH, t, Ind).
3.Расчет величины навески и взятие трёх навесок.
4.Описание методики титрования.
5.Расчет результатов титрования: Н, Т KMnO4 и статистическая обработка результатов титрования.
6.Вывод.
Реагенты:
KMnO4 ~ 0,05н раствор4 ;
Н2С2О4∙2Н2О, кристаллическая;
H2SO4, 2н раствор.
Методика. На аналитических весах берут три навески щавелевой кислоты, близкие по массе к теоретической и растворяют в колбах для титрования в произвольном объёме воды, удобном для титрования. Разработаны две методики стандартизации KMnO4.
1. К раствору Н2С2O4 добавляют 15-20 мл 2н H2SO4, нагревают до 60оС (Не кипятить! Это приводит к разложению щавелевой кислоты:
Н2С2O4 CO2↑+ CO↑ + 2H2O),
и титруют раствором KMnO4 из бюретки, сначала медленно, добиваясь обесцвечивания каждой капли титранта, затем, по мере образования Mn2+, быстрее. Титрование считается законченным, когда от одной капли KMnO4 раствор окрасится в бледно-розовый цвет, устойчивый 20-30 с.
Результаты занести в таблицу:
q экспер.. |
V KMnO4 |
H KMnO4 |
H E |
T KMnO4 |
|
|
|
|
|
и обработать с использованием методов математической статистики. Вывод.
2. К приготовленному раствору Н2С2O4 добавляют 15-20 мл 2н H2SO4, прибавляют из бюретки ~ 90-95% раствора KMnO4, нагревают до 60оС и заканчивают титрование, добавляя титрант до получения устойчивой розовой окраски, не исчезающей 30с.
Работа 2. Определение железа (II) в растворимых солях.
Определение железа (II) основано на реакции:
5Fe2++MnO4--- +8H+→5Fe3++Mn2++ 4H2O (I)
Титрование проводят в сильнокислой среде. Присутствие Cl --- приводит к перерасходу перманганата и получению нечёткого конца титрования. Связано это с тем, что реакция (I) индуцирует реакцию между MnO4--- и Cl ---. Индуцированной реакции не возникает при достаточных концентрациях Mn(II). Перед титрованием хлорида железа (II) в раствор добавляют смесь Рейнгарда-Циммермана, состоящую из H2SO4, MnSO4 и H3PO4. Присутствие H2SO4 обеспечивает нужный pH среды, MnSO4 предотвращает окисление Cl ---; H3PO4 связывает образующиеся Fe3+ в бесцветный комплекс (иначе хлоридные комплексы Fe3+ затрудняют определение к.т.т.). Если надо определить содержание Fe(III) в солях, то оно предварительно восстанавливается металлическим цинком (см. работу по дихроматометрии).
План работы.
Записать уравнения реакции, объяснить направленность.
Рассчитать и построить кривую титрования при pH = 0 и pH = 4.
Обосновать условия титрования (рН; влияние мешающих ионов; использование смеси Рейнгарда-Циммермана).
Методы восстановления Fe(III) Fe(II).
Титрование. Расчёт Н и Т раствора соли Fe(II). Статистическая обработка результатов.
Контрольная экспериментальная работа по определению массы (объёма) соли Fe(II). Расчёт и статистическая обработка результатов.
Реагенты:
KMnO4, 0,05М стандартный раствор;
сульфат железа (II) FeSO4/ раствор или кристаллогидрат/.
H2SO4, 2Н раствор.
Методика. Из исходного раствора соли железа (II) с концентрацией ~1,0 – 0,5М приготовить в мерной колбе на 100мл. раствор с концентрацией ~0,05М. Методом пипетирования установить точную концентрацию исходного раствора. При этом каждый раз в колбу для пипетирования добавлять 10мл. 2Н H2SO4 и 50мл. дистиллята. Титровать до бледно-розовой окраски, устойчивой 30с.
Если исходный раствор содержит Fe(III), то его предварительно надо восстановить. Для этого в колбу для титрования добавить 1-2 гранулы металлического цинка, аккуратно подогревая до обесцвечивания раствора. Полноту восстановления можно контролировать тиоцианатной реакцией на Fe3+. Затем пинцетом достать остаток Zn, ополаскивая над колбой дистиллятом, охладить, добавить 10мл. 2Н H2SO4 и титровать. При необходимости добавить 3-5мл. H3PO4 1:10. Определение провести 3-4 раза. Результаты занести в таблицу и сделать расчёты. Затем аналогично провести контрольную работу и определить V(m) выданного образца Fe(II) ±E .
ДИХРОМАТОМЕТРИЯ.
Данный метод определения веществ основан на реакции их окисления дихромат-ионами. Раствор K2Cr2O7 можно приготовить по точной навеске, т. к. он удовлетворяет всем требованиям к первичному стандарту.
В кислой среде дихромат является сильным окислителем:
Cr2O72- + 14H+ + 6е 2Cr3+ + 7H2O; EoCr2O72-- / Cr3+ =1,33В.
Кислую среду можно создавать, используя не только серную кислоту, но и соляную, фосфорную. При CHCl < моль/л ионы Cl --- не окисляются дихромат-ионами.
Прямым титрованием можно определить ряд восстановителей: железо(II), включая [Fe(CN)6]4 ‾; сульфиты; иодиды; арсениты; аскорбиновую кислоту; метанол и т.д. Методом обратного титрования определяют некоторые окислители: нитраты, хлориты и др.
Работа 1. Определение железа (II)
Определение основано на реакции:
6Fe2+ + Cr2O72-- + 14H+ → 6Fe3+ + 2Cr3+ + 7H2О;
В процессе титрования концентрация Fe3+ повышается, следовательно, возрастает EoFe3+ /Fe 2+ , что приводит к преждевременному изменению цвета дифениламина (Ео = 0,76В.). Поэтому вводят H3PO4, которая связывает Fe3+ в устойчивый бесцветный комплекс, тем самым, понижая потенциал пары Fe3+/Fe2+.
Растворы Fe(II) часто содержат Fe(III), поэтому перед титрованием проводят их восстановление до Fe(II). Восстановление можно проводить с помощью SnCl2; Zn метал. и т.д. В первом случае происходит реакция:
2[FeCl4] -- + [SnCl4]2-- 2Fe2+ + [SnCl6]2-- + 6Cl ---
Скорость реакции возрастает при нагревании в присутствии конц. HCl, которая создаёт нужную среду и образует комплексы с Fe(III), интенсивно окрашенные в жёлтый цвет и могут служить индикатором.
Избытка восстановителя нужно избегать, небольшие оставшиеся количества окисляют с помощью HgCl2:
[SnCl4]2-- + HgCl2 [SnCl6]2-- + Hg2Cl2
Реагенты:
K2Cr2O7, стандартный раствор (0,05н);
HCl, конц.;
H2SO4, конц.;
SnCl2, раствор;
HgCl2, раствор;
H3PO4, конц. раствор;
дифениламин, 1% раствор в конц. H2SO4;
План работы.
Записать уравнения реакций, лежащие в основе определения Fe(II).
Рассчитать кривую титрования Fe(II) – Cr2O72- - H+ при условиях: рН = 2; С реагирующих веществ = 0,05моль-экв./л.
Обоснование выбора индикатора.
Титрование. Занесение результатов в таблицу.
Рассчитать Н ±E , и Т раствора соли Fe(II).
Методика. Приготовить 0,05н раствор K2Cr2O7 по точной навеске. Рассчитать точно Н и Т приготовленного раствора.
В колбу для титрования ёмкостью 100мл. пипеткой внести 10мл исследуемого раствора, добавить 5мл. HCl конц., нагреть почти до кипения и к горячему раствору добавить 1–2 капли SnCl2 до обесцвечивания (избыток восстановителя не добавлять!).
Раствор охладить, добавить 2мл. H2SO4 конц., 3мл. раствора HgCl2; 4-5мл. H3PO4 конц.; 15мл. H2O; 2 капли (С6H5)2NH и медленно титровать дихроматом при сильном перемешивании (особенно в конце) до появления сине-фиолетовой окраски.
Восстановление металлическим цинком.
Реагенты:
K2Cr2O7, стандартный раствор (0,05н);
HCl, конц.;
H2SO4, конц.;
H3PO4, конц.;
Zn металлический, гранулы;
Дифениламин, 1% в H2SO4, конц.
Методика. В коническую колбу для титрования отобрать пипеткой 10мл. исследуемого раствора, добавить 5мл. HCl конц., внести 3 – 4 гранулы металлического цинка, сразу закрыть маленькой воронкой и нагреть на песочной бане (реакция не должна идти очень бурно) до обесцвечивания раствора и растворения цинка (не прореагировавший цинк пинцетом убрать и последний сполоснуть Н2О).
Охладить под струёй воды, добавить 3-4мл. H2SO4 конц., вторично охладить, добавить 5мл. H3PO4 конц., 15мл. Н2О; 2 капли индикатора и медленно титровать дихроматом калия до появления синей или грязно-синей окраски раствора.
Расхождение между результатами титрования не должно превышать 0,1мл.
Работа 2. Определение железа в рудах.
Реагенты:
K2Cr2O7, стандартный раствор (0,05н);
HCl, конц.; 1:4; 0,05н;
HNO3, конц. ( = 1,17);
H2SO4, конц. ( = 1,84);
H3PO4, конц. ( = 1,70);
Zn металлический, гранулы;
Дифениламин, 1% в H2SO4, конц.
Методика. Точную навеску анализируемой руды (0,7-1г.) поместить в химический стакан ёмкостью 300мл., добавить 40мл. HCl (1:4). Закрыть часовым стеклом и растворить при нагревании на песочной бане. Затем добавить 2-5мл. HNO3 и продолжать нагревание. Растворение считается законченным, когда на дне стакана остаётся серовато-белый осадок SiO2nН2О. Раствор выпарить досуха, остаток обработать 10мл. HCl конц. и снова выпарить. Повторить 2 раза, затем добавить 4мл. HCl конц. и разбавить горячей водой до 50мл. Раствор отфильтровать через фильтр «белая лента» в мерную колбу ёмкостью 200мл. Хорошо все промыть горячей 0,05М HCl для количественного перенесения ионов железа, охладить, и при перемешивании довести до метки.
Далее для определения железа поступить как в предыдущей задаче (п.б).
Раствор оставить для определения ионов железа в комплексонометрии.
Иодометрия.
Этот метод основан на реакции:
I3-- + 2e 3I--; Eo I3-/ 3I- =0,545В.
Иод является окислителем средней силы, поэтому данную систему используют как для определения как окислителей, так и восстановителей.
Методы, основанные на прямом окислении веществ раствором иода, иногда называют иодиметрическими, а методы, в которых окисляется иодид с последующим титрованием выделившегося иода – иодометрическими.
Индикатором в иодометрии служит 1% свежеприготовленный раствор крахмала, который образует с иодом соединение синего цвета комплексно-адсорбционного типа. Чувствительность крахмала высока, но резко падает с повышением температуры. Крахмал следует добавлять в конце титрования, иначе он даёт настолько прочные соединения с иодом, что идет перерасход титранта Na2S2O3.
Работа 1. Стандартизация раствора Na2S2O3 по дихромату калия.
Прямое титрование тиосульфата дихроматом провести нельзя, т.к. реакция с сильными окислителями идет нестехиометрично. Поэтому используется метод замещения. Сначала проводят реакцию между дихроматом и иодидом:
Cr2O72- + 6I-- + 14H+ 2Cr3+ + 7H2O + 3I2 (1)
Выделившийся йод в количестве, эквивалентном дихромату, оттитровывают тиосульфатом натрия:
I2 + 2S2O32- 2I-- + S4O62- (2)
Для протекания реакции (1) необходима сравнительно высокая концентрация Н+ (для повышения потенциала системы Cr2O72-- /2Cr3+), большой избыток иодида (для растворения I2 и понижения потенциала системы I3-/ 3I-). Так как скорость реакции (1) невелика, нужно некоторое время.
Реакция (2) протекает гораздо быстрее, чем
S2O32- + 2H+ H2S2O3 H2O + SO2 + S,
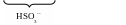
Все же перед титрованием иода нужно понизить концентрацию Н+ разбавлением.
План работы.
Записать уравнения реакций, обосновать направленность.
Обосновать условия реакций (1) и (2).
Методика работы.
Титрование. Расчёт и обработка результатов. Все результаты должны быть занесены в таблицу.
Реагенты:
K2Cr2O7, кристаллический, х.ч.;
Na2S2O3, 0,05н раствор;
H2SO4, 2н раствор;
KI, 10% раствор;
крахмал, 1% свежеприготовленный раствор.
Методика. Работу можно выполнить методом отдельных навесок или методом пипетирования. В первом случае рассчитывают величину теоретической навески K2Cr2O7, чтобы на её титрование шло ~ 15мл. титранта и берут три экспериментальные навески, близкие по массе к теоретической.
В коническую колбу для титрования ёмкостью 200-250мл. внести с помощью мерного цилиндра 15-20мл. 2н H2SO4, 10мл. 10% KI (раствор должен оставаться бесцветным) и количественно перенести навеску K2Cr2O7.
Перемешать и поставить в тёмное место на 3-5мин., прикрыв часовым стеклом. Затем добавить 100мл. дистиллята и быстро оттитровать раствором тиосульфата до бледно-желтой окраски; добавить 1-2мл. 1% крахмала и дотитровать до исчезновения синей окраски. В к.т.т. окраска бледно-зелёная (Cr3+). Повторить три раза и сделать расчёты.
Работа 2. Определение меди (II).
Иодометрическое определение меди основано на реакциях:
2Cu2+ + 4I-- 2CuI + I2 (1)
I2 + S2O32- 2I-- + S4O62- (2) Eo Cu2+/ CuI =0,86B.
Для протекания реакции (1) в растворе надо создать слабокислую среду (рН~2-4) для предотвращения гидролиза Cu2+ и таким путём повысить потенциал системы Cu2+/ CuI; и брать большой избыток иодида (для понижения потенциала системы I2 / 2I-- и растворения иода).
Для подкисления не следует применять HCl, т.к. образование хлоридных комплексов меди (II) затрудняет восстановление Cu(II); используют растворы H2SO4 или CH3COOH. К снижению результатов приводит адсорбция I3— осадком CuI. Уменьшить влияние этого фактора можно, вводя KSCN, который реагирует с CuI:
CuI(тв.) + SCN-- = CuSCN(тв.) + I-
Растворимость тиоцианата меди (I) более, чем в 10 раз меньше растворимости CuI, что обеспечивает протекание реакции, по крайней мере, на поверхности осадка (CuSCN не адсорбирует I3--).
План работы.
Записать уравнения реакций, обосновать их направленность.
Обосновать условия титрования.
Методика определения.
Титрование. Расчёт Н ±E , и Т соли меди (II).
Решение контрольной экспериментальной задачи. Расчёт V контр. Cu2+ ,±E и определения.
Реагенты:
Na2S2O3, 0,05н, стандартный раствор;
H2SO4, 2н раствор;
KI, 10% раствор;
крахмал, 1% свежеприготовленный раствор;
CuSO4, исходный раствор, ~0,5М.
Методика. В мерной колбе на 100мл. приготовить ~0,05М раствор из исходного раствора CuSO4 . В колбу для титрования внести аликвоту приготовленного раствора соли меди (II), 2-4мл. 2н Н2SO4, 10-15 мл. 10% KI, прикрыть и поставить в тёмное место на 5 минут. Затем титровать раствором Na2S2O3 до жёлтой окраски суспензии, добавить 1мл. 1% крахмала и дотитровать при тщательном перемешивании до белой (телесной) окраски суспензии. Сделать расчёты.
Аналогично провести титрование контрольной задачи и определить выданный исходный объём CuSO4 или массу CuSO4.
Работа 2. Определение сахаров.
Методика основана на восстановлении сахарами Cu(II) до Си(I) из тартратного комплекса:
C6H12O6 + 2K2Cu(C4H4O6)2 + 5KOH = C6H11O7K + Cu2O + 4K2C4H4O6 + 3H2O
Соль меди и тартрат берут в 2-3 кратном избытке по отношению к глюкозе. После реакции, избыток ионов меди восстанавливают иодидом в кислой среде и выделившийся иод титруют тиосульфатом натрия.
2Cu2+ + 5I-- 2CuI + I3--
Обязательно проводят контрольный опыт для определения концентрации меди (II) в исходном растворе. Количество меди, пошедшее на реакцию с сахарами находят по разности.
Реагенты:
CuSO45H2O, 0,04М (10г. / 1л. раствора);
K2C4H4O6 (115г K2C4H4O6 растворяют в воде, прибавляют 40г NaOH, разбавляют водой до 1л.);
KI, 5% раствор;
Na2S2O3, 0,05М стандартный раствор;
H2SO4, 1М раствор;
крахмал, 1% свежеприготовленный раствор.
Методика. Из анализируемого раствора, содержащего 50-100мл. глюкозы, приготовить в мерной колбе на 100мл раствор. Пипеткой отобрать 10,00мл. приготовленного раствора в колбу для титрования, добавить точно 10,00мл. раствора CuSO4, мензуркой 3мл раствора тартрата калия и хорошо перемешать. Образовавшийся тёмно-синий раствор нагреть и прокипятить 2-3мин. на плитке. При этом выделяется жёлтый осадок, переходящий в красный.
Раствор хорошо охладить, добавить 20мл. KI, 10мл. раствора H2SO4 и немедленно титровать желтоватую суспензию раствором тиосульфата натрия до бледно-жёлтой окраски. Ввести 3-5 капель раствора крахмала и титровать при перемешивании до исчезновения синей окраски(V1).
Аналогично провести контрольный опыт. Для этого в колбу для титрования ввести точно 10мл. раствора CuSO4, 3мл раствора тартрата калия (мензуркой), 20мл 5% KI и 10мл 1М H2SO4. Полученный раствор оттитровать раствором Na2S2O3 (аналогично).
Массу глюкозы (г.) рассчитывают по формуле:

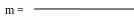 (V2
– V1)CMVk
(V2
– V1)CMVk
где 2V01000
V0 – объём анализируемого раствора;
V2 – объём Na2S2O3, пошедший в контрольном опыте;
V1 – объём Na2S2O3, на титрование сахара;
C – молярная концентрация Na2S2O3;
M – молярная масса сахара (180,0 для глюкозы);
Vk – объём мерной колбы, из которой отбирали аликвотную часть.
КОНТРОЛЬНЫЕ ВОПРОСЫ И ЗАДАНИЯ ПО ТЕМЕ «ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ ТИТРОВАНИЕ».
Требования к ОВР, используемым в титриметрии.
Перечислить основные ОВ методы объёмного анализа. Какие рабочие растворы и индикаторы применяются в каждом из этих методов?
В каких координатах строятся кривые титрования? От каких факторов зависит величина скачка титрования? В каких случаях кривая титрования симметрична, ассиметрична относительно точки эквивалентности? Рассмотреть влияние рН, ионной силы, конкурирующих реакций комплексообразования, осаждения на форму кривых.
Фиксирование точки эквивалентности. ОВ индикаторы, их характеристика. Интервал перехода и показатель титрования. Ошибка титрования.
Предварительное окисление и восстановление. Назвать окислители и восстановители, применяемые с этой целью:
газообразные.
гомогенные.
твёрдые.
Перманганатометрия:
Приготовление, устойчивость, хранение раствора KMnO4.
Стандартизация раствора KMnO4. Первичные стандарты.
Определение Fe(II), Mn(II), H2O2, NO2--, MnO2.
Иодометрия:
Характеристика окислительно-восстановительной способности пары иод – иодид-ион.
Характеристика стандартных и рабочих растворов метода (I2, Na2S2O3).
Фиксирование т.э.
Характеристика условий определения окислителей и восстановителей.
Определение Fe(III), Cu(II), H2O2, NO2--, , арсенитов и арсенатов, кислот, Br2 и Cl2, Cr(VI) и Cr(III), воды и функциональных групп органических соединений.
Бихроматометрия:
Характеристика метода. Сравнение достоинств и недостатков метода по сравнению с другими ОВ методами.
Условия; индикаторы метода.
Примеры применения бихроматометрии для определения восстановителей (прямого и косвенного) и окислителей.
Условия определения Fe(II), Fe(III). Рассчитать ошибку титрования Fe(II) в отсутствии и присутствии фосфорной кислоты.
Другие ОВ методы титрования (броматометрия, цериметрия, ванадатометрия и др.).