

Тема 8. Химическая идентификация
АНАЛИТИЧЕСКАЯ ХИМИЯ - наука о методах определения химического состава веществ. Вопросы, которые решает аналитическая химия, можно свести к следующим: что представляет собой данное вещество, из каких компонентов оно состоит, каковы их количество и распределение? Чтобы ответить на эти вопросы, проводят самые разнообразные химические реакции, применяют широкий спектр химических, физических, физико-химических, биологических методов, разрабатывают новые методы анализа и совершенствуют уже существующие. Число методов аналитической химии чрезвычайно велико и постоянно растет.
ОСНОВНЫЕ ПОЛОЖЕНИЯ
Стадии анализа.
Решение аналитических задач включает несколько стадий.
Постановка задачи. Предположим, нужно определить количество ртути в водоеме. А что именно подразумевается под словом «ртуть»? Это может быть вся ртуть, независимо от конкретной химической формы, или все органические соединения ртути (например, диметилртуть), или все ее неорганические соединения, или вся ртуть в определенной степени окисления, или идентификация всех ртутьсодержащих соединений и определение их количества.
Выбор метода. Метод анализа выбирают исходя из поставленной задачи, размеров объекта и образца, содержания определяемых веществ, наличия примесей, требуемой точности результатов и имеющегося оборудования; учитывают также возможную продолжительность и стоимость анализа.
Отбор образца. Для разных аналитических методов требуются, конечно, и разные по величине образцы – в количестве от нанограммов (1 нг = 10–9 г) до нескольких граммов. Вряд ли возможно целиком проанализировать объект, который весит намного больше, чем требует выбранная для анализа методика. В этих случаях отбирают образец, или пробу, вещества.
Подготовка образца к анализу. Если количественные измерения проводят в растворе, образец растворяют в подходящем растворителе; при этом концентрацию образца подбирают так, чтобы она находилась в пределах применимости метода. Иногда приходится выделять определяемое вещество из смеси, поскольку многие методы анализа неспецифичны и даже неселективны. Специфичным называют метод, при помощи которого определяется только конкретное вещество, а селективным – предпочтительный для данного вещества метод, пользуясь которым можно определять и другие вещества. Специфичных методов очень мало, селективных – значительно больше. Например, высоко-селективны масс-спектрометрия и иммунологический анализ.
Измерения. Чтобы определить количество анализируемого вещества или его состав, измеряют какую-либо его физическую величину: количество вещества, израсходованного или образовавшегося в результате химической реакции; скорость реакции; интенсивность поглощения, испускания или рассеяния света; ток, возникающий в ходе окислительно-восстановительных процессов; количество выделившегося или поглощенного тепла и т.д. Зная связь между результатами измерений и теми величинами, которые интересуют исследователя, а также сравнив эти результаты с соответствующими стандартами, устанавливают количество определяемого вещества или его состав.
Интерпретация результатов. Когда результаты уже получены, может возникнуть ряд вопросов: решена ли поставленная задача? как проводить дальнейшие исследования? Не исключено, что для получения более точных результатов нужно усовершенствовать методику анализа.
Рабочие кривые.
Рабочая кривая – это графическая зависимость, связывающая концентрацию определяемого вещества с тем параметром, который измеряется в ходе анализа (оптической плотностью, интенсивностью флуоресценции, электродным потенциалом, скоростью реакции и т.д.). Масштаб координатных осей – линейный или логарифмический – выбирается в зависимости от конкретного эксперимента.
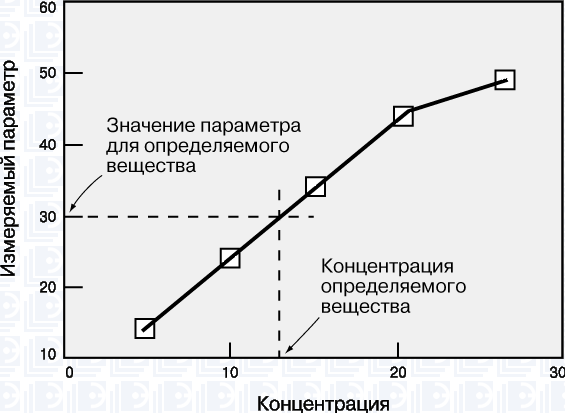
Для построения рабочей кривой сначала готовят стандартные образцы известной концентрации. Затем для каждого из них измеряют тот или иной параметр и откладывают его значение в виде точки против соответствующей концентрации. По точкам проводят плавную кривую, на которую точки ложатся наилучшим образом. Для этого используют какую-либо подходящую математическую функцию или эмпирическую зависимость.
У каждого метода есть свои рабочий диапазон, чувствительность, фон, порог обнаружения.
Рабочий диапазон – это диапазон концентраций, в пределах которого применима данная методика. Линейный участок кривой отвечает области концентраций, в которой результаты наиболее надежны. При близких к предельным высоких и низких концентрациях рабочие кривые обычно становятся нелинейными. Это обусловлено ограниченными возможностями используемых методов анализа и оборудования. Если концентрация определяемого вещества попадает в нелинейную область высоких значений, то образец следует разбавить и анализ повторить.
Чувствительность метода характеризуется величиной изменения измеряемого параметра при данном изменении концентрации. Она равна угловому коэффициенту (тангенсу угла наклона) рабочей кривой. Как правило, чем выше чувствительность, тем надежнее результаты и тем ниже порог обнаружения.
Результат измерения часто включает составляющую, не связанную с определяемым веществом, – ее называют фоном. Наличие фона может быть связано с особенностями оборудования или влиянием матрицы, в которую включен образец. Чтобы оценить величину фона, проводят контрольный опыт. Для этого готовят контрольный образец, в котором нет определяемого вещества, а есть только все посторонние примеси, имеющиеся в матрице, а также реагенты, добавляемые в процессе анализа. Контрольный образец подвергают той же аналитической процедуре, что и определяемое вещество. Значение измеряемого параметра для этого контрольного образца считают равным фону.
Порог обнаружения – это наименьшая концентрация определяемого вещества, при которой сигнал заметно отличается от фона. Величина порога обнаружения зависит от чувствительности и точности метода: чем они выше, тем ниже минимальные определяемые концентрации.
Матрица. Термин «матрица» относится к окружению определяемого вещества. Это все вещества, присутствующие в образце, в т.ч. и определяемые, отличные от данного. Так, хлор определяют в плазме крови, консервированной моркови, питьевой или морской воде. Эти образцы различаются по своим химическим и физическим свойствам, а следовательно, их матрицы тоже различны. Простейшая матрица – питьевая вода: она содержит относительно немного веществ, концентрация которых к тому же невелика.
Качественный анализ
Основные понятия и терминология
Химический анализ (определение химического состава) веществ и материалов имеет целью обнаружение, идентификацию, разделение и определение химических элементов и их соединений, а также выяснение химического состава веществ.
Идентификация компонентов и определение качественного состава вещества или смеси веществ является предметом качественного анализа. При этом утверждение об отсутствии данного компонента («не обнаружен») подразумевает, что его содержание ниже некоторого предела. Таким образом, деление на качественный и количественный (определяющий содержание компонента) анализ в определенной степени условно.
Качественный анализ может быть проведен различными методами. Использование физических методов для установления качественного состава объекта будет рассмотрено в соответствующих разделах, посвященных физическим и физико-химическим методам. В данном разделе рассматриваются химические методы качественного атомно-ионного анализа.
В качественном химическом анализе аналитический сигнал получают в результате проведения химической реакции.
Химическое превращение анализируемого вещества при действии аналитического реагента с образованием продуктов с заметными аналитическими признаками (сигналами) называют аналитической реакцией, частной реакцией или реакцией открытия. Вещество, которым действуют на открываемый компонент, называют аналитическим реагентом или реактивом на открываемый ион.
Аналитическим сигналом может быть цвет и его изменение, запах, выделение газообразных продуктов, окрашивание пламени, образование люминесцирующих соединений, выпадение или растворение осадка.
В случае образования осадка, кроме самого факта выпадения, аналитическим сигналом может служить его цвет, форма (кристаллическая или аморфная), а также характерная форма кристаллов.
Аналитические реакции, согласно рекомендации ИЮПАК, подразделяют на специфические и избирательные (селективные) методы, реакции и реагенты.
Специфическими называют те методы, реакции или реагенты, с помощью которых в данных условиях можно обнаружить только одно вещество; избирательными — методы, реакции и реагенты, позволяющие обнаружить небольшое число веществ.
Специфические реагенты и реакции позволяют обнаружить данное вещество или ион в присутствии других веществ или ионов.
Селективные реагенты и реакции позволяют обнаружить несколько веществ или ионов. Таких реагентов и реакций известно существенно больше, чем специфических.
Избирательность достигается правильным выбором и установлением соответствующих условий реакции. К факторам, определяющим условия протекания реакции, относят рН, температуру, концентрации открываемого и посторонних ионов, природу растворителя (вода, органические или водно-органические среды). Реакции или реагент можно сделать более избирательными или даже специфичными варьированием рН, концентраций, маскированием, изменением степени окисления элементов, температуры.
Реагенты по их избирательности можно разделить на три группы:
1. Специфические реагенты — например: крахмал для обнаружения I2.
2. Избирательные (селективные) реагенты — например: диметилглиоксим в аммиачной среде реагирует с Fe(II), Co(II), Ni(II), Zr(IV) и Th(IV).
3. Групповые реагенты — например: HCl осаждает нерастворимые хлориды Ag(I), Hg(I), Tl(I), Pb(II).
Групповые и селективные реагенты, образующие малорастворимые соединения с ионами, используют при разделении катионов на аналитические группы. Ионы, осаждаемые данным групповым реагентом, образуют аналитическую группу ионов. Используемый групповой реагент и условия осаждения определяют состав аналитической группы ионов.
Анализ смеси ионов — трудная аналитическая задача, поскольку посторонние ионы могут препятствовать открытию интересующего нас иона. Такие ионы называют мешающими. Помехи со стороны сопутствующих ионов начинают проявляться при определенном соотношении открываемых и мешающих ионов и усиливаются с увеличением концентрации последних. Причинами мешающего влияния при открытии ионов могут быть:
1. Образование (мешающими ионами) при действии реагента соединений, аналогичных аналитической форме открываемого иона, и генерирование ими такого же сигнала, что и аналитический сигнал открываемого иона.
2. Образование (мешающими ионами) соединений, препятствующих наблюдению аналитического сигнала.
3. Вероятность проведения аналитической реакции открытия невелика, например, в результате связывания реагента мешающими ионами.
Для устранения мешающего влияния сопутствующих ионов используют два пути:
-
избирательное распределение компонентов анализируемой системы между двумя разделяющимися фазами (методы разделения или отделения). В практике качественного анализа наибольшие значения имеют осаждение, экстракция и хроматография;
-
маскирование мешающих ионов. Для этого используют химические реакции, протекающие в той же фазе, что и реакции обнаружения, и приводящие к уменьшению концентрации мешающего иона или реагента. Для маскирования и демаскирования применяют реакции комплексообразования, кислотно-основные и окислительно-восстановительные.
Реальные объекты всегда многокомпонентны, часто и многофазны. Для открытия каждого иона необходимо:
-
создать условия протекания частной реакции;
-
устранить мешающее влияние сопутствующих компонентов;
-
зарегистрировать аналитический сигнал.
Возможны два пути решения этой задачи: проведение систематического качественного химического анализа или дробное обнаружение ионов.
Последовательное разделение ионов на отдельные аналитические группы методом осаждения групповыми реагентами (или иным образом) называют систематическим ходом анализа.
Метод дробного обнаружения основан на применении специфических и селективных реагентов без разделения на группы. Мешающее влияние сопутствующих ионов устраняют, используя приемы маскирования, а также применяя более селективные или специфические реагенты.
Таким образом, процедура качественного химического анализа представляет собой последовательное отделение аналитических групп с дальнейшим открытием входящих в них ионов систематическим или дробным методами. В ходе выполнения анализа как систематическим, так и дробным методами аналитик управляет поведением ионов в растворе, прежде всего их концентрациями. Такое управление возможно на основе равновесных реакций путем смещения равновесий. В распоряжении аналитика два типа равновесных процессов — гомогенные и гетерогенные равновесия. Гомогенные равновесия — это диссоциация — ассоциация, окисление — восстановление, гидролиз, нейтрализация, комплексообразование. Количественное описание этих равновесий основано на законе действующих масс и уравнении Нернста для окислительно-восстановительного потенциала системы. К гетерогенным равновесиям относятся, прежде всего, растворение и осаждение осадков, экстракционное распределение между двумя жидкими фазами и хроматографические процессы. Расчеты положения гетерогенного равновесия возможны на основе констант межфазных распределений, в первую очередь правила произведения растворимости.
Характеристика чувствительности аналитической реакции выражается в том, что аналитические реагенты и аналитические реакции позволяют обнаруживать определяемое вещество в пробе, если его содержание превышает некоторый минимальный предел. Если концентрация определяемого вещества ниже этого предела, то и концентрация аналитической формы окажется настолько незначительной, что невозможно будет зарегистрировать аналитический сигнал.
Предел обнаружения — минимальная концентрация или минимальное количество вещества, которое может быть обнаружено данным методом с какой-то допустимой погрешностью. Его обозначают сminP, где Р — доверительная вероятность. Чувствительность характеризует изменение сигнала с изменением концентрации и выражается коэффициентом чувствительности, который численно равен тангенсу угла наклона линейной зависимости аналитического сигнала от концентрации.
Предел обнаружения в качественном анализе традиционно называли открываемым минимумом. В настоящее время в качественном анализе используется большое число реагентов и частных реакций с низкими пределами обнаружения. Обычно для открытия ионов применяют реакции с пределом обнаружения 10–7 г (0,1 мкг) в 1 мл раствора. Физические методы позволяют открыть элементы в твердых образцах с пределом обнаружения менее 10–15 г. Предел обнаружения, наряду с избирательностью, является важнейшей характеристикой реакции и метода анализа.
Приемы и техника выполнения реакций
Реакции в пробирке — в полумикрометоде реакции проводят в специальных конических пробирках, отмеряя объемы по каплям и используя центрифугирование для отделения осадков.
Микрокристаллоскопические реакции проводят на предметном стекле и наблюдают образование осадка и форму кристаллов под микроскопом в 75-кратном увеличении (приблизительно). Капли исследуемого раствора и реагента помещают рядом и стеклянной палочкой образуют между ними перемычку. Образование осадка происходит в результате взаимной диффузии реагента и определяемого иона, что обеспечивает формирование правильных относительно крупных кристаллов.
Капельные реакции проводят на полосках фильтровальной бумаги. Сначала концом капилляра с раствором реагента касаются бумаги до образования пятна диаметром около 3 мм. В центр полученного пятна аналогичным образом наносят каплю исследуемого раствора.
Обнаружение с использованием экстракции — в пробирку с притертой пробкой помещают по несколько капель анализируемого раствора, реагента и органического растворителя. Для понижения предела обнаружения соотношение объемов органической и водной фазы обычно берут равным 1:4. В закрытой пробирке смесь взбалтывают 1–2 мин и после расслаивания наблюдают окраску или люминесценцию органический фазы.
Обнаружение с использованием флотации использует ту же методику, что и экстракция. Предел обнаружения можно существенно понизить, если осадок флотируется на поверхности раздела органической и водной фаз. Так, пределы обнаружения Ni2+ по реакции с диметилглиоксимом составляют при открытии его:
в пробирке — 1,4 мкг
капельным методом — 0,16 мкг
флотацией на границе вода — эфир — 0,002 мкг.
Люминесцентные реакции обычно проводят на фильтровальной бумаге или на предметном стекле, реже — в пробирках. Свечение (флуоресценция или фосфоресцирование) существенно зависит от присутствия примесей, концентрации реагирующих веществ, природы растворителя, температуры, поэтому при выполнении этих реакций необходимо проводить контрольный опыт. Каплю контрольного раствора, содержащего все компоненты, кроме определяемого, наносят на фильтровальную бумагу рядом с анализируемым раствором. Техника нанесения растворов на бумагу обычная. Влажные пятна высушивают на воздухе и наблюдают люминесценцию при освещении ультрафиолетовым светом. Часто люминесцентные капельные реакции проводят при низкой температуре. Для этого пинцетом осторожно погружают бумагу в жидкий азот на 20–30 с, и сразу же после ее извлечения рассматривают в ультрафиолетовом свете.
Каталитические реакции проводят обычно в пробирках, наблюдая резкое увеличение скорости протекания реакции в присутствии определяемого иона
Количественный анализ
В аналитической химии есть несколько методов, основанных на определении положения химического равновесия. К ним относятся, в частности, классические гравиметрия и титриметрия, а также сравнительно новый иммунологический анализ.
Гравиметрия (весовой метод). В гравиметрии определяемое вещество переводят в химически чистое состояние или превращают в весовую форму – соединение с точно известным постоянным составом, которое можно легко выделить и взвесить. Количество анализируемого вещества рассчитывают исходя из массы весовой формы и уравнения реакции, связывающей это вещество с весовой формой. Химические стандарты не требуются. Весовые методы анализа очень точны, их часто используют в сомнительных случаях в качестве контроля. Точность анализа ограничивается точностью определения массы и полнотой образования и выделения чистого вещества. Гравиметрия – продолжительная процедура, поскольку и перевод определяемого вещества в весовую форму, и выделение ее из смеси требуют времени. Кроме того, необходимо убедиться в том, что весовая форма – это вещество точно известного постоянного состава, не содержащее примесей.
Большинство весовых определений основано на образовании и выделении из раствора (как правило, водного) твердых нерастворимых осадков. Задача состоит в том, чтобы осадить по возможности максимальное количество определяемого вещества (по крайней мере, 99,99%), поэтому осадок (чаще всего соль) должен обладать как можно меньшей растворимостью. Растворимость соли определяется величиной константы равновесия реакции растворения, в которой образуются ионы. Количественное осаждение обычно осуществляют, добавляя к раствору с определяемым веществом стехиометрический избыток осаждающего реагента. Растворимость соли в присутствии избытка одного из ионов, входящих в ее состав, снижается. Для уменьшения влияния других равновесных реакций, приводящих к увеличению растворимости соли, необходимо контролировать состав раствора.
Для разных анализируемых веществ применяют разные осаждающие реагенты. Некоторые из них приведены в таблице:
|
НЕКОТОРЫЕ ОСАЖДАЮЩИЕ РЕАГЕНТЫ, ИСПОЛЬЗУЮЩИЕСЯ В ГРАВИМЕТРИИ |
|
|
Анализируемое вещество |
Реагент |
|
Ag+ |
Cl– (из KCl или NaCl) |
|
Al3+ |
8-гидроксихинолин |
|
Ca2+ |
C2O42–, CO32– |
|
Ni2+ |
Диметилглиоксим |
|
Галогениды |
Ag+ (из AgNO3) |
|
SO42– |
Ba2+ (из BaCl2) |
Одно из основных преимуществ весовых определений заключается в том, что не нужно калибровать приборы или готовить стандартные растворы. Результат получают, взвесив осадок и зная состав участвующих в реакции соединений.
Титриметрия (объемный метод). В титриметрии концентрацию определяют, измеряя объем стандартного или титрованного реагента (титранта), израсходованного в химической реакции с определяемым веществом в растворе (или газовой фазе). Измерение проводят с помощью процедуры титрования. Это простой, относительно быстрый, универсальный и точный метод.
При титровании титрант добавляют порциями или непрерывно с небольшой постоянной скоростью и измеряют его объем до тех пор, пока не будет достигнута точка эквивалентности, отвечающая объему титранта, при котором в реакцию вступает все определяемое вещество. Точку эквивалентности находят, непрерывно следя за изменением тех или иных свойств титруемого раствора (цвета, оптической плотности, электрохимических свойств и т.д.) при помощи специальных приборов или визуально.
Чтобы данную химическую реакцию можно было использовать в титровании, участвующие в ней вещества должны находиться в строго определенных количественных (стехиометрических) соотношениях. Реакция должна протекать быстро и практически до конца, а точка эквивалентности точно фиксироваться. Чаще всего используют реакции нейтрализации (кислотно-основные), комплексообразования и окислительно-восстановительные. Реакции нейтрализации распространены наиболее широко; именно их мы и рассмотрим для пояснения ключевых моментов всех реакций титрования.
Кривые титрования. Кривая титрования – это график зависимости pH, оптической плотности или каких-либо других характеристик титруемого раствора (ось ординат) от объема добавленного титранта (ось абсцисс).
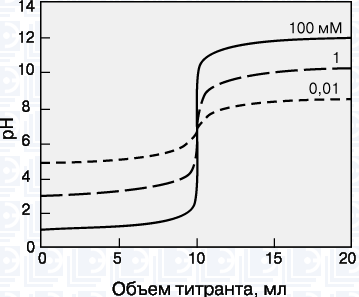
--------------------------------------
На рисунке: Кривые титрования cильнoй киcлoты cильным ocнoвaниeм для
paзныx кoнцeнтpaции oпpeдeляeмoгo вeщecтвa.
------------------------------------------------------------------------------
Масштаб оси абсцисс всегда линейный, а оси ординат может быть линейным или логарифмическим. Линейный масштаб удобен для тех методов контроля за титрованием (спектрофотометрия, амперометрия), в которых контролируемый параметр меняется с концентрацией линейно, а логарифмический – в случае логарифмического изменения (например, при потенциометрии с ионоселективным электродом). Логарифмический масштаб часто используют при визуальном определении конечной точки титрования, поскольку именно в этом масштабе наиболее наглядно проявляется резкое изменение свойств раствора вблизи точки эквивалентности.
Зависимость кривых титрования от концентрации и константы равновесия. Для точного определения конечной точки титрования необходимо, чтобы на кривой титрования вблизи точки эквивалентности наблюдался перегиб (скачок). Это требование устанавливает пределы как для минимальной определяемой концентрации, так и для минимальной константы равновесия, приемлемой для реакции титрования.
Цветные индикаторы. Цветной индикатор – вещество, которое меняет свою окраску при взаимодействии с одним из компонентов титруемого раствора.
Инструментальное определение конечной точки титрования. Непрерывный контроль процесса титрования с помощью приборов позволяет получить данные о его ходе как до, так и после точки эквивалентности. Эти данные можно представить в виде графика и определять конечную точку графически или путем вычислений. Чаще всего используют спектрофотометрическое (измерение оптической плотности), амперометрическое и потенциометрическое (измерение электродного потенциала) титрование.
Кулонометрическое титрование. Кулонометрическое титрование обычно проводят при постоянном токе. Титрант образуется в результате электрохимических процессов на рабочем электроде в сосуде для титрования. Число молей анализируемого вещества равно произведению силы тока на время, необходимое для образования титранта в количестве, достаточном для достижения конечной точки титрования с учетом стехиометрии. Химические стандарты не требуются. К титрантам, которые образуются в ходе электрохимических процессов, относятся H+, OH–, Br2 и I2.
Прямое и обратное титрование. В простейшем варианте титрования анализируемое вещество взаимодействует непосредственно с титрантом. Количество анализируемого вещества рассчитывают исходя из молярной концентрации титранта, его объема, требуемого для достижения точки эквивалентности, и стехиометрии реакции между определяемым веществом и титрантом.
В обратном титровании анализируемое вещество взаимодействует не с титрантом, а с другим реагентом, присутствующим в избытке. Избыток затем определяют титрованием. Если известно исходное количество реагента и определен его избыток, то разность между ними – это количество реагента, пошедшее на реакцию с определяемым веществом.
Обратное титрование используют, например, когда константа равновесия реакции прямого титрования слишком мала.
Кроме того, для кач. анализа используются:
Спектроскопические методы основаны на взаимодействии электромагнитного излучения с веществом, т.е. на определении характеристик поглощаемого, испускаемого или рассеянного излучения. Часто параллельно со спектроскопическими методами используют масс-спектрометрию; хотя с ее помощью и не изучают взаимодействие излучения с веществом, результаты измерений обычно представляют в виде спектра.
Поглощение в УФ- и видимой областях. Спектры поглощения в УФ- и видимой областях содержат как качественную, так и количественную информацию о поглощающем веществе. Последнее и позволяет использовать их в аналитической химии.
Люминесценция. В люминесцентной спектроскопии измеряется интенсивность излучения, испускаемого атомами или молекулами вещества при их переходе из возбужденного состояния в основное. Люминесценция бывает двух типов: флуоресценция и фосфоресценция. При флуоресценции атом или молекула переходит в основное состояние из короткоживущего возбужденного состояния. Она наблюдается почти сразу после поглощения, быстро спадает и исчезает в результате столкновений излучающей молекулы с другими молекулами в растворе (тушение флуоресценции). Фосфоресценция наблюдается при переходе молекулы в основное состояние из относительно долгоживущего возбужденного состояния, так что между поглощением света и испусканием может пройти относительно много времени. Для фосфоресценции характерны бóльшая длина волны излучения, меньшая высота пиков и большее влияние матрицы. Флуоресцентные измерения более избирательны, чем спектрофотометрические, поскольку зависят сразу от двух длин волн: поглощаемого и испускаемого света.
Инфракрасная (ИК) спектроскопия. Спектры поглощения в видимой и УФ-областях, о которых шла речь выше, возникают в результате электронных переходов в атомах и молекулах. Поглощение же в ИК-области обусловлено переходами между колебательными уровнями, отвечающими разной колебательной энергии функциональных групп. В ИК-спектроскопии чаще всего используют среднюю часть ИК-области, 4000–200 см–1.
Ядерный магнитный резонанс (ЯМР). Метод ЯМР основан на резонансном поглощении электромагнитной энергии, обусловленном магнетизмом ядер. Это поглощение наблюдается в сильном магнитном поле, под действием которого энергетические уровни ядер, обладающих магнитным моментом, расщепляются. Наложение небольшого по величине и изменяющегося по частоте электромагнитного поля вызывает переходы между уровнями, проявляющиеся в виде линий поглощения в спектрах ЯМР. Метод ЯМР – один из наиболее эффективных методов структурных исследований. Он позволяет получить информацию о строении молекул, о том, какие ядра присутствуют в определяемом веществе и в каком количестве, каково их окружение.
Масс-спектрометрия (МС). Масс-спектрометрия – один из наиболее эффективных и широко применяющихся аналитических методов. Его отличают высокая селективность, чувствительность и точность.
Принцип метода состоит в том, что определяемое вещество переводят в газообразное состояние, ионизируют и образовавшиеся ионы (заряженные фрагменты исходных молекул) разделяют в магнитном поле по величинам отношения массы к заряду. Любой масс-спектрометр состоит из системы напуска образца, ионизационной камеры и системы разделения ионов. В приборе поддерживают высокий вакуум (~10–6 мм рт.ст.). Методы регистрации отработаны настолько хорошо, что позволяют без труда производить подсчет отдельных ионов.
А так же электрохимические методы, хроматографические методы и т.д. и т.п.