

Физические методы исследования в химии
.pdf6.Ангармонический осциллятор. В чем отличия колебаний ангармонических от гармонических?
7.Запишите выражение для энергии стационарного уровня колебаний ангармонического осциллятора. Поясните смысл величин, входящих в него.
8.Основное и специфическое правила отбора для разрешенных переходов ангармонического осциллятора.
9.Каков вид спектра ангармонического осциллятора? Зависит ли энергия колебательного кванта ангармонического осциллятора от квантового числа? Что такое обертоны?
10.Какие характеристики двухатомных молекул можно определить из их колебательных спектров?
11.Понятие о нормальных координатах, форме колебания. Что такое характеристические частоты?
Задания к теме «Колебания молекул. Определение структуры и свойств двухатомных молекул»
Задание 1. Часто энергию стационарных уровней и линий в колебательных спектрах выражают в см-1. Найдите коэффициент соответствия между см-1и Дж.
Задание 2. Покажите, что колебательный спектр гармонического осциллятора состоит из одной полосы поглощения.
Задание 3. Что такое нулевой колебательный квант. Чему равна энергия этого кванта.
Задание 4. Молекула НСl имеет фундаментальную частоту колебаний 2988.9 см-1. Найдите силовую постоянную связи этой молекулы. На основании полученного результата определите фундаментальную частоту ее дейтерированного аналога (DCl).
Задание 5. Найдите в общем виде выражение для энергии колебательного кванта гармонического и ангармонического осцилляторов.
Задание 6. В колебательном спектре двухатомной молекулы, колеблющейся по закону ангармонического осциллятора, наблюдают три соседних полосы с энергиями ω1>ω2>ω3. Показать, что энергия гармонического кванта равна ωг= ω1+ω2- ω3.
Задание 7. Колебательным энергетическим уровням молекулы 23Na127I соответствуют волновые числа 142.81, 427.31, 710.31, 991.81 см- 1.Определите силовую постоянную связи NaI, энергию разрыва связи и энергию нулевого кванта.
41
Задание 8. Частоты колебаний в спектре 23Na35.5Сl расположены на 85.8×1011, 85.5×1011, 85.2×1011Гц. Найдите силовую постоянную и энергию диссоциации связи NaСl.
Задание 9. Инфракрасный спектр испускания возбужденной молекулы 1H19F состоит из серии линий 3958.38 v=1→v=0, 3778.25 v=2→v=1, 3598.10 v=3→v=2 см-1. Определите силовую постоянную и энергию диссоциации связи НF, а также постоянную ангармоничности и энергию нулевого кванта колебания этой молекулы.
Задание 10. Галогениды водорода имеют следующие фундаментальные частоты колебаний (см-1): 1H35.5Cl - 2988.9, 1H81Br - 2649.7, 1H127I -
2309.5. Определите силовые постоянные связей H-Gal.
Задание 11. Первые пять колебательных энергетических уровней молекулы 1H 35.5Cl находятся на 1331.84, 3917.44, 6398.94, 8776.34,11049.60 см-1. Вычислите по этим спектральным данным частоту гармонического кванта молекулы и энергию диссоциации связи НCl.
ГЛАВА 7. Электронные спектры поглощения и излучения молекул. Внутримолекулярные фотофизические процессы
7.1. Электронные состояния и спектры двухатомных молекул
Как было показано при рассмотрении вращательных и колебательных спектров двухатомных молекул многие особенности свойств вращательного и колебательного движений находят объяснение на основе классической теории взаимодействия света с веществом. Электронное движение и электронные спектры достаточно строго могут быть рассмотрены только в рамках квантово-механических представлений. Известно, что без помощи ЭВМ уравнение Шредингера решено для молекулы Н2, имеющей всего два электрона. Нахождение волновых функций и энергии электронных состояний других даже двухатомных молекул, не говоря о электронных спектрах многоатомных молекул, можно осуществить только с помощью ЭВМ по специальным программам в различных приближениях.
Согласно квантово-химическим представлениям каждое электронное состояние характеризуется полными орбитальным и спиновым моментами количества движения - L и S, соответственно. Двухатомная молекула обладает аксиальной симметрией, поэтому проекция L на выделенное направление имеет важное значение. Проекция задается величиной соответствующего орбитального квантового числа Ʌ, принимающего следующие значения: Ʌ= 0, 1, 2, 3.... Электронные состояния Ʌ= 0, 1, 2, 3....,
42
которым отвечают эти значения, обозначаются символами Σ, Π, ∆, Φ....
Общему спиновому моменту отвечает спиновое квантовое число S, которое определяет мультиплетность данного электронного состояния: g = (2S+1). Мультиплетность определяет число энергетических подуровней, на которое может расщепиться данное электронное состояние во внешнем поле. Состояния с S=0 называют синглетными, состояния, для которых S =1, - триплетными.
Электронные состояния двухатомных молекул могут различаться также по свойством симметрии. В основе этого лежит поведение волновых функций молекулы при операциях симметрии. Для операции отражения в плоскости симметрии, проходящей через ось молекулы, электронные состояния делятся на положительные (+) и отрицательные (-) в зависимости от того меняет или нет знак волновая функция молекулы при этой операции симметрии. Двухатомные молекулы, состоящие из двух одинаковых атомов имеют центр симметрии. По отношению к отражению в центре симметрии электронные состояния таких молекул подразделяются на четные (g) и нечетные (u). Так, например, основное состояние молекулы водорода обозначают 1Σ+g, что означает: синглетное, положительное и четное состояние.
Квантово-химическая теория дает следующие правила отбора для радиационных электронных переходов в двухатомных молекулах:
∆Ʌ=0, ±1, ∆S=0, - ↔ -, + ↔ +, u ↔ g.
При этом надо помнить, что квантовый переход между электронными состояниями будет запрещенным, если не будет выполнено хотя бы одно из правил отбора.
Поскольку при любых электронных переходах происходит изменение свойств электронной оболочки, это сказывается на структуре и свойствах молекулы. При возбуждении меняется равновесная геометрия молекулы (чаще увеличивается, реже - уменьшается) и энергия диссоциации (чаще уменьшается). При этом чаще потенциальные кривые электронновозбужденных состояний имеют минимум, т.е. соответствуют устойчивому состоянию молекулы, но при значительном увеличении равновесного расстояния потенциальная кривая может оказаться без минимума, т.е. возбужденное состояние неустойчиво. Если при электронном возбуждении молекула попадает в неустойчивое состояние, она распадается на атомы или ионы. Поскольку при диссоциации кинетические энергии продуктов распада могут быть любыми, частота электронных переходов не квантуется, что приводит к излучению сплошного спектра.
Таким образом можно констатировать, что каждому электронному состоянию отвечает своя потенциальная кривая соответствующих парамет-
43
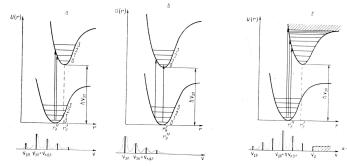
ров (равновесное межъядерное расстояние, квазиупругая постоянная связи, энергия диссоциации и т.д.). Кроме того каждое электронное состояние обладает своей системой колебательных и вращательных подуровней, при переходе между которыми образуется сложный электронно - колебательно - вращательный спектр (рис. 3.1). Так как электронный спектр - это комбинация электронных состояний, то неизбежно его вид будет отражать природу и взаимное расположение этих комбинирующих состояний.
7.2. Принцип Франка-Кондона для внутримолекулярных процессов
Распределение интенсивности в электронно-колебательной полосе спектра можно объяснить с помощью принципа Франка-Кондона. В его основе лежат 3 положения. Молекула состоит из двух связанных подсистем: совокупность электронов и совокупность ядер. Скорости движения этих двух подсистем существенно различаются. Частота колебания электронов составляет 1014-1015 Гц, что равно периоду колебания 10-14-10-15с. В то же время частота колебаний ядер составляет 1012-1013 Гц и период колебаний - 10-12- 10-13с. Изменение свойств электронной оболочки при поглощении или излучении кванта света происходит так быстро, что скорости и положения ядер не успевают измениться. Отсюда следует первое положение: переходы между потенциальными поверхностями происходят вертикально, т.е. межъядерное расстояние при переходе не изменяется (рис. 7.3 б).
Рис. 7.3. Схема электронно-колебательных переходов и спектров для различных случаев изменения равновесного межъядерного расстояния при оптическом ком возбуждении: а - rE'
> rE''; б - rE' ≈ rE''; в - rE' ˂ rE''
Второе положение. При колебательном движении скорость прохождения положения равновесия максимальна, а вблизи пересечения с по-
44
тенциальной кривой скорость движения минимальна, иначе говоря наибольшей будет вероятность застать колеблющуюся частицу именно в точке поворота. Поэтому наиболее вероятными будут те переходы, которые начинаются и заканчиваются в этих областях (исключение составляет нулевой колебательный уровень основного состояния).
Третье положение. При комнатной температуре большая часть молекул находится на нулевом колебательном уровне. Исключение составляют невозбужденные колебательные уровни (υ =0), на которых молекулы преимущественно находятся на расстояниях, близких положению равновесия.
Дополнительно следует помнить, что при электронных переходах квантовая механика не накладывает ограничений на изменение колебательных квантовых чисел.
Какую информацию можно извлечь из вида электронноколебательной полосы спектра поглощения? Рассмотрим это на следующих примерах. Первый случай - наиболее распространенный: при электронном возбуждении межъядерное равновесное расстояние в молекуле увеличивается. В этом случае потенциальная кривая возбужденного состояния сдвинута вправо, как это показано на рис.7.2 а. В случае хорошего разрешения колебательной структуры электронной полосы видно, пик максимальной интенсивности соответствует переходу с нулевого колебательного уровня основного состояния на колебательный уровень возбужденного состояния с υ ' > 0. На рис. 7.2 а это соответствует колебательному переходу υ = 0→υ '= 1. Интенсивности других колебательных переходов ниже интенсивности колебательного перехода υ = 0→υ '=1. При большем увеличении равновесного расстояния в возбужденном состоянии по сравнению с основным состоянием наибольшая интенсивность может приходиться на другие колебательные переходы, например, υ =0→υ '= 2, υ = 0→υ '= 3 и т.д. А электронное возбуждение в этом случае происходит с увеличением запаса колебательной энергии в возбужденном состоянии.
Второй случай - менее распространенный: при электронном переходе величина равновесного расстояния практически не изменяется. В этом случае понятно, что наиболее интенсивный чисто колебательный переход имеет место при колебательном переходе υ = 0→υ '= 0 (рис.7.2 б). В этой ситуации при возбуждении не происходит изменения запаса колебательной энергии в возбужденном состоянии по отношению к основному состоянию.
45
Наконец, возможен и такой случай, когда при электронном возбуждении равновесное межъядерное расстояние в возбужденном электронном состоянии существенно больше равновесного расстояния в основном электронном состоянии (третий случай, рис. 7.2 в). В такой ситуации возможен переход, в результате которого электрон оказывается в возбужденном электронном состоянии на колебательном уровне, энергия которого выше энергии диссоциации, что приводит к разрыву связи. Элек- тронно-колебательный спектр в такой случае помимо разрешенной колебательной структуры полосы со стороны высоких частот будет содержать участок сплошного спектра, который, как отмечено выше, свидетельствует о процессе диссоциации молекулы. Важно отметить, что в этом случае молекула диссоциирует не по причине перехода на фотодиссоциативную кривую электронного состояния, а за счет значительного изменения межъядерного расстояния при переходе в возбужденное электронное состояние.
Таким образом, из вида электронно-колебательной полосы в спектре поглощения двухатомной молекулы можно заключить как меняется равновесное расстояние в двухатомной молекуле и имеет ли место при возбуждении процесс диссоциации. Следует отметить, что принцип ФранкаКондона является одним из наиболее фундаментальных принципов, на которых базируется современная молекулярная спектроскопия. Анализ электронно-колебательных спектров многоатомных молекул показывает, что в некоторых случаях из их спектра поглощения также можно сделать заключение о направлении изменения равновесного межъядерного расстояния той из связей многоатомной молекулы, колебания которой проявляются в электронно - колебательной полосе.
7.3. Электронные спектры поглощения многоатомных молекул. Закон Ламберта-Беера
Теоретическое рассмотрение электронных спектров многоатомных молекул представляет значительные трудности из-за наличия у таких молекул большого числа колебательных степеней свободы Именно поэтому не существует единой сколько-нибудь последовательной и строгой теории удовлетворительно описывающей спектры различных классов молекул. Ряд общих закономерностей электронных состояний, описанных для двухатомных молекул, справедлив и для многоатомных молекул, например, симметрия волновых функций. Также, как в случае двухатомных молекул, переходы между электронными состояниями подчиняются правилам отбора. Так, например, по-прежнему запрещены электронные
46

переходы между электронными состояниями различной мультиплетности. В случае больших спин-орбитальных взаимодействий в молекуле могут разрешаться переходы между синглетными и триплетными состоянии, однако интенсивность таких переходов крайне низка.
Существует два главных закона поглощения света. Первый из них - закон Бугера-Ламберта. Он гласит, что относительное количество поглощаемого пропускающей средой света не зависит от интенсивности падающего излучения, а все последующие слои среды поглощают равные доли проходящего через них света. Влияние концентрации поглощающих световую энергию частиц учитывает другой важный закон - закон Беера: поглощение света пропорционально числу молекул вещества, через которое проходит свет.
Пусть тонкий слой однородной среды поглощает некую долю падающей на него монохроматической световой энергии (рис.7.1). После прохождения слоя толщины dx интенсивность света уменьшилась на величину dI. Математически законы поглощения Бугера-Ламберта и Беера запишутся в виде:
dI = - αСI0 dx, где I0 и I - интенсивности падающего и прошедшего света, соответственно, С- мольная концентрация поглощающего вещества, α-
коэффициент поглощения, зависящий от свойств молекулы и частоты падающего света. Найдем поглощение всего объема
Il
∫DI =−∫αCL , откуда следует I - I0 = exp(-αCl) или
I 0 |
0 |
ln I = - αCl. Здесь l -толщина поглощающего слоя.
I0
На практике предпочтительнее иметь дело с десятичными логарифмами, поэтому полученное выражение перепишем в виде:
lg I = - αCl/2.303 = - εсl,
I0
где ε = α/2.303 - молярный коэффициент экстинкции, зависящий от свойств поглощающей молекулы и частоты падающего света.
47

Отношение интенсивности светового потока, вышедшего из поглощающего слоя I, к интенсивности входящего потока I0 называется пропусканием Т. Тогда можно записать:
lg |
I |
= lgT = D = - εСl. Здесь D - оптическая плотность. |
(7.1) |
|
|||
|
I0 |
|
|
Величины, характеризующие интенсивность поглощения или пропускания вещества (D, ε, Т) на данной частоте (длине волны) при графическом изображении в некотором спектральном диапазоне имеют вид спектральной кривой поглощения (или пропускания), изображенной на
рис. 7.2.
Для получения величины поглощенной энергии во всей полосе поглощения необходимо проинтегрировать полосу поглощения по всей области частот. Площадь под кривой поглощения называют интегральной интенсивностью А. Учитывая, что площадь под кривой поглощения с удовлетворительной точностью можно заменить площадью прямоугольника со сторонами εmax и ∆σ1/2 можно записать:
А = ∫2.303ε (ν )Dν = |
∫C2.303ε (σ )Dσ ≈ 2.303 с εmax ∆σ1/2. (7.2) |
ν |
σ =1/ λ |
Здесь с - скорость света, εmax- молярный коэффициент экстинкции в максимуме полосы поглощения, ∆σ1/2- полуширина полосы (ширина полосы на половине высоты) в см-1.
Знание интегральной интенсивности полосы поглощения позволяет сравнить рассчитанные квантово-химическими методами спектры поглощения молекул с экспериментальными данными. В расчетах интенсивность электронного перехода оценивают как силу осциллятора f, которая связана с интегральной интенсивностью следующим образом:
f = ( |
4MECε0 |
) A ≈ 2.303 с ( |
4MECε0 |
) εmax ∆σ1/2, |
(7.3) |
|
|
||||
|
NAE |
|
NAE |
|
|
где me, е ̶ масса и заряд электрона, соответственно,с - скорость света, ε0
-диэлектрическая проницаемость вакуума, NA- число Авогадро.
Вотличии от закона Бугера-Ламберта, который строго соблюдается, закон Беера имеет много исключений. Эти исключения чаще связаны с изменением состава раствора при изменении концентрации вещества. Например, многие кислоты, основания и соли в растворах не подчиняют-
48
ся закону Беера только потому, что с увеличением степени разбавления они полнее ионизуются, а нейтральные молекулы и заряженные их части поглощают по-разному. Другими причинами нарушения закона может быть флуоресценция раствора, рассеяние света и т.д. Поэтому прежде, чем применять спектрофотометрический метод, необходимо убедиться, что закон Бугера-Ламберта-Беера не нарушается, только в этом случае можно доверять величине измеренной интенсивности и ее изменениям, к которым приводит, например, межмолекулярное взаимодействие.
7.4. Классификация электронных переходов
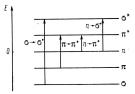
Для органических молекул характерны три основных типа молекулярных одноэлектронных орбиталей: σ-, π- и n-орбитали (рис. 7.3). Определяют природу одноэлектронных орбиталей (занятых и вакантных) из анализа результатов квантово-химического расчета.
σ-Орбиталью называют орбиталь, участвующую в образовании одиночной связи; π- орбиталью, орбиталь, образующая вторую связь в группе с кратной связью; n-орбиталью называют атомную орбиталь, занимаемую неподеленной парой электронов, т.е. электронов, не участвующих в образовании химической связи. Орбитали σ- и π-типа, образующие химические связи, называют связывающими. При поглощении молекулой кванта света один из σ-, π- или n-электронов с заполненной одноэлектронной орбитали переходит на сво-
бодную орбиталь более высокой энергии σ*- или π*-типа. Электронные переходы в молекуле обозначают, указывая тип орбиталей, между которыми происходит переход, например, n→π*, π→π*, σ→σ* и т.д.
n→π*- Переходы имеют место в молекулах с кратными связями, содержащими группы С=О, NO2, -N=N- и т.п. Поскольку орбитали n- электронов, как и σ-орбитали плоских молекул, чаще располагаются в плоскости молекулярного остова, переход электрона с них на π*- орбитали запрещен, в реальной ситуации переходы этого типа имеют малую интенсивность. То же можно сказать и об интенсивности переходов σ→ π*- или π→ σ*- типа. Комбинация между одноэлектронными молекулярными орбиталями одного типа разрешена, поэтому переходы π→π*- и σ→σ*- типов могут иметь высокую интенсивность, если нет запретов по
49
симметрии. В результате этих переходов возникают различные орбитальные типы электронно-возбужденных состояний: ππ*, σσ*, nπ*, σπ* и т.п.
Для грубой оценки интенсивностей электронных переходов, образующих спектр поглощения, воспользуемся полученным в предыдущем разделе соотношением между силой осциллятора и молярным коэффициентом экстинкции (7.3):
f = 6.26×10-19 εmax ∆σ1/2. |
(7.4) |
Обычно полоса поглощения, сформированная одним электронным переходом, имеет полуширину ∆σ1/2 ≈ 5000 см-1. Измерив в экспериментальном спектре коэффициент экстинкции в максимуме полосы, можно вычислить силу осциллятора электронного перехода, образующего данную полосу поглощения. Из экспериментального спектра можно определить и полуширину полосы поглощения данного соединения, взяв вместо среднего значения ∆σ1/2, измеренное экспериментально.
Анализ данных по молярным коэффициентам экстинкции и силам осцилляторов большого числа органических соединений показал, что полосы поглощения, образованные разрешенными π→π*- переходами, имеют величину ε ≈ 103-106л/моль см и соответствующую ей силу осциллятора от f = 1 до нескольких единиц. Полосы запрещенных π→π*- переходов имеют силу осциллятора 0.01- 0.001. Полосы поглощения n→π*-переходов обладают малой интенсивностью с ε ≈ 10-100 л/моль см, что соответствует f = 10-4-10-3. Владея этими сведениями, можно в первом приближении, определить тип электронного перехода, образующего полосу в экспериментальном спектре поглощения. А сопоставление эксперимента и результатов квантово-химического расчета по энергиям и силам осциллятора позволяют уточнить тип электронного перехода и определить локализацию электронно-возбужденных состояний, переходы в которые из основного образуют данный спектр поглощения.
7.5. Процессы дезактивации поглощенной энергии. Диаграмма энергетических уровней
Световая энергия, поглощенная молекулой, приводит молекулярную систему в «неустойчивое» состояние, из которого она стремится выйти путем дезактивации избытка энергии над энергией основного состояния. Процессы дезактивации поглощенной энергии по своей природе могут быть излучательными и безызлучательными. При этом эти процессы могут быть обусловлены как внутри-, так и межмолекулярными взаимодействиями. Здесь мы остановимся на внутримолекулярных причинах, которые, как показали многочисленные исследования, являются определяю-
50