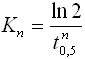2.6. Кристаллизация полимеров
Под кристаллизацией понимают процесс образования кристаллов при переходе вещества из термодинамически менее устойчивого состояния в более устойчивое, сопровождающийся выделением энергии - энтальпии кристаллизации (ΔHкр, обычно равной по величине энтальпии плавления - ΔНпл). Процесс, как правило, протекает в среде переохлажденного пара или жидкости и является фазовым переходом вещества из нестабильного некристаллического состояния в стабильное кристаллическое. Наиболее термодинамически устойчивой структурой является монокристалл, формирование которого обычно затруднено и требует специальных технологических усилий. Поэтому реальной практически достижимой структурой является поликристаллическая, находящаяся в менее термодинамически устойчивом состоянии, нежели монокристалл.
При теоретическом рассмотрении процесса кристаллизации различают две его независимые стадии: зародышеобразование и рост кристаллов. Зародышеобразование по механизму возникновения первичного зародыша делят на гомогенное (самопроизвольное зарождение зародышей путем случайного, флуктуационного соединения многих молекул или групп молекул при тепловом движении их в растворе или расплаве) и гетерогенное (ограничение подвижности молекул и образование зародышей индуцируется внесением извне в раствор или расплав, например, коллоидных частиц, способных адсорбировать на себе кристаллизуемое вещество). Часто для инициирования процесса кристаллизации в подготовленную среду извне вносят готовые зародыши. Однако в любом случае процесс кристаллизации может начаться только при достаточном переохлаждении, не меньшем некоторого порогового значения (Tпор):
|
|
(2.8) |
где Tпл - температура плавления кристалла.
После зарождения первичного зародыша при достаточно малом переохлаждении начинается рост кристалла, приобретающего свойственную ему многогранную форму. При этом молекулы захватываются поверхностями роста, мигрируют по этим поверхностям, пока не находят места для окончательного встраивания в кристаллическую структуру. Чем медленнее растет кристалл, тем ближе его состояние к устойчивому. Быстро закристаллизованный материал, как правило, далек от равновесного состояния и с течением времени или при внешнем воздействии (тепловом, механическом) способен к рекристаллизации с увеличением размеров и устойчивости крупных кристаллитов за счет более мелких или к полиморфному переходу.
Условием термодинамического равновесия фаз является равенство их химических потенциалов. Если потенциал некристаллической фазы μж, а кристаллической μкр, то при фазовом равновесии (вблизи плоской поверхности раздела) μж = μкр. Термодинамической движущей силой фазового перехода является отклонение от равновесия. Мера этого отклонения - разность μж - μкр.
При μж - μкр > 0 растут кристаллы, при μж - μкр < 0 идет декристаллизация (плавление, растворение, испарение и т.д.). При кристаллизации из расплава

где Т
- истинная
температура;
![]() -
равновесная температура плавления, qкр
- теплота кристаллизации на один элемент.
Аналогично рассматривается переход
одной кристаллической модификации
в другую (полиморфные переходы).
-
равновесная температура плавления, qкр
- теплота кристаллизации на один элемент.
Аналогично рассматривается переход
одной кристаллической модификации
в другую (полиморфные переходы).
В гомогенной среде при условии μж > μкр происходит образование более выгодной кристаллической фазы. Объединение атомов или молекул в агрегат с объемом V и поверхностью Ω ведет при неизменном давлении и температуре к изменению термодинамического потенциала Гиббса на величину:
|
|
(2.9) |
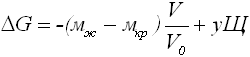
где V0 - удельный объём одной частицы в кристалле, σ - удельная свободная энергия поверхности (в первом приближении не зависящая от ориентации этой поверхности). Первый член отражает энергетический выигрыш, сопровождающий образование устойчивой кристаллической фазы, и пропорционален кубу линейного размера зародыша (b). Второй - проигрыш из-за появления границы раздела.
Тогда ΔG при малых b растет с его увеличением, а при больших b - уменьшается, проходя через максимум, характеризующийся критическим размером зародыша b0.
В гетерогенной системе примеси, снижая поверхностную энергию, уменьшают работу по образованию зародыша, способствуя его образованию. Однако примеси частично ограничивают доступ молекул к поверхности нуклеации, поэтому с ростом концентрации примесей скорость образования зародышей, как правило, проходит через максимум.
Изотермическая кристаллизация Первой моделью, которая описывала структуру частично-кристаллического полимера, была модель «бахромчатой мицеллы» (рис. 2.12), предложенная в 1930 г. Гернгроссом и Германом. Предполагалось, что отдельные участки соседних макроцепей при сближении могут образовывать микрокристаллиты, которые играют роль узлов в общей аморфной сетке. Такая модель не допускает возможности формирования больших полимерных монокристаллов, что противоречило эксперименту.

Только в 1957 г. независимо Келлером и Фишером получены неопровержимые доказательства складчатой, ламелярной структуры полимерных кристаллов (рис. 2.13).

Из многочисленных теорий, в которых делаются попытки количественной оценки процессов, происходящих при кристаллизации полимеров со складывающимися цепями, наиболее широкую известность приобрели равновесная и кинетическая. В основе обоих видов этих теорий лежит одно из фундаментальных свойств полимеров: зависимость толщины ламелей, а, следовательно, и периода складывания l от температуры кристаллизации Ткр. При этом равновесная теория исходит из определяющей роли Ткр и степени приближения к равновесному состоянию системы при этой температуре. Период складывания определяется минимумом плотности свободной энергии кристалла при данной температуре кристаллизации.
Кинетическая теория также рассматривает свободную энергию системы, но приводит к выводу, что l зависит от обратной величины степени переохлаждения 1 / ΔT. Наиболее вероятные значения периода складывания определяются кинетическими требованиями максимальной скорости роста кристалла при выбранном ΔT, а весь процесс контролируется энергетикой образования зародышей кристаллизации, которые характеризуются линейными размерами и значениями поверхностной свободной энергии боковых (σs) и торцевых (σe) граней.
Если рассмотреть первичный зародыш длиной l со стороной квадратного сечения a, то уравнение (2.2) примет вид:
|
|
(2.10) |
где Δgпл - термодинамический потенциал единицы объёма без учета вклада поверхности.
Исследование на максимум дает значения критических размеров первичного зародыша (а* и l*) и энергетический барьер его образования (ΔG*).
|
|
(2.11) |
|
|
(2.12) |
|
|
(2.13) |



где Δhпл - энтальпия плавления единицы объёма.

То есть критический размер зародыша пропорционален обратной величине переохлаждения.
Для вторичного зародыша, формирующегося на поверхности растущей ламели, нет прироста поверхностной энергии для граней параллельных поверхности роста. Уравнение 2.9 принимает вид:
|
|
(2.14) |
где b0 = na, n - число складок.
Соответственно, уравнения (2.11 - 2.13) принимают вид:
|
|
(2.15) |
|
|
(2.16) |
|
|
(2.17) |



Согласно кинетической теории кристаллизации в ходе последующей кристаллизации высота растущего ламелярного кристалла вначале повышается до значения:
|
|
(2.18) |

где

а затем остаётся постоянной. Значение параметра y может меняться в пределах 1 > ψ > 0.
Конечная толщина кристаллита оказывается пропорциональной первичной:
|
|
(2.19) |
и для малых степеней переохлаждения, считая 2σe / Δg >> δl, имеем линейную зависимость:
|
|
(2.20) |

Прямая (2.20) должна пересекать прямую Tпл = Ткр в точке, соответствующей равновесной температуре плавления полимера. Следовательно, экспериментально установленная зависимость (2.20) может быть применена для нахождения .
Скорость кристаллизации может быть описана биэкспоненциальным уравнением:
|
|
(2.21) |

где первый экспоненциальный член отражает вероятность образования зародышей критических размеров, а второй – вероятность реализации процесса переноса через границу раздела расплав - кристалл (ΔF* - энергетический барьер этого процесса). Температурная зависимость энергетического барьера переноса предполагается аналогичной температурной зависимости энергии активации вязкого течения. При высоких температурах она почти постоянна, а при низких температурах быстро увеличивается с приближением к температуре стеклования (Tс).
Иногда в расчетах прибегают к аппроксимации следующего вида:
|
|
(2.22) |

где Т0 - температура, ниже которой становится невозможным перемещение молекул через границу раздела фаз. Часто используют также универсальное уравнение для энергии активации вязкого течения Вильямса-Лендела-Ферри:
|
|
(2.23) |

Тогда (2.21) принимает вид:
|
|
(2.24) |

Уравнение 2.21 успешно используется при анализе процесса изотермической кристаллизации полимеров. В частности, оно трансформируется к виду:
|
|
(2.25) |

где
 Аn
- параметр, связанный с числом гетерогенных
центров зародышеобразования и максимальной
скоростью роста зародышей; Kn
и n
- константа
валовой скорости кристаллизации и
показатель Аврами соответственно в
уравнении Аврами, описывающем переход
вещества из некристаллической фазы в
кристаллическую, как функцию времени
(t):
Аn
- параметр, связанный с числом гетерогенных
центров зародышеобразования и максимальной
скоростью роста зародышей; Kn
и n
- константа
валовой скорости кристаллизации и
показатель Аврами соответственно в
уравнении Аврами, описывающем переход
вещества из некристаллической фазы в
кристаллическую, как функцию времени
(t):
|
|
(2.26) |

Здесь хt и x∞ - доля вещества, претерпевшего фазовый переход в момент времени t и в бесконечности соответственно.
Если провести анализ
экспериментальных данных в координатах
суммы нормированных констант скоростей
валовой кристаллизации и транспортного
члена (левая часть уравнения 2.25) и
температурного параметра
![]() аппроксимируя
зависимость линейной, можем получить
значение σsσe.
Используя эмпирическое соотношение:
аппроксимируя
зависимость линейной, можем получить
значение σsσe.
Используя эмпирическое соотношение:
|
|
(2.27) |
рассчитываем значение σe.
Дважды логарифмируя обе части уравнения (2.26), получаем линейную зависимость:
|
|
(2.28) |

Аппроксимируя экспериментально полученные данные линейной регрессией, получают значения n и Kn.
Часто константу скорости кристаллизации выражают через t0,5 - полупериод кристаллизации, определяемый соотношением
|
|
(2.29) |
|
|
|