

XAXANINA
.pdfПример контрольного теста
1.Потенциометрия основана на установлении зависимости между: 1) потенциалом и концентрацией иона; 2) потенциалом и электропроводностью иона;
3) ионной силой раствора и концентрацией иона;
4) ПР и концентрацией ионов.
2.B каких реакциях в ГЭ стеклянный электрод выполняет роль индикаторного? 1) в реакциях окисления-восстановления; 2) в реакциях нейтрализации; 3) в реакциях осаждения;
4) в реакциях комплексообразования.
3.Уравнение Нернста для электродов, обратимых по катиону (электродов 1-го ро-
да), имеет вид:
1)E E0 RTnF ln[Men ] ;
2)E E0 RTnF ln[Men ] ;
3)E E0 RTnF ln[an n ] ;
4)Е = Е0 – 0,059/n lg[Men+].
4. Укажите схему ГЭ, составленного из электродов: стеклянного (индикаторного) и
хлоридсеребряного (электрода сравнения):
1)(–) Pt, H2│H+║стекло│KCl, AgCl│Ag (+);
2)Ag│AgCl│KCl (насыщ.)│стекло│[H+]║KCl (насыщ.)AgCl│Ag;
3)(–) Pt, H2│H+│стекло│║KCl, Hg2Cl2│Hg(+);
4)2) Ag│KCl (насыщ.)│стекло│[H+]║KCl (насыщ.) Hg2Cl2│Hg.
5. Вычислите концентрацию [H+], если [ОH–] (в моль/л) равна 4·10–10:
1) 2,5·10–6; 2) 2,5·10–5; 3) 1,5·10–4; 4) 1·10–7.
81

Вольтамперометрия
Вольтамперометрия (ВА) - метод анализа, основанный на исследовании зависи-
мости тока поляризации I от напряжения E, прикладываемого к электролитической ячей-
ке. Графическое изображение такой зависимости называется вольтамперограммой (ВАГ).
Основные виды ВА: прямая, осциллографическая, переменно-токовая, инверсион-
ная, амперометрическое титрование.
Электролитическая ячейка в ВА включает два электрода - индикаторный микро-
электрод (поляризуемый) и неполяризуемый электрод сравнения. Например, в инверси-
онной вольтамперометрии (ИВА) электролитическая ячейка представляет собой кварце-
вый стаканчик с электролитом (фоном), в который опущены рабочий электрод (ртутно-
пленочный, стеклоуглеродный, золотографитовый или др.) и электрод сравнения (хло-
ридсеребряный). Часто для уменьшения омического сопротивления в цепи в раствор по-
мещают третий электрод (хлоридсеребряный, стеклоуглеродный или платиновый).
В зависимости от разновидности ВА, т.е. от вида развертки поляризующего напря-
жения и способа измерения тока, на ВАГ наблюдается либо S-образная волна, либо пик,
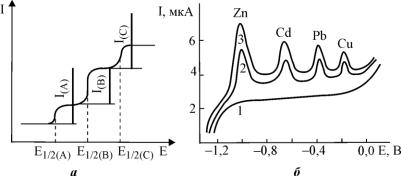
который может быть направлен либо вверх, либо вниз (рис.9).
Рис.9. Типичный вид ВАГ при проведении прямой ВА (а) и ИВА (б)
ВАГ позволяет одновременно получить и качественную, и количественную информа-
цию о веществах, восстанавливающихся или окисляющихся на микроэлектроде (деполя-
ризаторах). Положение волны (пика) на оси потенциалов определяется природой вещест-
ва, участвующего в электродной реакции, а величина тока линейно зависит от его концентрации.
Зависимость между током I и концентрацией определяемого элемента C описыва-
ется уравнением Ильковича
I = KC,
82
где K - постоянная, зависящая от условий проведения опыта. Численные значения
K и С часто определяют экспериментально методом калибровочного графика, методом добавок или методом стандартов. В данной работе используется метод добавок. При экс-
периментальных определениях нередко величину силы тока I можно заменить на пропор-
циональную ей величину высоты волны (пика) h.
Если в растворе присутствует несколько ионов, то на ВАГ наблюдается не одна волна (пик), а несколько - по числу восстанавливающихся ионов (рис.10). При этом важ-
но, чтобы потенциалы волн (пиков) различались в достаточной степени, что позволит из-
бежать их взаимного наложения.
Рис.10. Вид ВАГ при проведении прямой ВА (а) и ИВА (б) при наличии нескольких ионов в исследуемом растворе: 1 - фон; 2 - интегральная; 3 - дифференциальная
Особенностью ИВА является наличие дополнительной (по сравнению с прямой ВА)
стадии предварительного концентрирования определяемого элемента на поверхности или в объеме индикаторного электрода.
Стадии ИВА: 1) накопление анализируемого вещества из раствора на поверхности индикаторного электрода за счет электрохимического восстановления (индикаторный элек-
трод подключен в качестве катода), при этом к ячейке приложен постоянный потенциал,
измерений не проводится; 2) регистрация ВАГ; идет процесс электрохимического раство-
рения концентрата с поверхности электрода (индикаторный электрод подключен в качест-
ве анода), при этом к ячейке приложен линейно-меняющийся потенциал (рис.11).
83
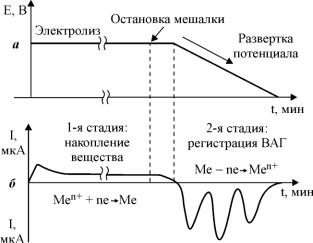
Рис.11. Развертка потенциала (а) и изменение тока (б) при регистрации ВАГ
Вследствие введения стадии концентрирования чувствительность ИВА на 2-3 по-
рядка выше, чем прямой ВА.
При проведении анализа важно избежать наложения пиков определяемых элемен-
тов и снизить действие факторов, искажающие аналитический сигнал. В связи с этим, раз-
работка методики ВА-анализа включает как оптимизацию сигнала, так и устранение ме-
шающих факторов. Например, наложение пиков определяемых элементов можно устранить выбором потенциала электролиза или подбором фона, содержащего лиганд, для маскировки одного из ионов.
Сильное влияние на характер появления пиков оказывают поверхностно-активные вещества (ПАВ), особенно органической природы, а также растворенный кислород.
Одним из методов устранения остатков органических веществ и кислорода являет-
ся облучение пробы ультрафиолетом в присутствии муравьиной кислоты (HCOOH).
Кислород можно удалять из растворов продувкой - пропусканием азота, а при pH ≥
7 - добавлением сульфита натрия
Na2SO3+1/2О2→Na2SO4.
Перед проведением непосредственно ВА-анализа необходимо провести пробопод-
готовку. Целями пробоподготовки являются: а) разложение органической основы; б) пе-
ревод определяемого элемента в электрохимически активную форму; в) удаление избытка окислителей и других мешающих реагентов; г) перевод пробы в раствор.
Измерения проводят с помощью ВА-анализаторов, где полученные данные обраба-
тываются с помощью компьютера, сохраняются в архиве прибора и могут быть распеча-
таны.
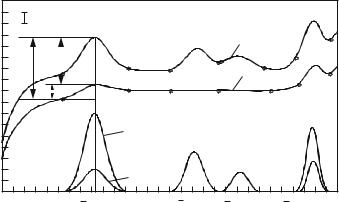
На рис.12 представлены ВАГ (интегральные и дифференциальные) при определе-
нии методом добавок ионов цинка, кадмия, свинца, меди.
84
I, мкА |
|
|
|
|
15 |
0,880 мкА |
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
Iдоб |
|
|
|
|
Iсум |
|
1 |
|
10 |
|
|
|
|
Ix |
|
|
|
|
|
|
|
|
|
5 |
|
2 |
|
|
|
|
|
|
|
|
|
1 |
|
|
0 |
0,9 |
0,6 |
0,4 |
0,05 E, В |
|
||||
Рис.12. ВАГ раствора, содержащего ионы цинка, кадмия, свинца, |
||||
|
меди (1), и пробы с добавкой (2) |
|
||
Кривые 1 соответствуют ВАГ пробы анализируемого раствора, кривые 2 - ВАГ пробы с добавкой.
Достоинства ВА-методов анализа очевидны: 1) экспрессность; 2) малый предел об-
наружения (осциллографическая, переменно-токовая до 10-8 моль/л, ИВА до 10-9 моль/л);
3) достаточная точность ≈ 3%; 4) возможность одновременного определения нескольких компонентов без их предварительного разделения; 5) возможность автоматизации.
85
Работа № 13
Определение содержания токсичных ионов тяжелых металлов в питьевой воде методом
инверсионной вольтамперометрии
Количественно определить содержание токсичных ионов тяжелых металлов (цин-
ка, кадмия, свинца, меди) можно методом инверсионной вольтамперометрии (ИВА).
Предельно допустимые концентрации токсичных металлов в питьевой воде состав-
ляют:
Элемент |
Zn |
|
Cd |
Pb |
Cu |
ПДК, |
5 |
|
0,001 |
0,03 |
1,0 |
мг/л |
|
||||
|
|
|
|
|
|
|
|
Оборудование и реактивы |
|
||
Комплекс аналитический вольтамперометрический в комплекте с компьютером.
Электрод рабочий ртутно-пленочный.
Электрод сравнения хлоридсеребряный.
Вода бидистиллированная.
Муравьиная кислота, ос.ч.
Кварцевые стаканчики на 20 и 25 мл.
Бумага фильтровальная.
Дозаторы с дискретностью доз до 1,0 мл или микробюретки на 1 и 2 мл.
Колбы мерные на 25 и 50 мл.
Стандартные растворы Zn2+, Cd2+, Pb2+, Cu2+ c концентрацией 10,0 или 1,0 мкг/мл.
Описание определения
Перед проведением измерений выполняют следующие работы: тщательную от-
мывку посуды, приготовление растворов, подготовку ВА-анализатора (в том числе подго-
товку электродов), подготовку проб.
Подготовка посуды и электродов. На ртутно-пленочный электрод нанесите плен-
ку ртути погружением серебряного стержня в ртуть. Хлоридсеребряный электрод запол-
ните 0,1 М раствором KCl. Электроды вставьте в соответствующие гнезда в приборе. В
три кварцевых стаканчика внесите по 10 мл бидистиллированной воды. На компьютере
86
запустите к выполнению файл отмывки загрязнений на электродах. Проведите минимум три цикла.
Определение фонового загрязнения. В три кварцевых стаканчика внесите по 10 мл бидистиллированной воды и добавьте по 4 - 7 капель концентрированной муравьиной ки-
слоты. Погрузите электроды в стаканчики. На компьютере загрузите файл "Определение
Zn, Pb, Cd, Cu в природных, сточных и питьевых водах". Проведите определение фона.
Усредните полученные данные. Если фон достаточно чистый, перейдите к анализу пробы.
Анализ пробы. Отберите пипеткой по 10 мл анализируемой пробы в кварцевые стаканчики (три параллельные пробы). Снимите вольтамперограммы для пробы в преде-
лах от –1,2 до +0,05 В. Усредните полученные данные. Найдите потенциалы пиков и по приведенным ниже данным определите, какие ионы присутствуют в растворе:
Катион |
Zn2+ |
Cd2+ |
Pb2+ |
Cu2+ |
Епика 0,07, В |
–0,9 |
–0,6 |
–0,4 |
+0,05 |
При необходимости проведите разметку пиков вручную. Если качественный анализ выполнен правильно, приступите к определению содержания ионов.
Анализ пробы с добавкой. Пипеточным дозатором введите в стаканчик определен-
ный объем стандартного раствора определяемых веществ. На компьютере заполните таб-
лицу "Количество". Проведите измерения. Усредните полученные данные.
При получении удовлетворительных результатов запишите документ в архив.
Вылейте анализируемый раствор и повторите на компьютере команды "Подготов-
ка посуды и электродов" практической части для отмывки электродов и кварцевых ста-
канчиков.
87
Работа № 14 Определение меди и цинка при их совместном присутствии
на катионите КУ-2
Ионный обмен часто применяют для разделения элементов при анализе растворов сложного состава. При определении примесей металлов в сточных промышленных водах в присутствии других ионов используется способность катионита КУ-2 сорбировать при-
меси, в то время как большие концентрации элементов, находящиеся в виде анионов, не сорбируются и переходят в фильтрат. Поглощенные примеси могут быть избирательно десорбированы.
В табл.16 приведены примеры раздельного вымывания сорбированных катионитом примесей при определении их в различных экологических объектах.
|
|
|
|
Таблица 16 |
|
Раздельное вымывание примесей с катионита КУ-2 |
|
|
|||
|
|
|
|
|
|
Элементы, не сорбируемые |
Элементы, десорбируемые соляной кислотой |
|
|||
катионитом |
до 1 н |
1 - 5 н |
|
5 - 10 н |
|
Теллур, селен, мышьяк, |
Висмут, |
Свинец, |
|
Медь, |
|
германий |
кадмий |
индий, сурьма |
|
кобальт |
|
Для раздельного вымывания примесей наряду с кислотами используются различ-
ные комплексообразователи.
Ионы меди и цинка образуют комплексные соединения с оксалат-ионом, имеющие различные константы нестойкости:
Kн([Сu(С2О4)2]2–) = 5,1·10–11; Kн([Zn(C2O4)3]4–) = 2,5·10–8.
При пропускании анализируемого раствора через катионит цинк и медь поглоща-
ются, а остальные примеси переходят в фильтрат. При промывании колонки катионита раствором оксалата аммония медь будет переходить в фильтрат, а оставшийся цинк мож-
но десорбировать соляной кислотой. Раздельно десорбированные медь и цинк можно оп-
ределить титрованием с трилоном Б.
Оборудование и реактивы
Соляная кислота, 3 н.
Оксалат аммония, 0,25 н.
Пероксид водорода, 6%-ный раствор.
Аммиак, 25%-ный раствор.
88
Титрованный раствор ЭДТА, 0,1 н.
Мурексид.
Эриохром черный Т.
Хлорид аммония, х.ч.
Метиловый оранжевый.
Хроматографическая колонка длиной 15 см, диаметром 1 см, заполненная катиони-
том КУ-2 в Н-форме.
Колбы конические на 250 мл, 2 шт.
Стаканы на 100 и 250 мл, 2 шт.
Бюретка на 5 мл.
Описание определения
Анализируемый раствор, содержащий примеси меди и цинка, пропускают через колонку с катионитом КУ-2 в Н-форме (предварительно подготовленную к работе). Ко-
лонку с катионитом промывают водой до нейтральной реакции по метиловому оранжево-
му и затем пропускают 40 - 60 мл 0,25 н раствора оксалата аммония. При этом медь пере-
ходит в раствор в виде анионного комплекса с оксалатом. Для титрования меди раствором ЭДТА оксалатный комплекс разрушают нагреванием в течение 10 мин с 6 - 7 каплями пе-
рекиси водорода. После охлаждения добавляют раствор аммиака до образования медно-
аммиачного комплекса (синее окрашивание). Полноту вымывания этого комплекса из ко-
лонки проверяют обработкой последних порций фильтрата пероксидом водорода при на-
гревании с последующим добавлением после охлаждения раствора аммиака.
Отсутствие синего окрашивания указывает на полноту вымывания меди. В случае наличия синего окрашивания в последних порциях фильтрата его приливают к основной массе фильтрата и продолжают промывать колонку раствором оксалата аммония до пол-
ного удаления меди. Весь фильтрат после обработки перекисью водорода и аммиаком титруют трилоном Б в присутствии мурексида. После добавления мурексида раствор ста-
новится желтым или коричневым, что зависит от количества мурексида, а также от кон-
центрации ионов меди. В точке эквивалентности окраска резко переходит в фиолетовую.
По количеству израсходованного на титрование трилона Б рассчитывают содержание ме-
ди в анализируемом растворе.
Оставшийся на катионите цинк удаляют промыванием 50 - 60 мл 3 н соляной ки-
слоты. К фильтрату добавляют ~ 40 мл аммиачного буфера, необходимого для определе-
ния цинка. Полученную смесь разбавляют дистиллированной водой до 100 мл и титруют
89
0,1 н трилоном Б в присутствии индикатора эриохрома черного Т до перехода винно-
красной окраски в синюю. По результатам титрования рассчитывают содержание ионов цинка в исследуемом растворе.
Примеры решения типовых задач
Метод добавок
Задача 1. Навеску минерала, состоящего из сульфата кобальта с примесью никеля,
массой 2,500 г растворили, добавили необходимые реактивы - HCl, желатин, пиридин - и
разбавили до 100,0 мл. Аликвоту раствора объемом 50,00 мл полярографировали и полу-
чили диффузионный ток (1,35 мкА). Затем в полярографическую ячейку добавили 5,00 мл стандартного раствора, содержащего 1,00·10-2 моль/л NiCl2, и получили диффузионный ток 3,80 мкА. Вычислить массовую долю (%) Ni в препарате.
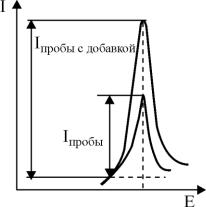
Решение. В соответствии с уравнением Ильковича
Ιпробы = ΚСпробы;
Ιпробы с добавкой = ΚСпробы с добавкой,
где Ιпробы, Iпробы с добавкой - диффузные токи до и после прибавления стандартного раствора; Спробы - начальная концентрация никеля; Спробы с добавкой - концентрация никеля после добавления стандартного раствора.
На рис.13 представлены ВАГ анализируемой пробы и пробы с добавкой.
Рис.13. Вольтамперограммы анализируемой пробы и пробы с добавкой
Разделим второе уравнение на первое
Iпробы с добавкой |
|
Спробы с добавкой |
. |
|
|
||
I пробы |
Спробы |
||
90 |
|
|
|