

posobie
.pdfПо определению мольного объема υυ = n(1r) , n(r) – число молей в кри-
сталле,
|
1 |
|
|
2 |
|
S |
|
1 |
|
l |
|
|
|
|
||||
μr − μ∞ = |
|
|
|
|
|
|
G |
|
(r) + |
|
|
G |
(r) . |
|
(5.9) |
|||
|
|
|
3 |
|
|
3 |
|
|||||||||||
Для одного моля |
n(r) |
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
2 |
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|||
|
|
|
|
|
|
S |
|
|
|
|
l |
|
, |
(5.10) |
||||
μr = μr − μ∞ = |
|
|
|
|
G |
моль(r) + |
|
|
G |
моль(r) |
||||||||
3 |
|
3 |
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
где Gмоль (r) – соответствующие избытки функции G в расчете на один моль дисперсной фазы, взятой в виде частиц размером r.
Для капель
|
2 |
G |
S |
|
> 0, |
(5.11) |
μr = μr − μ∞ = |
3 |
|
моль(r) |
|||
|
|
|
|
|
|
где GSмоль(r) – энергия, затраченная на образование поверхности моля кристалла радиусом r.
Уравнение (5.11) есть уравнение Гиббса. Физический смысл его: в жидкой дисперсной фазе химический потенциал вещества повышается по сравнению с бесконечной фазой на 2/3 от поверхностной свободной энергии 1 моля капель с радиусом r.
Если рассматривать равновесие дисперсной жидкой фазы и её пара, то повышение химического потенциала μ мелких капель приводит к повыше-
нию равновесной упругости пара: |
Pr |
|
|
|
μr = RT ln |
, |
(5.12) |
||
P |
||||
|
|
|
||
|
∞ |
|
|
где Pr – упругость пара для капель с размером r, Р∞ – упругость пара бесконечно протяженной или плоской поверхности жидкости.
5.1.2. Кристаллизация из растворов и газов
Два состояния системы, в котором I относится к жидкости, не содержащей кристалл, II – когда в объеме той же жидкости образуется кристалл с массой n(r):
G = GII −GI , |
(5.13) |
или G = Gкрυ −Gжυ . |
(5.14) |
Переход из I в II затрачивает только n(r) молей вещества, заключенных в объеме υ. С использованием (5.3) имеем
G = μ∞(кр) n(r) + Gs (r) + Gl (r) − μжn(r) . |
(5.15) |
Здесь три первых слагаемых относятся к объему, граням и ребрам кристалла, а последнее определяет G для n (r) молей жидкости.
Для системы, состоящей из новой фазы с линейным размером r и объемом υ, находящейся в равновесии с переохлажденной жидкостью или паром:
51
μж = μкр (r) . |
(5.16) |
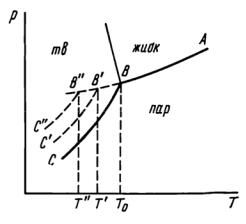
Пример (рис. 5.1).
Рис. 5.1. Метастабильные равновесия кристаллов различного размера. Рассматриваемое равновесие, отвечающее температурам T', T", представляет собой метастабильное равновесие кристаллов размером r с переохлажденной жидкостью. ВС, В′С′, В″С″ – кривые соответствуют давлению пара дисперсной твердой фазы, взятой в виде кристаллов различного размера
По мере уменьшения размера частиц r вся кривая р(Т) для твердой фазы смещается вверх, так как при каждой температуре
μr = RT ln |
Pr |
= |
2 GмольS |
> 0 , |
|
P |
|||||
|
|
3 |
|
||
|
∞ |
|
|
|
где Gsмоль – энергия, затраченная на образование поверхности 1 моля кристаллов. Она растет с дисперсностью.
Работа образования новой фазы из переохлажденной жидкости
G = −(μr (кр) − μ∞(кр) )n(r) + Gs (r) + Gl (r) , |
(5.17) |
||||||||||
где n (r) – число молей жидкости, из которых образуется кристалл. |
|
||||||||||
Учитывая что |
|
1 |
|
2 |
|
|
|
1 |
|
|
|
|
|
|
|
S |
|
l |
|
|
|||
μr (кр) − μ∞(кр) |
= |
|
|
3 |
G |
|
(r) + |
3 |
G |
(r) . |
(5.18) |
|
|
||||||||||
|
|
n(r) |
|
|
|
|
|
|
|||
окончательно для работы образования одной частицы получаем
G = |
1 GS (r) + |
2 Gl (r) , |
(5.19) |
|
3 |
3 |
|
где все величины G относятся к свойствам одной частицы с размером r. Точно такое же можно записать для мольных величин.
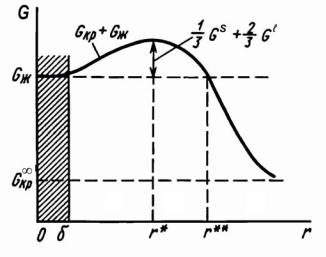
Физический смысл: в переохлажденной жидкости, которая термодинамически неустойчива по отношению к бесконечной твердой фазе, образование равновесного кристалла размером r* требует затраты энергии, равной 1/3 свободной энергии поверхности. Остальные 2/3 этой величины компенсируются выигрышем G при переходе от объема жидкости к объему твердой фазы.
Для капель жидкости уравнение Гиббса
52
G = |
1 GS (r) . |
(5.20) |
|
3 |
|
Эти соотношения графически показаны на рис. 5.2. Здесь δ – толщина поверхностного слоя. В области r ≈ δ и r < δ частицы не являются новой фа-
зой и скорее относятся к флуктуациям плотности жидкости. При r** = 32 r* за-
трата энергии на образование кристалла или капли полностью компенсируется выигрышем свободной энергии за счет выигрыша энергии образования объема более стабильной фазы, а переохлажденная жидкость становится неустойчивой по отношению к кристаллической фазе, которая при r > r** образуется с уменьшением свободной энергии системы.
Рис. 5.2. Работа образования частицы новой фазы
Возникновение и рост новой фазы. Когда новая фаза имеет размер меньше r**, причем для частиц < r*, рост кристалла сопровождается непрерывным повышением свободной энергии, а μ новой фазы выше, чем в жидкой μж < μкр .
Только при r > r * кристаллы могут расти в переохлажденной жидкости, так как μж > μкр (r* < r < r **) > μкр (r → ∞) . Обычно r* малы (1–10 нм). К этим явлениям применима теория флуктуаций, которая дополняет термодинамику. Термодинамическая теория используется для нахождения величин барьера ∆Gобр, с преодолением которого связано возникновение зародыша новой фазы, способного к самопроизвольному росту ( r ≥ r *). Вычисляется с помощью уравнения (5.20). Величина r* – критический размер зародыша. При обычном пересыщении Pr / P∞ ≈ 1.5. Это дает r* от 100 нм до 1 нм.
5.1.3. Гомогенное и гетерогенное зародышеобразование
Гомогенное зародышеобразование. Появление зародыша новой фазы в метастабильных системах связано с переходом вещества в термодинамиче-
53
ски стабильное состояние. В метастабильной фазе проходит гомогенное спонтанное образование ансамблей разного состава из атомов материальной фазы путем их последовательной обратимой ассоциации:
М1+М=М2 М2+М=М3
---------------
Мn-1+М=Мn
Метастабильная фаза находится в квазиравновесии: скорость образования спонтанно растущих ассоциатов мала.
∆G системы при образовании зародыша новой фазы в виде двух слагаемых. Первое отражает макрообъемные свойства системы, учитывает уменьшение энергии Гиббса системы (выигрыш энергии) при образовании зародыша новой фазы ∆Gr, состоящего из n атомов и характеризующегося разными ∆μ между метастабильной фазой среды (с) и стабильной новой фазой – зародышем (з).
G = n(μз − μс ) = ( G)макр = −n(μс − μз ) = −n μ < 0 . |
(5.21) |
В расчете на 1 моль
μ = RT ln(Pr / P∞ ) .
Второе слагаемое обусловлено микрообъемными свойствами – микроскопичностью зародыша и образованием поверхности, приводящей к увеличению энергии, (∆Gj)микр > 0.
|
|
4 |
|
3 |
2 |
|
G = ( Gj )макр +( |
Gj )микр = − |
3 |
πr |
|
μ /Vm + 4πr σ . |
(5.22) |
Здесь Vm – молярный объем.
Когда выигрыш в объемной энергии равен проигрышу в поверхностной энергии, получим
|
∂( |
Gj ) |
= 0 , |
|
(5.23) |
|||||||
|
|
|
|
|
|
|
||||||
|
|
∂rj |
|
|
|
|
|
|
||||
ri,кр |
= |
2Vmσ |
, |
|
|
(5.24) |
||||||
|
|
|
||||||||||
|
|
|
|
|
μ |
r |
|
|
||||
|
|
|
|
|
|
|
|
|
||||
Pr = P∞ exp( |
2σVm |
|
1) . |
(5.25) |
||||||||
|
|
|||||||||||
|
|
|
|
|
|
RT |
r |
|
||||
Уравнение (5.25) называется уравнением Гиббса–Томсона (Кельвина). |
||||||||||||
Гетерогенное зародышеобразование. Появление пространственных не- |
||||||||||||
однородностей повышает вероятность зародышеобразования. |
|
|||||||||||
( Gi )гет = ( Gi )макр |
+ ( Gi )микр |
+ ( Gi )зс . |
(5.26) |
|||||||||
Третий член отражает изменение энергии при возникновении поверх- |
||||||||||||
ности раздела зародыша со средой. |
|
2Vmσзс |
|
|
|
|
||||||
ri,кр = |
. |
|
(5.27) |
|||||||||
|
|
|||||||||||
|
|
|
|
|
μ |
r |
|
|
||||
|
|
|
|
|
|
|
|
|
||||
54

Графически зависимость поверхностного натяжения от радиуса частицы показана на рис. 5.3.
μr = μr − μ∞ . |
(5.28) |
Значение энергии Гиббса дисперсного металла отличается от значений массивной фазы. Удельная поверхностная энергия Гиббса рассматривается как функция радиуса мольного объема:
δG = |
A |
|
2σ |
−kT (cr −c∞ ) , |
(5.29) |
|
ρNA |
r |
|||||
|
|
|
|
где А – атомная масса металла, ρ – плотность, NА – число Авогадро, сr и с∞ – число точечных дефектов (вакансий) на атом в частице и концентрация вакансий в массивном металле. Первое слагаемое отражает вклад поверхностной энергии, второе – вакансий.
c |
= c |
exp |
2σ |
|
V |
, |
(5.30) |
|
r |
∞ |
|
|
|
r |
|
|
|
|
|
|
kT |
|
|
|
|
|
где ∆V – изменение объема при замене атома в узле решетки вакансией. Для частиц < 10 нм экспоненциальный фактор способен обеспечить значительное увеличение числа вакансий.
Рис. 5.3. Вид зависимости удельной энергии Гиббса (поверхностного натяжения) от радиуса малого объекта [Самсонов В.М. Условия применимости термодинамического описания высокодисперсных и микрогетерогенных систем // Журнал физ. химии, 2002. Т. 76, № 11. С. 2047–2051]
5.1.4. Химическое осаждение металлов
При оценке равновесных потенцилов ион-металлической пары необходимо учитывать изменение потенциала Е металлического компонента вследствие его дисперсного состояния. Физико-химические свойства малых частиц заметно отличаются от свойств плоских поверхностей за счет избыточных поверхностных характеристик.
Из уравнения (5.24), связывающего химический потенциал с кривизной поверхности, следует, что с уменьшением радиуса r частиц металла, находящихся в растворе, стандарный равновесный потенциал металла Е0 сдвигается к более отрицательным значениям:
55
Er0 = E∞0 − |
2σVm |
. |
(5.31) |
|
|||
|
zFr |
|
|
Здесь z – число электронов в реакции восстановления ионов металла, F – постоянная Фарадея.
На основании уравнений (5.24) и Нернста получено выражение, описывающее взаимосвязь равновесного потенциала Еr частицы и числа атомов в ней:
0 |
2σV |
2/3 |
4πN |
A |
1/3 |
|
|
z+ , |
(5.32) |
|
Er = E∞ − |
m |
|
|
|
+ RT ln a |
Me |
||||
|
|
|
||||||||
|
zF |
|
|
3m |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
где m – число атомов в частице.
E(r) = Er0 − E∞0 . |
(5.33) |
Дисперсностьможетоказатьсущественноевлияниеприr< 10 нм(рис. 5.4).
- |
E (r),В |
|
|
|
|
0.30 |
1 |
|
|
|
|
|
|
|
|
|
|
0.20 |
|
|
|
|
|
0.10 |
2 |
|
|
|
|
3 |
|
|
|
|
|
|
|
|
|
|
|
0.00 |
|
|
|
|
|
|
0.0 |
0.5 |
1.0 |
1.5 |
2.0 |
|
|
|
|
|
lg r [r, нм] |
Рис. 5.4. Смещение потенциала ион–металлической пары, связанное с дисперсностью частиц серебра (1), меди (2) и висмута (3)
Восстановление ионов металла
νMe Mez+ +νRed Re d +... →νMe Me0 +νOxOx +...
происходит с убылью энергии Гиббса
− G = zFE ,
где Е – разность равновесных потенциалов:
E = EMez+ / Me0 − EOx / Red .
(5.34)
(5.35)
Восстановленная форма вещества с более низким (электроотрицательным) редокс-потенциалом EOx/Red является восстановителем для окисленной формы металла с более высоким потенциалом EMez + /Me0 . При выборе восста-
новителя для осаждения металла необходимо учитывать концентрацию и
56
рН раствора, от которых изменяется величина редокс-потенциалов восстановителя в сравнении с Е0:
|
E |
= E |
0 |
|
|
RT |
ln |
aνOx |
|
|
|
|
+ |
zF |
Ox |
. |
|||
|
|
|
|
||||||
|
Ox/ Red |
Ox/ Red |
|
|
aRedνRed |
||||
Большинство восстановителей |
|
имеет |
более отрицательный Е, чем |
||||||
ЕН+ /Н2 |
, что позволяет восстанавливать не только благородные, но и небла- |
||||||||
городные металлы. К ним относятся гипофосфит, гидразин, формальдегид, боргидрид, дитионит натрия, соли щавелевой, винной кислот и др.
При химическом осаждении дисперсных металлов ситуация осложняется тем, что окислительно-восстановительный потенциал частиц металла зависит от числа атомов. С этой точки зрения химическое восстановление осуществляется в термодинамически и кинетически нестабильных системах. Наноструктуры метастабильны, но долго живучи. По мере уменьшения частиц металла EMez + /Me0 приближается EOx/Red: их равенству соответствует
критическая величина rкр, которое можно выразить через значение Е при условии, что окисление восстановителя не зависит от размера частиц.
rкр = 2Vmσ / zF(EMez+ /Me0 |
− EOx/Red ) = |
2Vmσ |
, |
(5.36) |
|
zFE |
|||||
|
|
|
|
где σ – поверхностное натяжение, Vm – молярный объем.
Для Cu, Ag, Ni критический радиус rкр = 0.3–3 нм. Частицы состоят из сотен и тысяч атомов. Рост частиц может происходить при r > rкр. Таким образом, при Е > 0 частица растет, Е = 0 – неустойчивое равновесие, Е < 0 – растворяется.
В начальной стадии реакции путем последующего укрупнения нестабильных мелких частиц формируются устойчивые частицы
Ag+ + Red ↔ Ag1 + Red+ Ag1 + Ag+ ↔ Ag+2
Ag+2 + Red ↔ Ag2 + Red+
− − − − − − − − − − − − − − −
Agn + Agm ↔ Agn+m
Частицы Ag1 и кластеры, состоящие из небольшого числа атомов, нестабильны. После достижения 1–5 нм частицы приобретают стабильность в реакционной среде. Затем следует рост первичных зародышей до перекрывания друг с другом за счет вакансий на их поверхности. После чего происходит рекристаллизация первичных зародышей в агрегатах с образованием плоских зерен Ме размером 15–30 нм и рост образующихся зародышей. Повторение процессов зародышеобразования, перекрывание зародышей, формирование из них зерен, их рекристаллизация и укрупнение осажденных частиц соответствуют увеличению редокс-потенциала.
57

Редокс-потенциал малых кластеров существенно более отрицательный, чем массивных металлов. Так, в соответствии с расчетами EAg0 +/Ag1 = –1.8 В,
ECu0 +/Cu1 = –2.7 В, в то время как EAg0 +/Ag∞ = +0.799 В, ECu0 +/Cu∞ = +0.52 В. С рос-
том числа атомов в частице Е0 сначала (при малом числе атомов) быстро
возрастает, затем рост его замедляется. Например, E0 |
+ |
= –0.62 В, |
|
|
Ag |
/Ag2 |
|
E0 + |
= +0.2 В. |
|
|
Ag |
/Ag12 |
|
|
5.2.Кинетика зарождения и роста наноразмерных частиц
5.2.1.Кинетика фазообразования при конденсации из паровой фазы
Зародыш с числом атомов j = jкр находится в метастабильном равновесии с окружающей средой. Чтобы сформировать зародыш новой фазы, системе следует преодолеть энергетический барьер Gj,кр . Для этого необхо-
димо, чтобы в результате случая было достигнуто такое состояние, при котором в течение некоторого времени число присоединившихся атомов к ансамблю из j атомов, имеющих размер меньше r < rкр , было больше числа
покидающих его. Согласно теории флуктуаций вероятность события, при котором термодинамический потенциал системы отличается от среднего на
величину Gj,кр , будет пропорционален exp(− GkTj,кр ). Число зародышей
N j,кр или плотность зародышей nj = N j,кр /V (V – объем системы) в предположении их термодинамического равновесия с одиночными атомами N1 с плотностью n1 согласно принципу Больцмана задается уравнениями
N |
j,кр |
= N exp(− |
Gj,кр |
), |
|
|||
|
|
|
||||||
|
|
1 |
|
kT |
(5.37) |
|||
|
|
|
|
|
||||
|
|
|
|
|
Gj,кр |
|
|
|
n |
j,кр |
= n exp(− |
|
). |
|
|||
|
|
|
||||||
|
1 |
|
kT |
|
||||
|
|
|
|
|
|
|||
Появление критических зародышей становится достаточно вероятным событием при низком Gj,кр , и присоединение к ним одного или несколь-
ких атомов приводит к их необратимому росту.
Скорость образования критического зародыша ϑj,кр (число зародышей,
формирующихся в единице объема в единице времени) пропорциональна их плотности n и частоте присоединения атомов к критическому заро-
дышу ν j,кр :
58

ϑ |
j,кр |
=ν |
j,кр |
n |
j,кр |
=ν |
n exp(− |
Gj,кр |
) . |
(5.38) |
|
||||||||||
|
|
|
|
j,кр 1 |
kT |
|
||||
|
|
|
|
|
|
|
|
|
||
Величина ϑj,кр зависит от частоты перехода ν атомов через границу раздела фаз и от плотности атомов ns , находящихся в окрестности зародыша:
ϑ |
j,кр |
=ν n C exp(− |
Gj,кр |
) = B exp(− |
Gj,кр |
) . |
(5.39) |
|
|
||||||
|
s 1 |
kT |
kT |
|
|||
|
|
|
|
||||
Здесь С1 – концентрация одиночных атомов в растворе.
Величина В представляет собой кинетический фактор и отражает скорость роста зародыша, обратная величина В-1 рассматривается как время жизни критического зародыша, т.е. время присоединения к критическому зародышу еще одного атома, переводящего его в закритическое состояние.
С макрокинетической точки зрения формирование зародышей – многостадийный процесс, который может протекать по разным механизмам, описываемым лимитирующей стадией зародышеобразования. По крайней мере два механизма определяют предельные режимы образования зародыша.
1.Кинетический (бездиффузионный) механизм: скорость роста ансамбля атомов определяется процессами на границе раздела фаз – адсорбцией, химической реакцией. Скорость роста таких зародышей пропорциональна концентрации атомов в окрестности зародыша и частоте встраива-
ния атомов в структуру зародыша. Линейный размер зародыша будет пропорционален времени его роста в период 10–5–10–3 с (рис. 5.5а).
2.Если характерные времена поверхностных процессов малы (велики их скорости), то режим формирования зародышей, поскольку в этом случае они являются эффективными стоками для атомов, будет определяться скоростью потока атомов к границе раздела, в частности, диффузией. С ростом зародыша его поверхность увеличивается, и пропорционально ей увеличивается протяженность диффузионного поля, линейный размер формирующегося ансамбля атомоввозрастаетпропорциональнокорнюквадратномуизвремени(рис. 5.5б).
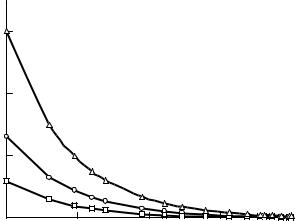
Рис. 5.5. Кинетика роста среднего радиуса частиц Pb после возникновения частиц в ударной волне при разных температурах T и пересыщениях s:
1 – T = 1181 K, s = 30; 2 – T = 1140 K, s = 54; 3 – T = 1140 K, s = 60; 4 – T = 1123 K, s = 94; 5 – T = 990 K, s = 683
59
Так, при изучении конденсации частиц свинца из пересыщенного пара рост частиц идет в основном за счет присоединения атомов к зародышу, а не в результате коагуляции зародышей, что подтверждается гауссовой формой кривой распределения частиц по размерам. При малых t функция r(t) линейна, далее r пропорциональна корню квадратному от t из-за падения концентрации атомов в окрестности зародышей.
5.2.2. Кинетика фазообразования при наличии химической реакции (газовая фаза)
Зарождение и рост кластеров продуктов в случае гомогенной химической реакции (газообразной) протекает по схеме
|
→ * |
ks |
|
M j |
kas |
→M j+m+ M1 |
(5.40) |
+ Mm ← M j+m |
|||
|
kd |
+M1 |
|
где kas, kd, ks – константы скорости образования (ассоциации), мономолекулярного распада (деструкции) и стабилизации метастабильного кластера
M *j+m , несущего на себе энергию возбуждения, выделяющуюся в результате
образования новых внутримолекулярных связей.
На ранних стадиях формирования предполагается, что рост кластеров происходит путем присоединения одиночных атомов М1 (мономеров). По мере их укрупнения возрастает роль ассоциативных процессов типа кла-
стер–кластер (m > 1). Вероятность распада M *j+m уменьшается вследствие
увеличения числа внутренних степеней свободы образующегося ассоциата. Для анализа кинетики процесса образования и диссоциации кластеров применяется один из подходов – квазихимическая модель кластерообразования (есть еще статистическая), описываемая уравнениями, аналогичными уравнениям формальной кинетики. В начальной стадии кластерообразования.
|
|
k1 j |
|
|
|
→ |
(5.41) |
M j+ M ← M j+1 |
|||
|
|
- k2 j |
|
Скорость образования мономера равна скорости распада j кластера за |
|||
вычетом скорости распада мономера: |
(5.42) |
||
|
dN j |
= −∑(I j – 2 I1 ). |
|
|
|
∞ |
|
|
dt |
j=2 |
|
Скорость образования j кластера равна скорости присоединения мономера к (j – 1) кластеру за вычетом скорости диссоциации j кластера:
|
|
dNi = I j-1 – I j , |
(5.43) |
||||
|
|
dt |
|
|
|
|
|
I |
1 |
= k N 2 |
– k' N |
2 |
, |
(5.44) |
|
|
1 |
1 |
2 |
|
|
||
I j = k j N1N j – k'j+1N j+1 , |
(5.45) |
||||||
60