

IV. Нарушение платочного метаболизма липидов. Жировая инфильтрация органов.
Жировая инфильтрация - избыточное отложение жиров в тканях, не относящихся к жировой.
На рисунке 2.1 4.3. схематически представлен патогенез жировой инфильтрации печени.
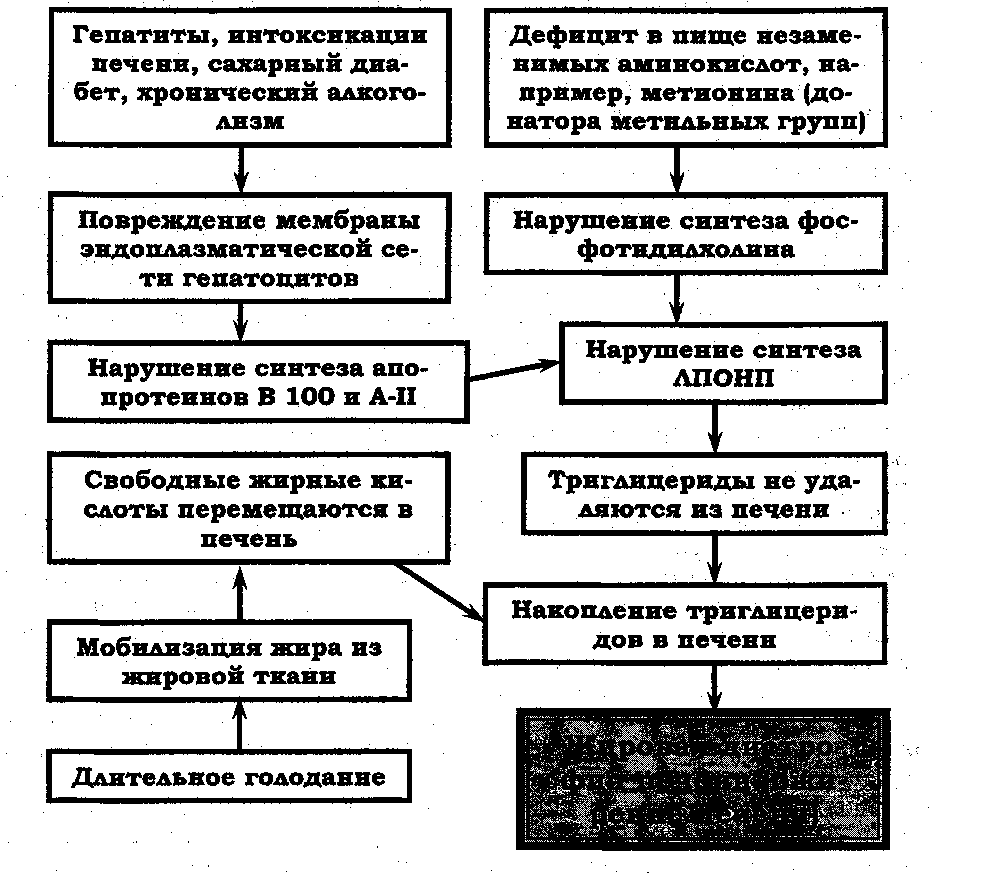
Рис. 2.14.3. Схема патогенеза жировой инфильтрации печени.
V Этиология и патогенез атеросклероза. Стадии развития атеросклероза и осложнения.
Атеросклероз («athere» - кашицеобразная масса (греч.), «sclerosis».- твердый) -хроническое заболевание, характеризующееся поражением стенки артериальных сосудов, в основе которого лежит нарушение липидного обмена, отложение в сосудистой стенке жировых масс с утолщением и деформацией стенки.
Атеромы вызывают стенозы (сужение просвета сосуда) и окклюзии (закупорку сосуда) артерий, что приводит к нарушениям гемодинамики в дистальном артериальном русле, ишемии и гипоксии органов и тканей. При осложненных атеромах развиваются тромбозы и эмболии.
125
Осложнения атеросклероза являются основной причиной смерти: ишемическая болезнь сердца (стенокардия) и инфаркт миокарда (60% мужчин и 41% женщин), ишемическая болезнь мозга с нарушением мозгового кровообращения, ишемическим и геморрагическим (кровоизлияние в мозг) инсультом (25% мужчин и 39% женщин, 1991). Частым проявлением атеросклероза является ишемия нижних конечностей, проявляющаяся симптомом «перемежающей хромоты» (ограничение дистанции ходьбы из-за болей в конечности с восстановлением ходьбы и снижением болевого синдрома после отдыха). Грозным осложнением атеросклероза является аневризма аорты (расширение аорты с деградацией ее стенки), которая может привести к разрыву стенки аорты с несовместимым с жизнью кровотечением. Атеросклеротическое поражение почечных артерий приводит к развитию стойкой вазоренальной артериальной гипертензии, а атеросклероз брыжеечных артерий приводит к ишемии кишечника (ишемический абдоминальный синдром)
Атеросклероз поражает аорту и крупные артерии эластического типа, преимущественно в местах их бифуркацию. Стадии развития атеросклероза.
1. Долипидная стадия - создаются условия для проникновения липидов в интиму, обнаруживаются очаговые микроповреждения эндотелия (повышения проницаемости), изменения интерстициальной ткани (расширение межклеточных каналов), клеточныхструктур (набухание), эластолиз (деструкция эластических мембран с накоплением холестерина). Все это способствует в дальнейшем липоидной инфильтрации внутреннейповерхности артерий. Продолжительность стадии зависит от способности протеолити-ческих и липолитических ферментов, находящихся в интиме, растворять и выводитьпродукты нарушенного обмена.
Долипидная стадия объективно проявляется снижением эластичности артерий (увеличение скорости распространения пульсовой волны) и дисфункцией эндотелия (в основном снижение выработки эндотелием оксида азота).
2. Стадия липоидоза (жировые пятна и полоски) начинается с накопления в интимеЛПНП в комплексе с иммуноглобулинами, а также фибрина, образования комплексоватерогенных липопротеинов с глюкозаминогликанами интерстициальной ткани, что сочетается с изменением аминокислотного состава эластина и ведет к набуханию эластических волокон, фрагментации внутренней эластической мембраны, разволокнению инабуханию внутренней оболочки сосудов. При этом активизируются гладкомышечныеклетки интимы и макрофаги, которые начинают поглощать липиды и трансформироваться в ксантомные (пенистые) клетки.
Стадия липидных пятен и полосок может быть установлена по утолщению комплекса «интима - медиа» дистального сантиметра общей сонной артерии, измеренного при помощи ультразвукового исследования.
Стадия липосклброза проявляется разрастанием молодой соединительной ткани в участках отлоения липопротеидов, созревание соединительной ткани ведет к образованию фиброзных бляшек. Вследствие нарушений эндотелия на поверхности интимы в местах скопления липидов может произойти агрегация тромбоцитов или выпадение нитей фибрина, что, является началом фибропластического процесса. Дальнейшее прогрессирующее накопление липидов сопровождается выраженым измением метаболизма гладко-мышечных клеток сосудов. Они теряют способность утилизировать поступающие жиры, которые по мере гибели клеток попадают в окружающие ткани, где располагаются в основном веществе, вдоль эластических волокон. Высвободившиеся липиды и фибриноген пропитывают бляшку, последний оказывает фибропластическое и склерозирую-щее действие. Возникает типичная фиброзная бляшка, состоящая из бедной клеточными элементами грубой соединительной ткани и жировых масс.
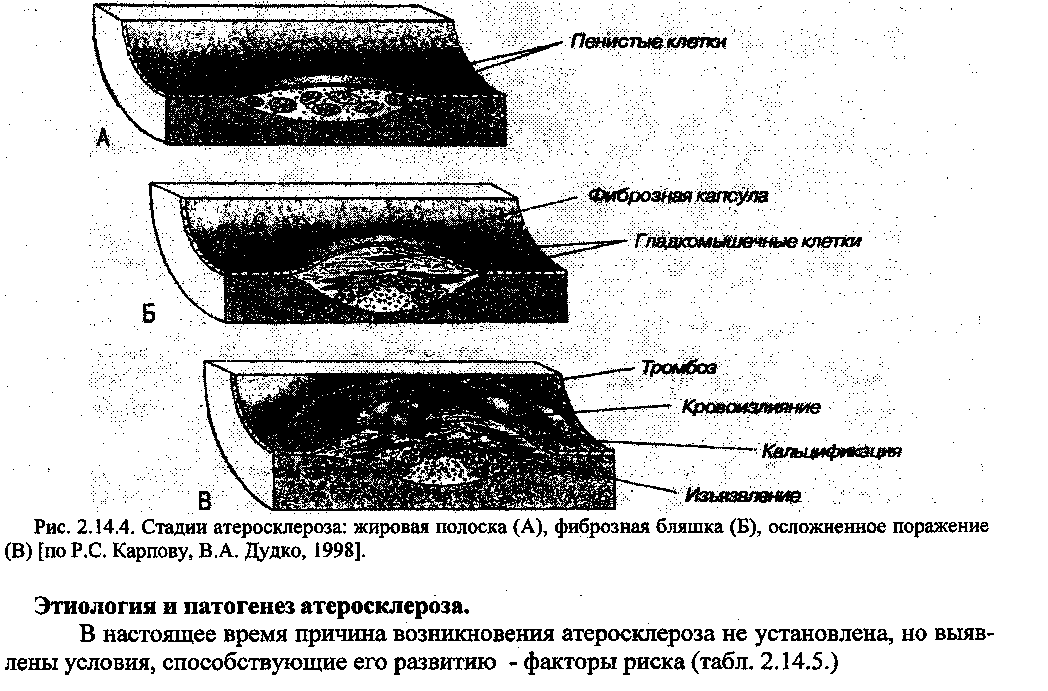
Стадия атероматоза (осложненная бляшка) характеризуется распадом в зоне бляшки липоидов, коллагеновых волокон, а также мышечных и ксантомных клеток. В результате образуется полость, содержащая жиро-белковый детрит (атероматозные массы) и от-
126
деленная от просвета сосуда прослойкой соединительной ткани, которая играет роль покрышки бляшки. Прогрессирование атероматоза приводит к осложненным поражениям сосудов -кровоизлияниям в бляшку, разрушению ее покрышки и изъязвлению. Выпадающий при этом в просвет сосудов детрит может стать источником эмболии, а сама атероматозная язва - служить основой для образования тромбов. В процессе разрушения клеток и кровоизлияния в атерому, в ней может накапливаться кальций, что обозначается как кальцинирование бляшки и имеет характерный вид. 5. Стадия атёрокальцйноза - отложение солей кальция в атероматозные массы, межуточное вещество и фиброзную ткань (петрификация). На рисунке 2.14.4. схематически представлена последовательность морфологических изменений в сосудистой стенке при атеросклерозе.
|
Таблица 2.14.5. Факторы, повышающие риск заболеваний, связанных с атеросклерозом [Россия, 1996] | |||
|
Модифицируемые факторы |
Немодифицируемые факторы | ||
|
|
биохимические и физиологические |
| |
|
образ жизни |
факторы |
личностные параметры | |
|
Высококалорийное питание с |
Дислипопротеидемия с гиперхоле- |
Возраст | |
|
1 повышенным содержа- |
стеринемией за счет ЛНП, низ- |
Пол | |
|
нием животного жира и |
ким уровнем ЛВП, гипертриг- |
Наличие у близких родственников | |
|
ХС |
лицеридемией |
клинических проявлений атеро- 1 | |
|
! Курение |
Артериальная гиперто ния |
склероза (для мужчин - до 55 | |
|
Избыточное потребление ал- |
Ожирение, метаболический X - |
лет, для женщин — до 65 лет) | |
|
коголя |
синдром |
Наличие в анамнезе семьи больного | |
|
Сниженная физическая ак- |
Сахарный диабет |
гиперлипидемий | |
|
тивность |
Гипергомоцистеинемия Стресс-коронарный тип личности (тип А по Дженкинсону) Дисфункция эндотелия Увеличение скорости распространения пульсовой волны |
| |
127
Существует большое многообразие теорий развития атеросклероза. Многие из них имеют историческую ценность. В настоящее время общепризнано, что основными звеньями патогенеза атеросклероза являются: нарушения метаболизма липидов, повреждение эндотелия и воспаление.
Первой научной теорией развития атеросклероза является инфильтрационно-комбинационная (холестериновая) теория атеросклероза Н.Н. Аничкова [1915,1935]. Она базируется на положении, согласно которому основная часть энергетических потребностей артериальной стенки, особенно ее бессосудистых структур (интимы и внутренней трети медии), восполняется за счет липидов плазмы крови. Делается допущение, что плазменные липиды поступают в сосудистую стенку путем просачивания (инфильтрации) плазмы в направлении от эндотелия к адвентиции. Предполагается также, что в норме липиды просачивающейся плазмы проходят без задержки в адвентицию и удаляются через систему лимфатических сосудов. Однако, когда количество липидов велико, они накапливаются в сосудистой стенке, вызывая развитие липидоза.
Весомым подтверждением холестериновой концепции заболевания являются случаи гомозиготной гиперхолестеринемии, при которой тяжелейший атеросклероз развивается в юношеские и даже детские годы, и только снижение уровня холестерина в крови, каким бы путем оно ни достигалось, спасает больных от инфаркта миокарда и неминуемой гибели.
В настоящее время эта теория получила дальнейшее развитие. Прежде всего, установлено, что развитию атеросклероза способствует нарушение метаболизма в организме липопротеидов с преобладанием в крови атерогенных липопротеидов - ЛПНП и ЛПОНП, снижение концентрации ЛПВП. Дело в том, что физиологическая функция ЛПНП заключается в транспорте холестерина в клетки, а ЛПВП наоборот выводит избытки холестерина из клеток На этом основании для оценки атерогенности плазмы крови
был предложен коэффициент атерогенности Климова (КА), который в норме должен быть ниже 3,5 у.е., а его увеличение отражает возрастание риска развития атеросклеротическо-го поражения артерий. В приведенной ниже формуле для расчета КА, ОХС - концентрация в плазме крови общего ХС, ХС ЛПВП - холестерина ЛПВП.
ОХС-ХСЛПВПКА =
хслпвп
[ : I
Установлено так же, что белковая часть молекулы липопротеидов (апопротеины) играет важную роль во взаимодействии этих молекул с клетками, обеспечивая связь молекулы с соответствующим рецептором клетки для ЛПНП, или ЛПВП. Поэтому избыток апопротеина В (апо В), входящего в состав ЛПНП, способствует избыточному транспорту ХС в клетки, а недостаток апо А, входящего в состав ЛПВП, препятствует его выведению из клеток и из организма в целом.
Существенный вклад понимание природы атеросклеротического процесса внесла аутоиммунная теория А.Н. Климова и др. (1980-1995), Согласно этой теории инициацию атеросклероза вызывают не столько липопротеины, сколько аутоиммунные комплексы, содержащие липопротеины в качестве антигена. Аутоиммунные комплексы вызывают повреждение эндотелия, ускоряют проникновение липопротеидов в сосудистую стенку; продлевают циркуляцию липопротеинов в крови и задерживают окисление и экскрецию холестерина с желчью, способствуют развитию гиперлипопротеинемии; откладываясь и фиксируясь в стенке артерий, оказывают цитотоксическое действие.
128
В настоящее время популярной является воспалительная теория атеросклероза. В ее
основе лежит сходство атеросклеротического процесса с воспалением. При атеросклерозе отмечена выраженная лейкоцитарная инфильтрация сосудистой стенки; активация макрофагов, фагоцитоз липопротеидов, холестерина, аутоиммунных комплексов, обломков клеточных структур; выделение лейкоцитами провосполительных цитокинов; гипергидратация сосудистой стенки и пролиферация гладкомышечных клеток. Воспалительная теория атеросклероза позволяет понять, что агрессивное развитие атеросклероза с формированием осложненных тромбо- и эмболоопасных бляшек связано с преобладанием провоспалительных цитокинов и гиперэргическим течением воспаления в сосудистой стенке, а стабильное течение атеросклероза - с преобладанием противовоспалительных цитокинов и гипоэргическим воспалением.
Важную роль в патогенезе атеросклероза играет повреждение эндотелия артериальной стенки. Эндотелий играет роль защитного барьера в отношении атеросклероза. Повреждение эндотелия вследствие воздействия гемодинамических факторов (кровяное давление, турбулентность потока крови, ее боковое давление), токсинов (никотин, некоторые лекарственные препараты), воспалительных процессов (вирусных, бактериальных, иммунологических), образования тромбов способствует проникновению макромоле-кулярных соединений из плазмы крови в артериальную стенку. Повреждение эндотелия сопровождается развитием дисфункции эндотелия, которая проявляется нарушением выработки в эндотелии биологически активных соединений. Важное значение при этом имеет снижение выработки эндотелиальными клетками оксида азота, оказывающего сосудорасширяющее действие, и цростациклина, снижающего тромборезистентность сосудистой стенки.
Кроме того, в процессе развития атеросклероза значимую роль играет акцивация пе-рекисного окисления линидов. Свободно-радикальное окисление ненасыщенных жирных кислот вызывает повреждение клеточных мембран в сосудистой стенке и способствует развитию атеросклероза.
Перекиси липидов ингибируют в эндотелиальных клетках артерий фермент про-стациклин-синтетазу в результате развивается локальная недостаточность простациклина при сравнительно высоком содержании тромбоксана, что усиливает агрегацию тромбоцитов на поверхности эндотелия, способствует развитию атеросклероза и его тромботиче-ских осложнений. В целом нарушение свертывающей системы крови с гиперкоагуляцией способствует развитию атеросклероза и атеротромбоза.
Существенное стимулирующее влияние на развитие атеросклероза оказывает нарушение метаболизма аминокислоты гомоцистеина - гипергомоцистеинемия. Гомоци-стеин - высокореакционная аминокислота, которая легко подвергается аутоокислению, что запускает в крови каскад свободно-радикального окисления, вызывающий повреждение эндотелия. Токсическое повреждение эндотелия сопровождается увеличением пролиферации гладкомышечных клеток, усилением адгезии и агрегации тромбоцитов, активацией коагуляционного гемостаза и торможением фибринолиза, дислипоппротеидемией, усилением адгезии моноцитов к эндотелию, увеличением продукции провоспалительных цитокинов (ФИО, ИЛ 6, 12). Все это приводит к ускоренному развитию атеросклероза с агрессивным течением. При гипергомоцистеинемии атеросклероз дебютирует на 5-10 лет раньше обычного срока (у людей 30-40 лет) и протекает с высокой склонностью к тромбо-тическим осложнениям.
Гомоцистеин образуется в организме из метионина, который поступает с пищей. Избыток гомоцистеина превращается в цистеин и выводится с мочой, и (или) вновь превращается в метионин. Превращения метионина протекают под влиянием ферментов, кофакторами которых служат витамины Вб, В12 и фолиевая кислота. Поэтому основной причиной гипергомоцистеинемии является витаминодефицит при нарушении питания, пищеварения и алкоголизме. Значительную роль играет аутосомно-рецессивная наследственная
129
ферментопатия энзимов метаболизма гомоцистеина. К гипергомоцистеинемии приводит так же почечная недостаточность.