

Факторы, определяющие конформации полимерных цепей.
Хотя число возможных конформаций полимерных цепей невообразимо велико, это не означает, что любые произвольно выбранные двугранные углы между связями могут быть реализованы. Первая причина этого достаточно очевидна: невозможно возникновение таких конформаций, в которых происходит пространственное наложение атомов, т.е. их сближение на расстояние меньшее суммы Ван-дер-Ваальсовских радиусов. При этом следует учитывать наложение близко расположенных атомов (ближнее взаимодействие) и атомов, расположенных в удаленных друг от друга участках цепи (дальнее взаимодействие); особо важны ближние взаимодействия. Так, для полипептидной цепи (рис.3) к ближним, в частности, относятся взаимодействия радикала Ri и группы С3=О, радикала Ri+1 и группы С6=О; значения углов i и i , при которых эти группы будут “наезжать” друг на друга, являются “запрещенными”. Чем больше величина радикала R, тем больше углов будут запрещенными и тем меньше – разрешенными. Для фрагментов полипептидных цепей, а также для некоторых целых цепей рассчитаны разрешенные значения углов и. Результаты расчета могут быть наглядно изображены на специальных графиках (графики или карты Рамачандрана). На рис. 14 (А) изображен график для глицинового звена в полипептидной цепи: тёмные области соответствуют разрешенным величинам углов и при обычных межатомных расстояниях, более светлые – при предельных межатомных расстояниях (которые могут иметь место в кристаллической структуре), белые области – запрещенные значения углов. На рис. 14 (В) приведена аналогичная диаграмма для аланинового остатка; здесь область разрешенных углов намного меньше из-за большего объёма боковой группы (СН3 вместо Н); для валинового звена (R=i-Pr) область стерически разрешенных углов иеще меньше.
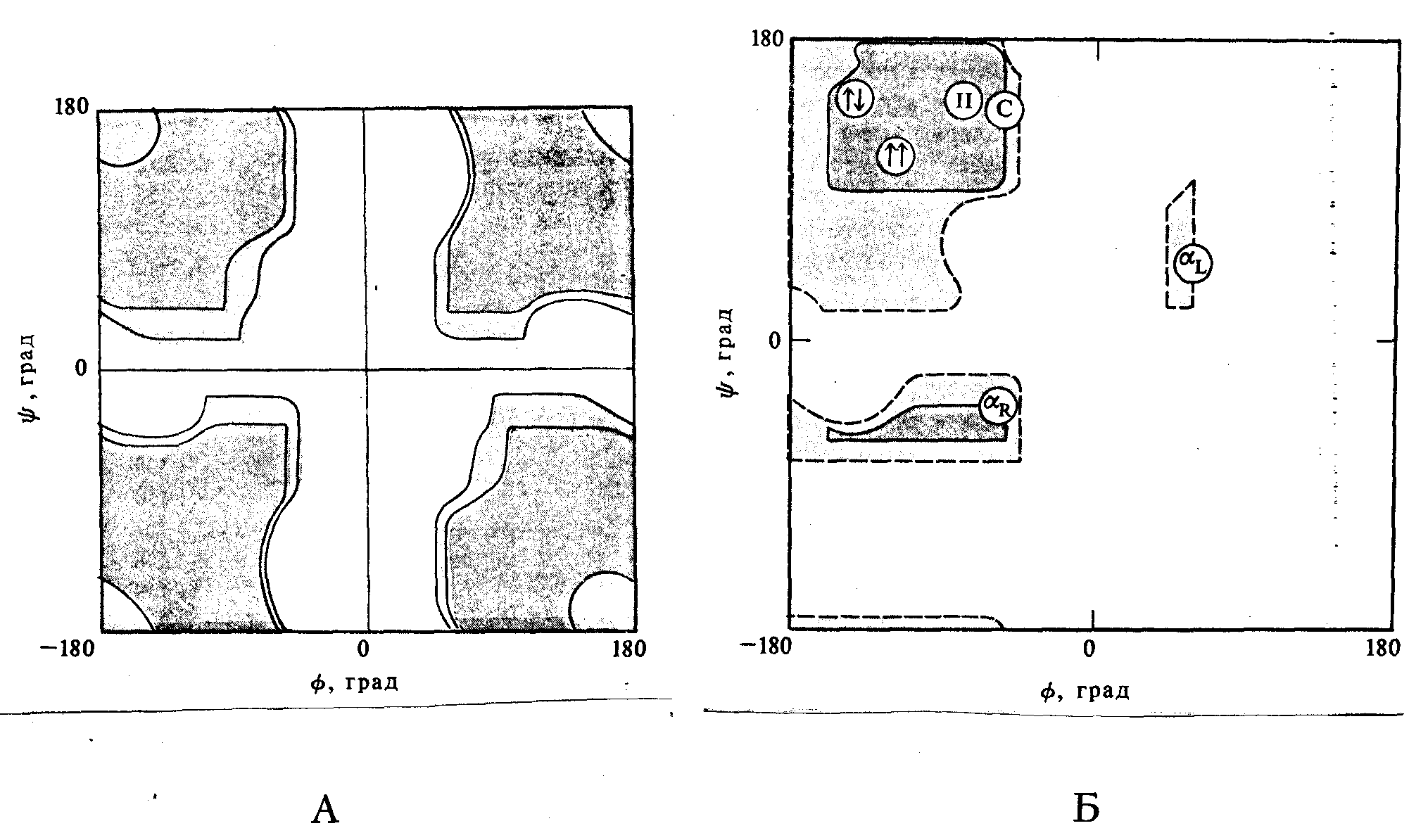
Рис.14. Стерические карты (карты Рамачандрана) для звеньев
полипептидной цепи: А – для глицинового; В – для аланинового
Рассмотренное выше прямое пространственное взаимодействие (стерическое наложение) существенно ограничивает область теоретически возможных конформаций полимерных цепей, однако и с учетом этих ограничений число “оставшихся” конформаций сколь угодно велико. Реальная пространственная форма полимерных цепей в рамках разрешенных конформаций определяется двумя основными факторами, действующими противоположно друг другу.
Первый фактор – тепловое движение, которое способствует вращению вокруг одинарных связей (полному повороту или частичным колебаниям) и, следовательно, изменениям конформаций макромолекул (их конформационной подвижности).
Второй фактор – силы внутри- и межмолекулярного взаимодействия; межмолекулярное взаимодействие включает как взаимодействие между полимерными макромолекулами, так и взаимодействие макромолекул с низкомолекулярными соединениями, например с растворителями (для биополимеров – обычно с водой). Эти силы, во-первых, делают конформации энергетически неравноценными; те, для которых силы взаимодействия максимальны, обычно имеют более низкую энергию, т.е. более устойчивы. Во-вторых они препятствуют вращению вокруг простых связей, т.е. изменению конформаций; такое вращение всегда требует затраты энергии, энергетический барьер вращения тем выше, чем значительнее силы взаимодействия. Иными словами, силы внутри- и межмолекулярного взаимодействия препятствуют изменениям конформаций и способствуют образованию стабильных долгоживущих конформаций.
Коротко рассмотрим основные виды этих сил взаимодействия.
Диполь-дипольные взаимодействия. Эти взаимодействия играют заметную роль при наличии в макромолекуле полярных групп и фрагментов, благодаря чему в ней возникают диполи. В частности, пептидные связи в белках обладают заметными дипольными моментами, достигающими 3,7 D (для сравнения: дипольный момент НС1 равен 1,03 D). Дипольные моменты параллельны связи N-H и направлены от N к Н:
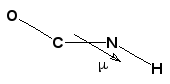
В зависимости от величин углов идиполи пептидных связей соседних звеньев могут принимать различную взаимную ориентацию и в зависимости от нее притягиваться или отталкиваться (диполи, ориентированные параллельно, отталкиваются, а ориентированные антипараллельно – притягиваются). Эти взаимодействия, естественно, влияют на энергию разных конформаций полипептидных цепей.
Водородные связи. Наиболее сильные водородные связи образуются в системах -X-H……Y- , где X, Y – атомы электроотрицательных элементов; в апротонных средах выигрыш в энергии при образовании таких связей весьма значителен – до 20 кДж/моль. Среди синтетических полимеров образование водородных связей характерно для поливинилового спирта (см. выше), полиамидов, полиакриловой кислоты и ряда других полимеров. В биополимерах возможности образования водородных связей весьма велики, и их роль трудно переоценить. В полисахаридах гидроксильные группы (рис. 5) обычно образуют межмолекулярные водородные связи; в полипептидах амидные группы образуют как внутримолекулярные (в спиралях), так и межмолекулярные (в складчатых структурах) водородные связи; в нуклеиновых кислотах также образуются межмолекулярные (межцепные) связи (в двойной спирали ДНК) и внутримолекулярные (внутрицепные) связи (в транспортных и рибосомальных РНК).
Если полимер находится в окружении протонодонорных молекул, например молекул протонных растворителей, картина может заметно меняться, так эти молекулы сами образуют водородные связи с макромолекулами полимера, что понижает энергию системы; поэтому образование водородных связей внутри полимерной системы приводит лишь к небольшому выигрышу в энергии – порядка 3-5 кДж/моль. Однако если образуется достаточно много водородных связей, выигрыш в энергии становится весьма существенным.
В последнее время обсуждается роль водородных связей в биополимерах с участием связей С-Н.
Ионные взаимодействия сводятся к отталкиванию одноименно заряженных и притяжению разноименно заряженных групп. Один из примеров ионного взаимодействия – полиакриловая кислота при рН>7 (см. ниже). Весьма характерны ионные взаимодействия для полипептидов и полинуклеотидов. В полипептидах наиболее характерно притяжение между разноименно заряженными ионизированными группами: СОО- (Asp, Glu) и –NH3+, =NH2+ (Lys , Arg, His). При сближении разноименно заряженных групп до расстояния порядка 0,3 нм между ними возникает ионная связь (солевой мостик):

 Lys-NH3+
-OOC-Asp
Lys-NH3+
-OOC-Asp
Ионные связи – сильные взаимодействия, тем не менее, при образовании этих связей в белках выигрыш в энергии относительно невелик (в среднем около 4 кДж/моль на одну связь). Дело в том, что белки находятся в водной среде; при этом ионизированные группы сильно гидратированы и поэтому имеют достаточно низкую энергию; образование солевого мостика лишь ненамного понижает ее.
В нуклеиновых кислотах весьма заметную роль играет отталкивание между одноименно заряженными ионизированными фосфатными группами; оно препятствует плотной упаковке НК; для такой упаковки требуется образование комплексов с белками, например для ДНК–с гистонами.
Силы Ван-дер Ваальса. Это – силы притяжения между близко расположенными атомами; они включают три эффекта, из которых наиболее универсальным является дисперсионный эффект – взаимодействие между мгновенными диполями, которые возникают при движении электронов вокруг ядер атомов. Силы эти неспецифичны, т.е. возникают между любыми близко расположенными атомами. Силы Ван-дер Ваальса весьма невелики и дают лишь малый выигрыш в энергии при контакте двух атомов; однако, если таких контактов много, то в сумме может получиться довольно значительное притяжение. В частности, это может иметь место при параллельной ориентации макромолекул полимеров или их боковых групп. Один из характерных примеров – стэкинг - взаимодействия в двойной спирали ДНК, когда между параллельно ориентированными (уложенными в “стопку”) гетероциклическими основаниями (особенно пуриновыми) возникают весьма значительные “вертикальные” взаимодействия.
Гидрофобные взаимодействия. Этот вид взаимодействия типичен для амфифильных молекул, помещенных в водную среду; среди низкомолекулярных соединений такие взаимодействия характерны для солей жирных кислот, для фосфолипидов и др.; среди высокомолекулярных соединений они наиболее типичны для белков, макромолекулы которых содержат как полярные, так и неполярные боковые группы, В водной среде неполярные группы предпочитают ассоциироваться друг с другом, избегая контакта с водой (напротив, полярные группы ассоциируют с водой).
Именно эти взаимодействия являются одной из важнейших причин, заставляющих многие пептидные цепи сворачиваться в глобулы. Гидрофобные взаимодействия в белках выгодны потому, что при ассоциации неполярных групп происходит увеличение энтропии. Этот неочевидный вывод становится понятным, если учесть, что неассоциированные неполярные группы окружены молекулами воды, которые образуют вокруг них упорядоченные структуры, например, в форме многогранников, в полостях которых находятся неполярные группы; эти многогранники могут объединяться, образуя кластеры. При ассоциации неполярных групп эти структуры разрушаются; это приводит к уменьшению упорядоченности системы и, следовательно, к увеличению ее энтропии. Энтальпия при этом растет, так что ассоциация гидрофобных групп эндотермична, а значит, повышение температуры благоприятствует гидрофобным взаимодействиям.
Соотношение противоположно действующих факторов - теплового движения и сил внутри- и межмолекулярного взаимодействия определяется:
а). Температурой, т.е. интенсивностью теплового движения: при достаточно низких температурах любые молекулы теряют конформационную подвижность, и любая конформация становится долгоживущей. Кстати, эта конформация не обязательно самая устойчивая - при быстром охлаждении можно “заморозить” и малоустойчивые конформации, например вытянутые конформации в растянутом образце каучука. Напротив, при достаточно высокой температуре большинство макромолекул приобретает подвижность (разумеется, речь идет о температурах ниже температуры разложения полимера). В качестве примера можно привести термическую денатурацию белков и нуклеиновых кислот. Для нас наибольший интерес представляют конформации макромолекул при температуре порядка 3000К, т.е. при “комнатной” температуре: в этой области наиболее часто эксплуатируются изделия из синтетических полимеров; практически при тех же температурах функционируют биополимеры.
б). Химическим строением макромолекул, от которого зависят силы внутри- и межмолекулярного взаимодействия. В зависимости от строения макромолекул полимеры подразделяются на гибкие (гибкоцепные) и жесткие (жесткоцепные). Гибкие макромолекулы в определенных (и не слишком жестких) условиях изменяют конформации достаточно легко, жесткие макромолекулы в тех же условиях изменяют конформации значительно труднее.
Гибкость (жесткость) макромолекул зависит от степени и характера внутри- и межмолекулярных взаимодействий в полимере: чем сильнее такие взаимодействия, тем более жесткими являются макромолекулы.
Гибкие макромолекулы не содержат объёмных, полярных и ионизированных групп, а также групп, склонных к образованию прочных водородных связей, т.е. силы взаимодействия в них невелики (разумеется, не по абсолютной величине, а по сравнению с жесткими макромолекулами). Примерами гибкоцепных полимеров являются полиэтилен (1), полипропилен (2), полибутадиен (3); для последнего вращение вокруг двойных связей, естественно, невозможно, однако вращение вокруг простых связей, соседних с двойными, происходит особенно легко.
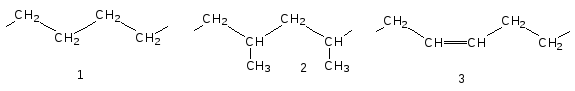
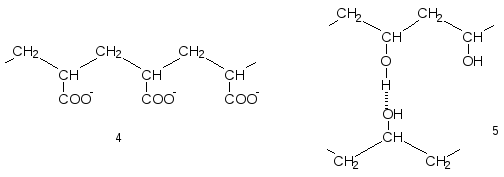
Появление в макромолекулах объёмных и особенно полярных групп увеличивают их жесткость, поскольку при этом возрастают силы внутри- и межмолекулярного взаимодействия. Примерами могут служить полиакриловая кислота в ионизированной форме, т.е. при рН>7 (4), в которой наблюдается отталкивание одноименно заряженных групп или поливиниловый спирт (5), гидроксильные группы которого образуют межмолекулярные водородные связи.
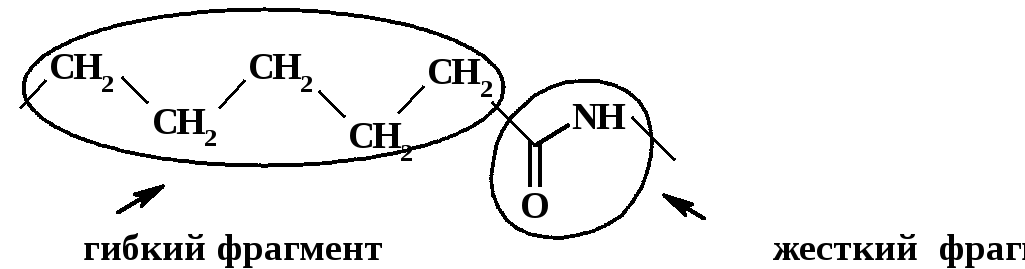 Некоторые
макромолекулы сочетают гибкие и жесткие
фрагменты; примером может служить
поликапролактам (6):
Некоторые
макромолекулы сочетают гибкие и жесткие
фрагменты; примером может служить
поликапролактам (6):
Амидная группа – типично жесткий фрагмент, т.к. а) вращение вокруг амидной связи затруднено (см. выше); б) амидная группа является полярной; в) она спо-
собна к образованию как внутри-, так и межмолекулярных водородных связей. В то же время пентаметиленовый фрагмент является гибким.
В ряде случаев жесткость макромолекул обусловлена не только силами внутри- и межмолекулярными взаимодействий, но и другими факторами:
А. Сопряжением: если в полимерной цепи имеются фрагменты, способные к
сопряжению, то наиболее устойчивыми будут конформации, в которых эти фрагменты будут копланарными, и их изменения будут энергетически невыгодными. Один из примеров такой ситуации был рассмотрен ранее – затрудненность вращения вокруг пептидной связи; это же относится ко всем полиамидам и полиэфирам, а также к полиуретанам, полимочевинам и ряду других полимеров.
Б. Наличием циклических фрагментов в полимерной цепи. В циклической структуре возможности вращения вокруг простых связей ограничены (в алициклах и неароматических гетероциклах) или отсутствуют (в ароматических циклах); поэтому включение циклов в основную цепь уменьшают ее конформационную подвижность. Типичным примером могут служить ароматические полиамиды; здесь к тому же система является высокосопряженной.
Ч резвычайно
жесткими являются макромолекулы
лестничных полимеров (рис.1,d).
Среди биополимеров циклические фрагменты
входят в структуру цепей полисахаридов
и нуклеиновых кислот.
резвычайно
жесткими являются макромолекулы
лестничных полимеров (рис.1,d).
Среди биополимеров циклические фрагменты
входят в структуру цепей полисахаридов
и нуклеиновых кислот.
В. Образованием агрегатов цепей. Наиболее известный пример – двойная спираль ДНК, в которой вращению вокруг простых связей любой из цепей препятствует другая цепь; это очень сильно повышает жесткость [однако полностью не исключает гибкости (см. ниже персистентную модель гибкости)].
Для всех важнейших биополимеров - белков, нуклеиновых кислот, полисахаридов – силы взаимодействия весьма значительны, а для белков они исключительно многообразны, пожалуй, в наибольшей степени среди всех известных полимеров; действуют и остальные факторы, повышающие жесткость. Следовательно, все эти биополимеры относятся к жестким (жесткоцепным) полимерам; их отличает высокая конформационная стабильность.
Сегмент как мера гибкости макромолекулы. Гибкость макромолекул проявляется в том, что в процессе теплового движения или при действии внешней силы отдельные участки длинных полимерных цепей могут проявлять кинетическую независимость, т.е. перемещаться в пространстве как бы независимо от остальных участков; такие кинетически независимые участки называются сегментами. Простейшей моделью такой ситуации является поведение обычной достаточно длинной гибкой нити: отдельные ее участки можно перемещать, не затрагивая положения остальной части.
В макромолекулах полимеров кинетическая независимость сегментов связана с конформационной подвижностью цепей. Допустим, длинная полимерная цепь находится в таких условиях, при которых возможно изменение ее конформаций путем поворота вокруг простых связей (для простоты возьмем полиэтилен с элементарным звеном СН2):
П![]() оложение
связи 3 по отношению к связи 1 определено
неточно, поскольку происходит поворот
вокруг простой связи 2, в результате
чего связь 3 может принимать новой
положение (например, 3’). Еще менее точно
определяется положение связи 4 относительно
связи 1 [(на это влияют повороты уже
вокруг двух связей (2 и 3)]; наконец,
достаточно удаленная связь (N) располагается
относительно связи 1 практически
произвольно. Расстояние между связями
1 и N можно считать величиной сегмента.
оложение
связи 3 по отношению к связи 1 определено
неточно, поскольку происходит поворот
вокруг простой связи 2, в результате
чего связь 3 может принимать новой
положение (например, 3’). Еще менее точно
определяется положение связи 4 относительно
связи 1 [(на это влияют повороты уже
вокруг двух связей (2 и 3)]; наконец,
достаточно удаленная связь (N) располагается
относительно связи 1 практически
произвольно. Расстояние между связями
1 и N можно считать величиной сегмента.
Естественно, чем более гибкой является макромолекула, тем меньше величина сегмента (чем гибче молекула, тем больше средний угол вращения вокруг простой связи и тем менее точно определяется положение удаленных связей относительно выбранной связи 1). Разумеется, величина сегмента зависит от условий, в которых находится полимер: в первую очередь от температуры, а также от того, находится полимер в растворе или в конденсированной фазе, от приложенной нагрузки и т.д. Поэтому сравнивать величины сегментов разных полимеров (а, следовательно, их гибкость) можно только в одинаковых условиях. При этом величины сегментов для гибких и жестких макромолекул могут отличаться в десятки раз. Например, в тех условиях, в которых сегмент гибкоцепного полимера (например, полибутадиена) включает 20-30 атомов основной цепи, сегмент жесткоцепного полимера (например, ароматического полиамида) содержит несколько сот атомов цепи.
Существование кинетически независимых частей молекулы (сегментов) характерно только для высокомолекулярных соединений (хотя и не в любых условиях) и приводит к появлению целого ряда специфических свойств; некоторые из этих свойств в практически очень важны (отметим хотя бы эластичность). Причиной этой специфики является своеобразное “расщепление” свойств многих полимеров: некоторые свойства определяются поведением целых молекул, а некоторые – поведением отдельных сегментов. Более подробно об этом будет сказано при дальнейшем рассмотрении. Существует несколько способов определения величины сегмента; при этом следует помнить, что сегменты не являются какими-то конкретными участками цепей, а представляют собой средние или эквивалентные величины (т.е. полимерная цепь ведет себя так, как если в ней в данных условиях содержится m сегментов в среднем по n звеньев каждый).
Термодинамическая и кинетическая гибкость. Термодинамическая гибкость характеризует способность полимерных цепей менять конформации в условиях термодинамического равновесия, в отсутствие изменений внешнего поля. В этом случае тепловое движение приводит к равновесному распределению конформаций: преобладают конформации с более низкой энергией. Мерой термодинамической гибкости является термодинамический сегмент (сегмент Куна); иногда его называют статистическим сегментом. Термодинамическая гибкость (и соответственно величина сегмента Куна) определяется в основном природой связей, образующих полимерную цепь. Кинетическая гибкость характеризует конформационную подвижность макромолекул при изменении внешнего поля, например, при действии механической нагрузки; при этом система неравновесна. Здесь наиболее важна скорость изменения конформаций (а не их энергии); скорость же зависит от величины потенциального барьера вращения: чем меньше эта величина, тем выше кинетическая гибкость. Мерой кинетической гибкости является кинетический сегмент; его величина определяется как величиной барьера вращения, так и характеристиками приложенного к полимеру воздействия (например, величиной механического воздействия на полимер). Кинетическая гибкость важна при практическом использовании ВМС, т.к. они часто эксплуатируются в динамическом режиме.
Персистентная гибкость. В отдельных случаях гибкость макромолекул обусловлена не способностью к изменению конформаций. Типичный пример – двойная спираль ДНК, где вращение вокруг простых связей невозможно из-за переплетения цепей. Поэтому изгибание цепи идет практически только за счет небольшой деформации валентных углов. При этом гибкость распределена вдоль двойной спирали равномерно, и макромолекула принимает вид упругой червеобразной нити. Такой механизм формирования пространственной структуры цепи называют персистентным, а пространственную модель полимерной цепи – персистентной моделью.