

Лекция 4. Анализ ппэ. Данные о переходном состоянии. Скорость определяющих в одно- и многоступенчатых химических реакциях.
Понятие переходного состояния (активированного комплекса) лежит в основе теоретического и экспериментального изучения механизмов химических реакций. Для того, чтобы получить определяющие навыки в использовании этого важнейшего в теоретической химической кинетики Понтия в данной лекции мы вначале рассмотрим на простейших примерах анализ одностадийных и многостадийных реакций. Кроме того, что обсудим некоторые общие закономерности химических превращений.
Для простого случая радикального замещения
H – H + D• → H• + HD
принято линейное расположение реагирующих атомов. Здесь общая энергия является функцией только расстояний rн-ни rH-D, таким образом «поверхность энергии» как функция определяется только этими параметрами [рис. 1(а)]. Перспективное изображение часто заменяют проекцией наr, r-плоскость [рис. 1 (б) ], по аналогии с линиями высот на географической карте. Поверхность энергии выглядит как перевал, через который возможен переход из одной энергетической впадины в другую.

Рис.1 Изменение потенциальной энергии при реакции:
H–H+D• → H–D+H•
(1)—кривая диссоциации связи Н—D; (2)—кривая диссоциации связи Н—Н
Точка, находящаяся на седловине, носит название переходного состояния (по-английски Transition state). Из этой точки система без всякого поглощения энергии может перейти либо в продукт реакции, либо вернуться в исходное состояние, причем оба процесса дают выигрыш в энергии. Этим объясняется низкая заселенность переходного состояния, для которого время жизни очень мало (≈10-12с). Кратчайший путь, по которому исходная система превращается в продукт с наименьшей затратой энергии [пунктирная линия на рис. 1(6)], можно представить на плоскости, если принять содержание энергии за ординату, тогда как по абсциссе будет изображен ход реакции от исходного состояния до продукта (рис. 2). Такую абсциссу называют координатой реакции. На приведенной энергетической диаграмме виден максимум, соответствующий наименьшей энергии, которую должна получить система для того, чтобы реакция прошла.
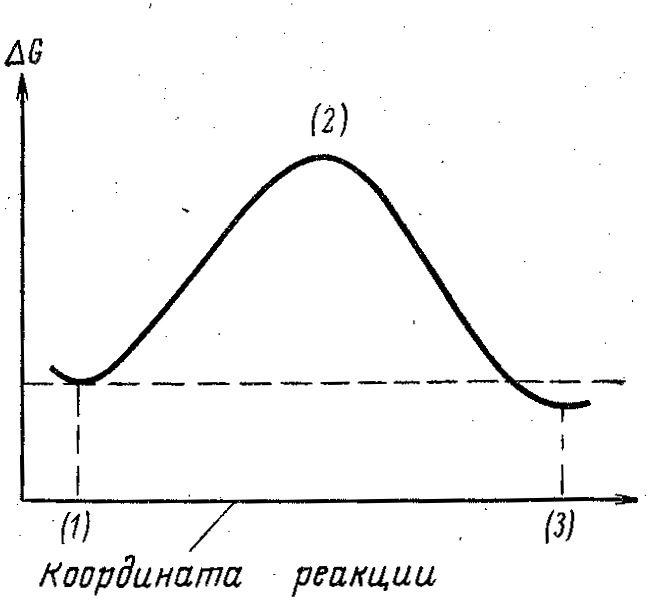
Рис. 2. Диаграмма изменения энергии при одноступенчатой реакции: (1) —исходные вещества; (2) — переходное состояние; (3) — продукты.

Рис. 3. Связь между изменением энергии реакции и потенциалами разрыва и образования связи:
(1) — кривая диссоциации связи Н—Н; (2) —кривая диссоциации связи Н—D.
Этому максимуму на поверхности энергии (см. рис. 1) соответствует седловина, через которую возможен переход из одной энергетической впадины в другую. В случае реакции
Н—H+D•→ H• +H—D
можно представить, что рис. 2 соответствует наложению потенциальных кривых связей Н—Н и Н—D(рис. 3).
Разница энергии между переходным состоянием и кривыми диссоциации проистекает вследствие связывающего взаимодействия двух Н-атомов с D-атомом в переходном состоянии.
Реакции более сложных молекул на отдельных стадиях должны иметь энергетическую диаграмму, похожую на рассмотренную выше. Расчеты в этом случае, разумеется, очень быстро усложняются, поэтому данные часто можно получить только при выборе определенного числа краевых условий.
Из-за многочисленных трудностей, которыми сопровождается определение поверхности энергии в случае реакций больших молекул, основные данные энергетических диаграмм желательно получить экспериментально. Если энергетическая диаграмма имеет максимум, как на рис. 2, то высота его определяет скорость реакции. Высота максимума соответствует энергии активации Еав уравнении Аррениуса
![]()
или свободной энтальпии активации ΔG≠в уравнении Эйринга
![]()
ΔG≠обозначает также свободную энергию активации Гиббса. Хотя разница междуΔG≠иΔF≠свободной энергией активации (Гельмгольца) для реакций в растворе численно невелика, обозначение свободной энергии активации какΔG≠, особенно в англосаксонской литературе, некорректно.
Значения ΔG≠или Еаполучают экспериментально из измерений скорости реакции или ее температурной зависимости (к — постоянная Больцмана,h— квантовая постоянная Планка,R— газовая константа, Т — абсолютная температура).
Для реакции первого порядка, например распада какой-либо молекулы на две части, существует следующая численная зависимость между скоростью реакции и свободной энтальпией активации:

Если энергия переходного состояния
определяется из скорости реакции по
уравнениям Эйринга или Аррениуса,
то, с другой стороны, положение максимума
по оси абсцисс, отвечающего положению
переходного состояния по координате
реакции, экспериментально определить
нельзя. Рис. 3 показывает, что большая
часть энергетической диаграммы
составляется из потенциальных кривых
разрывающихся и образующихся связей.
Если образующиеся и разрывающиеся связи
имеют различную прочность, то при реакции
наблюдается тепловой эффект ΔG(рис. 4). Соответственно изменяется
крутизна ветвей на энергетической
диаграмме. Если, например, образующаяся
связь существенно прочнее, чем
разрывающаяся, то, скорее всего,

 нисходящая
ветвь энергетической диаграммы будет
круче, чем восходящая.
нисходящая
ветвь энергетической диаграммы будет
круче, чем восходящая.
Исходя из упрощенного предположения, что обе ветви энергетической диаграммы обладают одинаковой крутизной, был выведен постулат Хэммонда.

Рис. 4. Диаграмма изменения энергии при экзотермической реакции: (1) — исходное состояние; (2) — переходное состояние; (3) —продукты.
Рис. 5. Диаграмма изменения энергии при эндотермической реакции: (1) — исходное состояние; (2) — переходное состояние; (3) — продукты.
В соответствии с этим постулатом переходное состояние экзотермической реакции (рис. 4) лежит ближе на координате реакции, т. е. расположение атомов в переходном состоянии и в исходном должно быть сходным. Наоборот, в случае эндотермической реакции (рис. 5) переходное состояние лежит дальше на координате реакции и его структура аналогична структуре продукта. О возможности оценки положения переходного состояния на координате реакции см. гл. 3.