


давлением, создаваемым пульсирующим насосом («искусственное сердце»), протекает в узком зазоре между двумя мембранами, омываемыми снаружи физиологическим раствором (физиологические растворы – это водные растворы, близкие по солевому составу, величине рН и другим свойствам к крови здорового человека, например, раствор, содержащий 0,9 % NaCl и 4,5 % глюкозы). Благодаря большой площади мембран (~15000 см2) из крови за 3-4 часа удаляются все вышеперечисленные «шлаки».
Размер пор мембран для ультрафильтрации составляет величину от 1 до 10 нм. Если использовать мембраны с более тонкими порами (менее 1 нм), то происходит задержка не только дисперсных частиц, но и относительно крупных молекул и даже ионов (размер ионов в водном растворе довольно значителен благодаря образованию гидратной оболочки). Правда для проведения такого процесса требуется рабочее давление большее, чем в случае ультрафильтрации. Этот баромембранный процесс называется гиперфильтрацией или
обратным осмосом.
Интересно отметить, что метод гиперфильтрации наряду с методом перегонки применяется в быту и промышленности для очистки и деионизации воды.
В результате диализа и ультрафильтрации из золей за счет избирательного переноса частиц через мембрану удаляются электролиты. Различия между этими процессами заключаются в механизме и движущей силе переноса вещества. В случае диализа очистка осуществляется за счет диффузии ионов или молекул, которые преимущественно имеют размер, существенно меньший, чем размер коллоидных частиц, а в случае ультрафильтрации разделение ионов, молекул и коллоидных частиц происходит по принципу сита. Движущая сила ультрафильтрации – градиент давления, а не градиент концентрации, как в случае диализа. В процессе очистки диализом золь разбавляется, а при ультрафильтрации – концентрируется.
17 Теории строения двойного электрического слоя
Строение коллоидных частиц может быть объяснено существованием двойного электрического слоя (ДЭС). При помещении коллоидного раствора в постоянное электрическое поле у дисперсных частиц обнаруживается заряд, который обусловлен или диссоциацией молекул, или избирательной адсорбцией ионов одного знака из дисперсионной среды. Поскольку коллоидная система электрически нейтральна, то в дисперсионной среде, окружающей дисперсные частицы, должны появиться электрические заряды противоположного знака и компенсирующие заряд частиц.
Существование ДЭС на границе раздела фаз играет важную роль во многих явлениях, имеющих место в дисперсных системах. Это электрокинетические и электрокапиллярные явления, а также электростатическое взаимодействие частиц, которое определяет устойчивость или неустойчивость дисперсных и коллоидных систем.
Первые представления о строении двойного электрического слоя были высказаны Гельмгольцем. Гельмгольц полагал, что ДЭС состоит из двух равномерно расположенных слоев зарядов противоположного знака. Это позволило рассматривать двойной слой как обычный плоский конденсатор, одна обкладка которого связана с твердой поверхностью (частицей, стенкой капилляра и т. д.), а другая, несущая противоположный знак заряда, находится в жидкости на очень малом расстоянии от первой. В этом случае падение потенциала в двойном слое происходит лишь линейно (рис. 8а), а плотность заряда
поверхности определяется по известной формуле плоского конденсатора: |
|
|||
|
ρ = θo / (4π δ) |
(17.1) |
|
|
где |
ρ - плотность заряда, δ - расстояние |
между |
обкладками |
конденсатора, |
- диэлектрическая постоянная среды между ними, θo |
- потенциал |
поверхности |
||
относительно раствора. |
|
|
|
|
Дальнейшее развитие теория двойного электрического слоя получила в работах Гуи и |
||||
Чэпмена. |
Они полагали, что тепловое движение оказывает влияние на распределение той |
|||
35
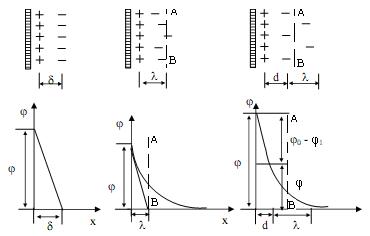
части зарядов, которая находится со стороны раствора. Согласно теории Гуи - Чэпмена, возле твердой поверхности, как и в любой другой части раствора, происходит тепловое движение ионов. Совместное воздействие электрического поля, с одной стороны, и теплового движения, с другой, приводит к тому, что двойной электрический слой оказывается не плоским, а размытым, диффузным. При возрастании температуры и диэлектрической постоянной среды диффузность двойного слоя увеличивается, а с увеличением концентрации электролита - падает. Строение двойного слоя по Гуи-Чэпмену и падение потенциала в этом слое схематически изображены на рисунке 8б. Потенциал по этой схеме падает не по прямой, а по кривой в связи с тем, что компенсирующие заряд поверхности противоионы распределены неравномерно.
Гуи и Чэпмен ввели ряд упрощений, предположив, что диэлектрическая постоянная не зависит от расстояния от поверхности твердой фазы и что собственный объем ионов равен нулю (т.е. двойной электрический слой можно рассматривать как систему точечных зарядов).
Кроме того, в этой теории не учитывалась возможность специфической адсорбции ионов. Вследствие этого некоторые экспериментальные факты не укладывались в рамках предложенной теории. Теория строения ДЭС, учитывающая специфическую адсорбцию и собственные размеры ионов, была предложена Штерном. Согласно этой теории, первый слой противоионов или даже несколько слоев удерживаются особенно прочно у поверхности за счет действия электростатических сил и сил специфической адсорбции. Эту часть двойного слоя называют адсорбционной или плотной частью. Строение ее зависит от того, сохраняется ли гидратная оболочка иона при его адсорбции или же ион частично дегидратирован. Толщину плотного слоя "d" определяют как расстояние от поверхности (точнее, от центра тяжести зарядов внутренней обкладки) до плоскости, проходящей через центры ближайших к поверхности противоионов. Эту плоскость называют плоскостью наибольшего приближения ионов. Толщина ―d‖ имеет порядок единиц ангстрем. Остальные противоионы образуют диффузную часть двойного электрического слоя.
Рисунок 8. Схема двойного электрического слоя (х – расстояние от электрода): а) ДЭС по Гельмгольцу, б) ДЭС по Гуи – Чэпмену, в) ДЭС по Штерну
Падение потенциала с расстоянием от твердой поверхности в этом случае имеет линейный характер лишь в плотном слое (рис. 8в). В области диффузного распределения ионов потенциал изменяется нелинейно.
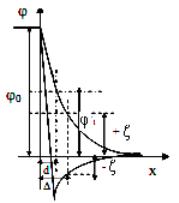
Теория ДЭС, как и всякая теория, нуждается в экспериментальной проверке. Однако нет возможности измерять потенциал на границе плотного и диффузного слоев (θ1) непосредственными, прямыми методами. Нельзя, например, поместить измерительный электрод точно в плоскость, проходящую через центры первого слоя ионов. Но можно измерять другую, близкую к θ1 величину - электрокинетический потенциал (δ), называемый
36

кратко дзета-потенциалом. Дзета-потенциал определяют как потенциал границы скольжения фаз, удаленной от границы раздела на расстояние (рис. 2). Граница скольжения устанавливается при относительном перемещении фаз в результате явлений, получивших название электрокинетических.
Рисунок 9. Соотношение между θ1 и δ - потенциалом (Δ - расстояние от поверхности до границы скольжения)
18 Электрокинетические явления
Существование двойного электрического слоя на границе раздела фаз, т.е. пространственного разделения разноименных ионов вблизи поверхности, обусловливает возникновение ряда характерных свойств дисперсных систем, в частности, электрокинетических явлений. К электрокинетическим явлениям относятся процессы взаимного смещения фаз под действием электрического поля. Это электрофорез - движение частиц дисперсной фазы в дисперсной среде и электроосмос - перенос дисперсионной среды через пористую перегородку под действием постоянного внешнего напряжения, а также обратные процессы - процессы возникновения разности потенциалов при взаимном смещении фаз - потенциалы течения и седиментации.
Величина ζ-потенциала связана со скоростью электрофореза заряженных частиц
уравнением Гельмгольца-Смолуховского:
k Ev (18.1)
где k – коэффициент, зависящий от формы частиц (для сфер k = 6, для цилиндров k = 4); v – линейная скорость перемещения частиц (или границы золя); ε – относительная диэлектрическая проницаемость; E – напряженность электрического поля.
Линейная скорость v изменяется пропорционально напряженности поля E, поэтому не может служить характеристикой частиц. В связи с этим введено понятие
электрофоретической подвижности:
|
v |
|
[U эф ] |
м |
|
м2 |
|
U эф |
|
; |
|
|
|
. (18.2) |
|
E |
с В м 1 |
|
|||||
|
|
|
|
В с |
|||
Величина Uэф не зависит от приложенного напряжения.
В системе SI значение δ -потенциала вычисляется по формуле:
|
|
|
|
v |
|||
|
|
|
|
|
(18.3) |
|
|
|
|
|
|
o E |
|||
Электрофоретическая подвижность различных частиц имеет величины порядка: для |
|||||||
золей Uэф = (0,4÷0,8)∙10-8 |
м2 |
, |
для эритроцитов животных Uэф = (1,0÷1,7)∙10-8 |
м2 |
. |
||
|
|
||||||
|
В с |
|
|
|
В с |
||
Экспериментально найденные значения электрофоретических подвижностей часто оказываются меньше расчетных. Здесь следует иметь в виду, что как единое целое перемещается не мицелла, а коллоидная частица. Поэтому на подвижность влияют два
37
явления: релаксационный эффект и электрофоретическое торможение. Первый из этих эффектов вызывается нарушением симметрии диффузного слоя ионов вокруг частиц. Второй эффект обусловлен добавочным трением электрической природы при движении частиц и противоионов в противоположные стороны. Описанная ситуация подобна той, когда лодка перемещается не в стоячей воде, а против течения воды.
Методы электрофореза имеют большое теоретическое и практическое значение. Величина δ -потенциала позволяет судить об устойчивости коллоидного раствора, поскольку изменение устойчивости, как правило, происходит симбатно с изменением электрокинетического потенциала. В настоящее время электрофорез является мощным средством для изучения фракционного состава сложных биологических систем – природных белков (метод Тизелиуса), а также используется для получения характеристики таких природных объектов, как энзимы, вирусы, бактерии, форменные элементы крови и др. С помощью электрофореза можно выделять из суспензий взвешенные частицы, а также производить покрытие твердых частиц или поверхностей слоем других веществ. Электрофорез применяют для очистки различных фармацевтических препаратов. В Фармакопеях (сборниках стандартов и положений, регламентирующих требования к качеству лекарственных средств) предусмотрено установление степени чистоты по электрофоретической однородности ряда антибиотиков, витаминов и других препаратов.
Скорость взаимного смещения фаз не зависит от деталей строения двойного электрического слоя (вида зависимости θ=f(x)), а определяется только потенциалом δ. Электрокинетический потенциал, таким образом, имеет смысл потенциала границы скольжения (рис. 2). При этом местоположение границы скольжения (расстояние Δ) по отношению к двойному электрическому слою остается неясным. Предполагается, что первый слой ионов со своими гидратными оболочками и первый слой молекул воды, смачивающих твердую фазу, не перемещаются относительно твердой фазы при течении жидкости через капилляр. Поэтому граница скольжения должна проходить либо на расстоянии "d" от поверхности (d = Δ) в этом случае δ = θ; либо смещена глубже в жидкую фазу, оставляя часть ионов диффузного слоя в неподвижном гидродинамическом слое жидкости; в этом случае δ < θ1. Различие между δ и θ1 должно быть тем менее заметным, чем больше диффузность ДЭС, т.е. уменьшается в области разбавленных растворов.
В соответствии с рассмотренной теорией строения ДЭС δ -потенциал всегда меньше общего скачка потенциала θ и должен быть одинаков с ним по знаку. Сжатие двойного электрического слоя при введении электролитов должно приводить к уменьшению δ - потенциала. Это соответствует результатам экспериментального изучения влияния электролитов на электрокинетический потенциал. Для электролитов с многозарядными, сильно адсорбирующимися ионами наблюдается явление перезарядки поверхности. Это связано со способностью таких ионов к специфической адсорбции - эти ионы адсорбируются в таких количествах, что не только нейтрализуют заряд поверхности, но и перезаряжают частицу. При этом общий потенциал θ остается постоянным, а δ - потенциал меняет знак на обратный (рис. 10).
Рисунок 10. Изменение потенциала в ДЭС при сверхэквивалентной адсорбции
38