

Частина третя хвороби мультифакторіальні і остеопороз
Глава 9. Асоціація онкогенетичних синдромів і остеопорозу
В.М.Лісовий, ю.Б.Гречаніна, л.В.Молодан
Зростання захворюванності й смертності від злоякісних новоутворень, пізня виявляємість, обмежена ефективність лікування, складність медико-соціальної реабілітації хворих диктують необхідність ранньої діагностики й профілактики цього виду патології.
За даними ВООЗ, кожна п'ята людина, що народилася в 1985 році, ризикує протягом життя перенести те або інше пухлинне ураження. Ризик розвитку раку для українців становить 28% для чоловіків і 18% для жінок.
На сьогодні прийнята класифікація pака, яка основана на етіологічному принципі, виглядає наступним чином. Рак ділиться на 4 групи:
Спадкові форми, які викликані мутацією.
Мультифакторіальні форми раку, в основі яких лежить взаємодія спадкової схильності та зовнішнього середовища.
Пухлини, що виникають у результаті спонтанних мутацій у соматичних клітинах.
Пухлини, які викликані впливом мутагенів і канцерогенів (фізичних, біологічних, хімічних).
Інтерес представляють спадкові форми pаку, оскільки вони можуть стати тією моделлю, яка навчить нас попереджувати і адекватно лікувати цю грізну патологію. Саме тому, ми вважали за доцільне розглянути онкогенетичні синдроми (ОГС) - такі моногенні, хромосомні та мультифакторіальні хвороби, при яких пухлина розвивається в результаті двох послідовних мутацій - геpмінальної і соматичної. ОГС - це генетично гетерогенна група захворювань, при яких ризик розвитку неоплазій у 10 - 100 разів вищий, ніж у популяції. Серед хворих з пухлинними ураженнями ОГС складають близько 7-10%.
Концепція розвитку раку при онкогенетичних синдромах полягає в тому, що рак - многостадійний процес, необхідно кілька генетичних подій для його розвитку. Для переходу нормальної клітини в малігнізовану, необхідні щонайменше дві мутації: перша - у статевих клітинах (гаметична), друга - у соматичних (соматична). Перша, гаметична мутація, робить клітину більш чутливою до вбудовування в її ДНК вірусу, до впливу різних мутагенів, тоді як друга (соматична) призводить до ракової трансформації.
У ґенезі розвитку раку має значення:
- імунна дефектність;
- хромосомна нестабільність;
- дефектність репараційних ферментів ДНК, яка вперше була показана на прикладі пігментної ксеродерми;
- порушення процесів метилювання.
При багатьох онкогенетичних синдромах з високою частотою спостерігається вторинний остеопороз та паранеопластичний рахіт і остеомаляція, які супроводжують гіпогонадизм, акромегалію, тиреотоксикоз, цукровий діабет, дефіцит кальцію внаслідок порушення його всмоктування (схема 1,2).
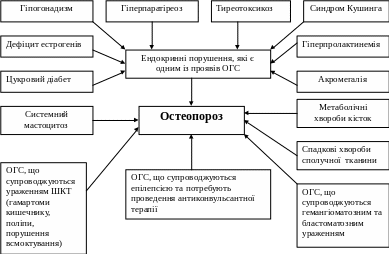
Схема 1. Можливі причини розвитку остеопорозу при ОГС
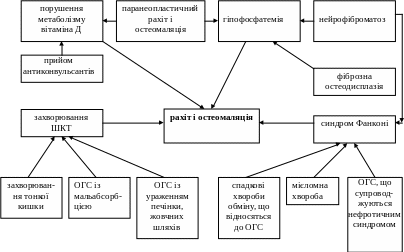
Схема 2. Можливі причини розвитку рахіту та остеомаляції при ОГС
Остеомаляція з прискореною елімінацією фосфату нирками часто виникає при пухлинах мезенхімального походження, в тому числі при гігантоклітинних пухлинах кісток (доброякісних та злоякісних), гемангіомах та фібромах, раку передміхурової залози, а також при репаративній гігантоклітинній гранульомі. Припускають, що пухлини секретують гуморальні фактори, що порушують транспорт фосфату в проксимальних канальцях та інгібують ниркову І-гідроксилазу. У ряді випадків видалення пухлини призводить до зниження елімінації фосфату нирками, підвищенню рівня фосфату в сироватці та виліковності остеомаляції. При описаних синдромах рівень 1.25(ОН)2D3 у сироватці дуже низький, але при цьому довгостроковий прийом досить високих доз кальцитріола не впливає на швидкість елімінації фосфату та його концентрацію у сироватці.
Серед хворих із моногенною або хромосомною детермінацією схильності до розвитку злоякісних пухлин описані стійкі сполучення зовнішніх проявів фенотипічних змін, що дозволили виділити більш ніж 300 онкогенетичних синдромів. Знання клінічних проявів цих синдромів і розуміння механізму розвитку злоякісних пухлин дає можливість лікарю будь-якої спеціальності при виявленні у хворого онкогенетичного синдрому вже в ранньому дитячому віці намітити таку тактику ведення, яка б попереджала розвиток пухлини та розвиток вторинного остеопорозу. Ми дозволимо собі лише коротко перелічити групи онкогенетичних синдромів, сподіваючись у майбутньому зупинитися на докладному їхньому описі й викладі питань реабілітації хворих.
До онкогенетичних синдромів відносяться:
Хромосомні хвороби - синдроми Дауна, Патау, Едварса, Шерешевського-Тернера, Клайнфельтера, 4 р-, 5 р-, 8q-, 15р+, 18 р- та інші;
Синдроми, пов'язані з мікроструктурними аномаліями хромосом - синдроми Беквіта-Відемана, Лангера-Гідіона, Ангельмана, Прадера-Віллі та ін.;
Гамартози (факоматози) - туберозний склероз, нейрофіброматоз, синдроми Штурге-Вебера, Луї-Бар, Гіппеля-Ліндау, Кліппеля-Треноне-Вебера; Рандю-Ослера, Блоха-Сульцбергера, Горліна-Гольця, множинних лентіго, нейрошкіряний меланоз, гіпомеланоз Іто, невус сальних залоз лінійний, розсіяний ангіоматоз та ін.;
Спадкові захворювання опорно-рухового апарата - множинна деформуюча суглобна хондродисплазія, хвороба Педжета, черубізм, синдром МакК'юна-Олбрайта, множинний хондроматоз, дерматоостеопойкелія, остеопетроз та ін.;
Спадкові захворювання шкіри - пігментна ксеродерма, альбінізм, іхтіоз, порокератоз Мебеллі, гідротична та ангідротична ектодермальна дисплазія, множинний ліпоматоз, епітеліома Брука, булльозний епідермоліз, вроджений дискератоз, невус Сеттона, синдром диспластичного невуса та ін.;
Спадкові захворювання ендокринної системи - дизгенезія гонад, тестикулярна фемінізація, множинний ендокринний аденоматоз I, II і III типів, синдром Вернера, сімейна феохромоцитома, нейро-ендокринно-кутанеальні синдроми та ін.;
Спадково обумовлені ураження шлунково-кишкового тракту - кератоз долонь, підошов та рак стравоходу; сімейний поліпоз кишечнику, синдром Гарднера, синдром Тюрко, генералізований юнацький поліпоз, поліпоз товстої кишки із множинними пухлинами сальних залоз, фіброматоз ясен та ін.;
Спадкові порушення обміну - тирозинемія, глікогенози, фруктоземія, целіакія, синдром Жильбера та ін.;
Ряд інших моногенних захворювань - синдроми Протея, Блума, Парі-Ромберга, Морган'ї-Стюарт-Мореля та ін.;
Ембріональні пухлини – нейробластома, ретинобластома, нефробластома;
Сімейний раковий синдром.
Ураження кісткової системи при хромосомній патології виявляється часто і представлено ВВР, СТД, остеопорозом, який детально розглянутий у відповідній главі.
При хромосомній патології часто зустрічаються пухлини різної локалізації. Нами були діагностовані:
гострий лейкоз у періоді новонародженості у двох випадках хвороби Дауна;
краніофарингіома, пухлина грудних залоз при синдромі Клайнфельтера;
пухлина гіпофіза, органів малого таза при синдромі Шерешевського-Тернера;
пухлина грудної залози у пацієнтки зі збалансованою транслокацією 18 хромосоми на 22;
множинні екзостози, мастопатія у хворого із частковою моносомією довгого плеча 8 хромосоми (рис. 1.)
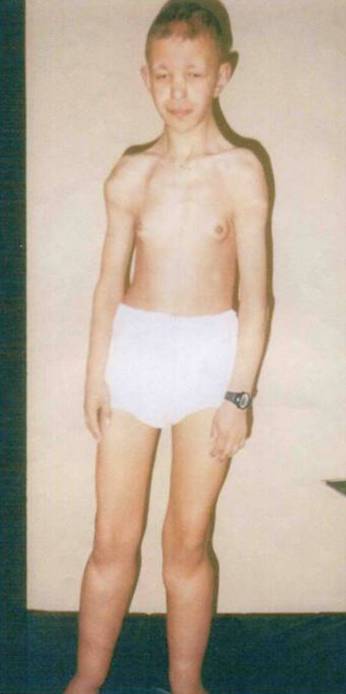
Рис. 1. 8q-.
Пухлинні ураження діагностовані нами при хромосомному поліморфізмі та екстремальних варіантах. Так, у дитини зі сутничним поліморфізмом діагностована кіста нирки, а у пацієнта з екстремальним варіантом по 9 хромосомі - злоякісна пухлина яєчка. Ці дані, на нашу думку, вимагають подальшого поглибленого дослідження.
Онкогенетичний характер хромосомної патології необхідно враховувати при призначенні терапії із приводу затримки темпів психомоторного розвитку, тонусних порушень, розумової відсталості, судорожного синдрому, гіпогонадизму, порушення менструального циклу, неплідності тому що застосування біостимуляторів, фізіотерапії, масивної гормонотерапії може призвести до другої соматичної мутації й розвитку раку.
Висока частота остеопорозу у хворих на хромосомні хвороби потребує його профілактики та корекції для запобігання розвитку ускладнень.
Серед моногенних захворювань високу питому вагу в нашому регіоні мають факоматози – група генетично гетерогенних і клінічно поліморфних моногенних захворювань, які характеризуються ураженням шкіри, нервової системи, судин, високим ризиком розвитку пухлин на тлі яких розвивається остеопороз, який носить вторинний характер.
Шкірні прояви представлені різноманітними пігментними та депігментованими плямами, невусами, нейрофібромами, аденомами сальних залоз, ангіомами, гемангіомами, ангі- і телеангіектазіями тощо.
Неврологічні розлади досить поліморфні та варіюють від чітко окреслених вогнищевих проявів до розсіяної мікросимптоматики та змін у психічній сфері. Більшість хворих мають високий інтелект, що обумовлено особливостями васкуляризації мозку.
Факоматози відносятся до категорії онкогенетичних синдромів і характеризуються високим ризиком розвитку пухлин.
Більшості цих захворювань властива імунна дефектність, хромосомна нестабільність і дефектність репараційних ферментів ДНК.
Факоматози поділяються на ангіоматозні, бластоматозні, раритетні та некласифіковані форми.
Класифікація факоматозів
І. Бластоматозні форми факоматозів.
1.Нейрофіброматоз.
Нейрофіброматоз І типу
Нейрофіброматоз ІІ типу
2. Туберозний склероз.
ІІ. Ангіоматозні форми факоматозів.
Енцефалотригемінальний ангіоматоз (хвороба Штурге-Вебера).
1.1 Кутанеальна форма
1.2 Нейро-кутанеальна форма
1.3 Нейро-окуло-кутанеальна форма
Синдром Луї-Бар
Синдром Кліппеля-Треноне-Вебера.
3.1 Гемігіпертрофічна форма
3.2 Круциатна форма
Хвороба Рандю-Ослера.
Хвороба Гиппеля-Ліндау.
ІІІ. Раритетні форми факоматозів*.
1.Синдром неутримання пігменту.
Синдром Блоха-Сульцбергера
спорадична форма
сімейна форма
1.2 Гіпомеланоз Іто
2. Синдром Горліна-Гольця
3. Нейро-шкіряний меланоз.
4. Невус Сеттона.
5. Синдром Маффуччі.
Синдром лінійного невусу сальних залоз.
Гемангіоматоз розсіяний.
Ангіоматоз дифузний кортико-менінгеальний.
Синдром множинних лентіго
Синдром диспластичного невуса
ІY. Некласифіковані форми факоматозів.
Деякі форми факоматозів, що зазначені у класифікації як раритетні у регіоні Східної України зустрічаються часто, зокрема: синдром Горліна-Гольця, невус Сеттона та ін.
Враховуючи ту обставину, що хворі на факоматози мають бластоматозні чи ангіоматозні ураження шкіри, нервової системи та внутрішніх органів, високу частоту розвитку неоплазій, розвиток остеопорозу, який носить вторинний характер для даної групи хворих є досить високим.
Вимоги до постановки діагнозу
Для ранньої діагностики факоматозів у групах високого генетичного ризику до програми обстеження родини повинні бути включені:
- загальноклінічне обстеження;
- детальна оцінка фенотипу пробанда та членів його родини;
- генеалогічний і синдромологічний аналізи;
- дослідження неврологічного статусу;
- дослідження офтальмологічного статусу;
-біохімічні, імунологічні, електрофізіологічні, цитогенетичні методи обстеження;
- ендоскопічні методи дослідження (за показаннями);
- ультрасонографія;
- ультразвукова та комп’ютерна денситометрія;
- комп'ютерна томографія та ЯМРТ.
Такий підхід дозволяє діагностувати захворювання в доклінічній або на ранній стадії, вчасно провести медико-генетичне консультування, призначити адекватну терапію, яка спрямована на попередження ускладнень у перебігу хвороби, на профілактику розвитку остеопорозу, пухлинного росту, розробити методи реабілітації та профілактики захворювання, а в разі необхідності провести пренатальну діагностику та вибрати адекватну тактику ведення вагітності та пологів.