

Контрольные вопросы по модулю «Врождённые пороки»
I. Внутриутробное развитие
Основные этапы внутриутробного развития, их последовательность и общая характеристика
Поясните понятия: что такое плюрипотентность и полипотентность, детерминация, морфогенез, гомейозисные гены? Каково значение гибели клеток в эмбриогенезе?
Периоды пренатального развития, их продолжительность
Мейоз
Сперматозоид. Строение, функционирование. Капацитация
Яйцеклетка. Строение клетки и её оболочек, значение оболочек
Что такое оплодотворение? Из каких событий оно состоит?
Взаимодействие сперматозоида с оболочками яйцеклетки в ходе оплодотворения. Акросомная реакция, ее значение. Кортикальная реакция, ее значение
Формирование оболочки оплодотворения и моноспермия
Зигота. Ее образование и характеристика (указать сроки)
Что такое дробление? Характеристика и ход дробления. Компактизация (указать сроки)
Формирование и строение бластоцисты (указать сроки)
Что такое эпибласт и гипобласт, каково их значение?
Образование, строение и значение первичной полоски
Что такое гаструляция? Как проходит гаструляция у человека?
что такое эмбриональная индукция? В чем суть и значение первичной эмбриональной индукции?
Перечислите зародышевые листки и их основные производные
Производные зародышевой эктодермы
Производные зародышевой энтодермы
Какова организация мезодермы? Перечислите основные производные различных отделов зародышевой мезодермы
Дорсальная (или пресомитная, параксиальная) мезодерма и образование сомитов. Строение сомитов и их производные
Начало и последовательность событий в ходе нейруляции (указать сроки)
Что такое нервный гребень, как и когда он образуется? Перечислите его производные
Понятие о мезенхиме. Производные мезенхимы
Как происходит формирование тела? (указать сроки)
Развитие эпителиальной и соединительной ткани. Связь эпителия с базальной мембраной. Как происходит образование коллагеновых и эластических волокон?
Хондрогенез, рост хряща и его регуляция
Прямой и непрямой остеогенез. Развитие костей скелета и черепа
Кроветворение у эмбриона и плода, стадии гемопоэза. Кровь новорожденного. Возрастные изменения гемограммы, физиологические перекресты
Развитие мышечной ткани
Развитие нервной ткани и системы
Развитие органа зрения, органа слуха
Развитие гипофиза, щитовидной и паращитовидных желез, надпочечника
Развитие сердца. Кровообращение плода и новорожденного
Развитие пищеварительной системы. Глоточный аппарат и его производные. Развитие лица и органов ротовой полости
Развитие органов дыхания
Развитие мочевыделительной системы
Развитие половых систем и дифференцировка пола
Что такое имплантация и как она происходит? (указать сроки)
Развитие провизорных органов — плаценты, желточного мешка, амниона, аллантоиса — и их значение
II. Врожденные пороки
Что такое врожденный порок? Каковы причины возникновения ВП?

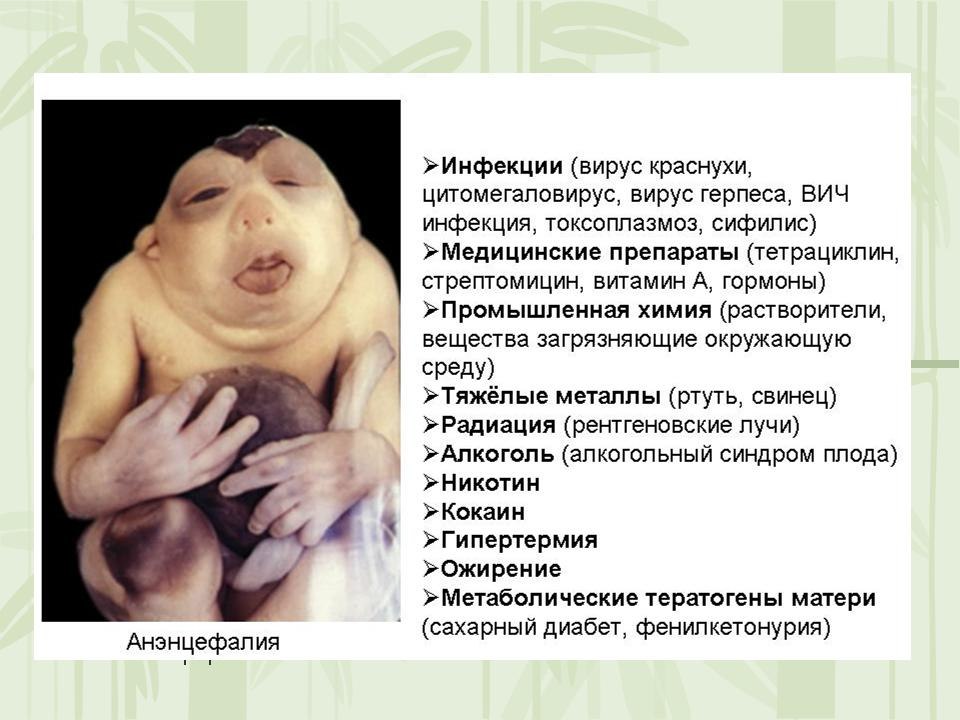
43.Классификация ВП. Что такое мальформация, дизрупция, деформация, дисплазия?

Мальформации и их причины (дефекты гомейозисных генов, генетические факторы)
Дизрупции и тератогены
Критические периоды возникновения врождённых пороков (указать сроки) и частота выявляемых аномалий

Гаметопатии — аномалии числа и структуры гамет. Синдром неподвижных ресничек, синдром круглой головки и др
Гаметопатия (gametopathia; Гамета + греч. pathos страдание, болезнь) 1) общее название нарушений в строении и функционировании гамет, а также повреждений зиготы на первых стадиях ее дробления; 2) общее название болезней, возникновение которых обусловлено изменениями генетического материала в процессе гаметогенеза, во время оплодотворения и на первых стадиях дробления оплодотворенной яйцеклетки.
Гаметопатии – поражение гаметпредков или родителей, а также повреждение зиготы и первых стадий ее дробления. Нек-рые исследователи понимают гаметопатии (см.) лишь как ненаследственные поражения гамет.
Причинами гаметопатии являются разнообразные влияния внешней среды (физические, химические и биологические) на половые клетки родителей или предков, вызывающие мутации, в т. ч.и так наз. спонтанные мутации.
Cиндром округлой головки-отсутствие способности к оплодотворению вследствие нарушенного формирования акросом сперматид и сперматозоидов. По разным оценкам эта патология обусловливает до 15% мужского бесплодия. Сперматозоиды имеют головку округлой формы, они подвижны, но невозможна акросомная реакция.

47.Что такое перезревание гамет?
Перезревание сперматозоидов происходит в половых путях женщин, когда увеличивается время от эякуляции до слияния гамет.
В основе возникновения феномена старения гамет лежит де-синхронизация заключительных этапов созревания, овуляции и оплодотворения. Наиболее изученным он является в отношении женских половых клеток. Возобновившие мейоз ооциты находятся в перманентном состоянии развития и преобразования, они не могут задержаться ни на одном из этапов этого процесса, и для его нормального продолжения «обречены» (и всегда готовы!) сделать «следующий шаг». При отсутствии такой возможности начинается процесс старения (точнее — перезревания) яйцеклеток, сопровождающийся рядом изменений морфофункционального характера, крайне неблагоприятно влияющих на «судьбу» репродуктивного процесса, в том числе на оплодотворение, состояние эмбриона и потомства.
Старение гамет можно рассматривать как универсальный процесс, возникающий при ряде нарушений регуляции репродуктивного процесса, а также в физиологических условиях при некоторых изменениях внешних факторов среды. Различают два типа перезревания женских гамет:
- преовуляторное (внутрифолликулярное)
- постовуляторное.
Первое обусловлено, как правило, задержкой (отсутствием) овуляции, второе — задержкой (отсутствием) оплодотворения.
Экспериментальные исследования и ряд клинических наблюдений показали, что результатом перезревания гамет может быть потеря ими способности к оплодотворению, а при происшедшем оплодотворении — формирование аномальных эмбрионов, в том числе с хромосомопатиями, возникновение других типов аномалий развития, когнитивные нарушения у потомства и т.п.
Что такое близнецы (монозиготные, дизиготные)? Причины из образования. Причины возникновения и типы сросшихся близнецов (пагов)
Монозиготные близнецы- образуются из одной зиготы (одной яйцеклетки, оплодотворенной одним сперматозоидом), разделившейся на стадии дробления на две (или более) части. Они обладают одинаковыми генотипами.
Причины из образования
разделение оплодотворенной яйцеклетки может происходить в результате задержки имплантации и дефицита кислородной насыщенности.
причиной полиэмбрионии может быть механическое разъединение бластомеров (на ранних стадиях дробления), возникающее в результате охлаждения, нарушения кислотности и ионного состава среды, воздействия токсических и других факторов.
возникновение монозиготной двойни связывают также и с оплодотворением яйцеклетки, имевшей два или более ядра. Каждое ядро соединяется с ядерным веществом сперматозоида, в результате чего образуются зародышевые зачатки. Описаны яйцеклетки и с тремя ядрами.
Дизиготные близнецы- Дизиготные близнецы развиваются в том случае, если две яйцеклетки оплодотворены двумя сперматозоидами. Естественно, дизиготные близнецы имеют различные генотипы.
Причины из образования
1 . мощная гормональная стимуляция яичников. Высокий уровень ФСГ может вызывать созревание и овуляцию одновременно нескольких фолликулов в одном или обоих яичниках или формирование в одном фолликуле двух яйцеклеток. Чаще всего две яйцеклетки исходят из одного фолликула.
2. Возможно также, что активация гипоталамических рилизинг-факторов приводит к стимуляции гипофиза и дальнейшей полиовуляции
.
Причины возникновения и типы сросшихся близнецов (пагов)
Сросшиеся (или сиамские) близнецы являются монозиготными, поэтому имеют одинаковый набор генов и всегда одного пола. Сросшиеся близнецы появляются, если это расщепление задерживается до 13 дня после зачатия. Таким образом, это монозиготные близнецы, которые не были разделены в утробе матери и остаются сросшимися после рождения.
1.Торакопаги - срастаются грудной клеткой - большинство близнецов (около 35%).
2.Омфалопаги – это близнецы, соединенные от талии до грудины (их около 30%).
3.Пигопаги – соединены спинами (19% близнецов).
4.Более редкий тип – это «близнец-паразит», маленький близнец, который присоединен к более здоровому со-близнецу и зависит от него. Зависимый близнец может представлять собой, к примеру, голову и руки, присоединенные к животу нормального близнеца.
5. Краниопаги (craniopagus): сросшиеся черепами, но имеющие раздельные туловища.
6. Дицефалы (dicephalus): две головы, одно туловище и две, три или четыре руки (дицефал дибрахиус, трибрахиус и тетрабрахиус, соответственно)
7. Илиопаги (iliopagus): срастание в подвздошных областях, спиной к спине, включая ягодицы.
8. Парапаги (parapagus): срастание боками, иногда сердце также затронуто.
Нарушения гаструляции: тератомы и сиреномелия
Тератома - эмбриома, дизэмбриома, опухоль человека и животных, возникающая в результате нарушения эмбрионального развития тканей. Встречается преимущественно в детском или молодом возрасте; локализуется в половых железах, реже в других органах и частях тела.
Как правило, состоит из многих тканей (соединительной, эпителиальной, мышечной, нервной и др.) с включениями дифференцированных дериватов этих тканей (например, зубов, волос). Наиболее сложные состав и строение у тератомы из ранних бластомеров или из первичных половых клеток, которые тотипотентны (способны давать начало любым тканям организма). Состав тератом, возникающих на более поздних стадиях эмбрионального развития (после гаструляции), ограничен формообразовательными потенциями того зародышевого листка или зачатка органа, от которых происходит данная тератома. От простых, относительно доброкачественных тератом отличают тератобластомы — злокачественные опухоли из тканей эмбрионального строения (без тенденции к дифференцировке), а также тератоиды— пороки развития, которые опухолями не являются, но могут послужить основой для их возникновения. Возможно перерождение тератомы в рак или саркому.

Сиреномелия
Данное заболевание является врожденной патологией, которая возникает вследствие нарушения кровоснабжения и характеризуется слиянием нижних конечностей в сочетании с агенезией почек, аплазией крестца, прямой кишки и мочевого пузыря. Ранее считалось, что эта патология представляет собой тяжелую форму синдрома каудальной регрессии.
При этом Часто отмечаются пороки сердца, почек, передней брюшной стенки, грудной клетки и нижних отделов позвоночника. Также часто выявляются единственная артерия пуповины, неперфорированный анус и агенезия половых органов.
Порок может касаться мягких тканей и некоторых трубчатых костей. Наряду с этим наблюдается гипоплазия или аплазия костей конечностей и таза, врожденный вывих бедра, сгибательные контрактуры тазобедренных и коленных суставов, косолапость. Могут быть сформированы две стопы ,одна или отсутствовать.
Синдактилия и полидактилия
Синдактилия
врожденное полное или неполное сращение пальцев кисти стопы в результате не наступившего их разъединения в процессе эмбрионального развития. Встречается одинаково часто у мужчин и женщин. Односторонняя С. отмечается примерно 2 раза чаще двусторонней. Нередко сочетается с другими пороками развития (Пороки развития). Различают простую и сложную, полную и неполную формы С. Простая форма включает кожную, многослойную и костную С.; сложная — кожную, многослойную, костную и сочетанную. Чаще обнаруживается сращение III и IV пальцев, реже III—IV—V, II—III и IV пальцев. Возможно сращение нескольких пальцев в единый конгломерат, при этом нередко имеются амниотические перетяжки. В большинстве случаев С. пальцы недоразвиты и деформированы.
Полидактилия
врожденная аномалия, представляющая собой увеличение количества пальцев на конечностях. Чаще всего встречается добавочный VI палец на одной или обоих кистях либо на стопах. Но количество их может быть и значительно большим. Добавочные пальцы могут быть сформированы достаточно хорошо и правильно, но чаще встречаются в виде рудиментов (небольшие деформированные части пальцев, свисающие на тонкой кожной ножке). Иногда добавочные пальцы срастаются с V пальцем (синдактилия).
50,Поясните значение терминов: эмбриопатия, фетопатия, агенезия, аплазия, гипоплазия, гиперплазия, атрезия, стеноз, эктопия, персистенция, диссинхрония, амелия, фокомелия, эктромелия


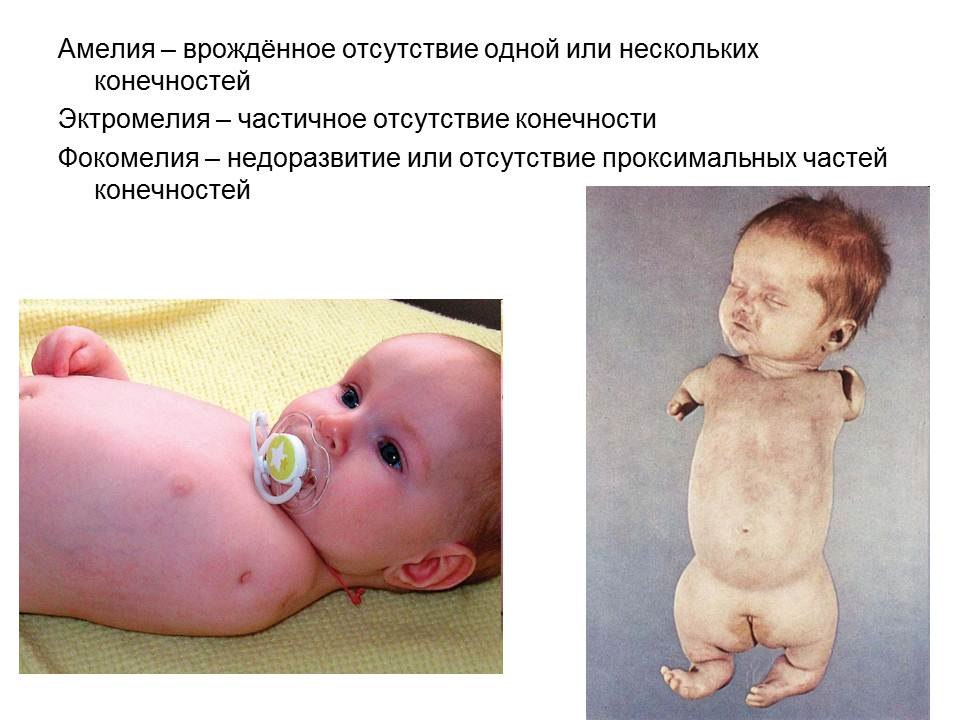
Фетопатия
(fetopathia; фето- (Фето‑) + греч. pathos страдание, болезнь)
общее название болезней плода, возникающих с начала 4-го лунного месяца внутриутробного развития, проявляющихся аномалиями развития или врожденными болезнями, нередко заканчивающихся асфиксией плода и обусловливающих преждевременные роды.
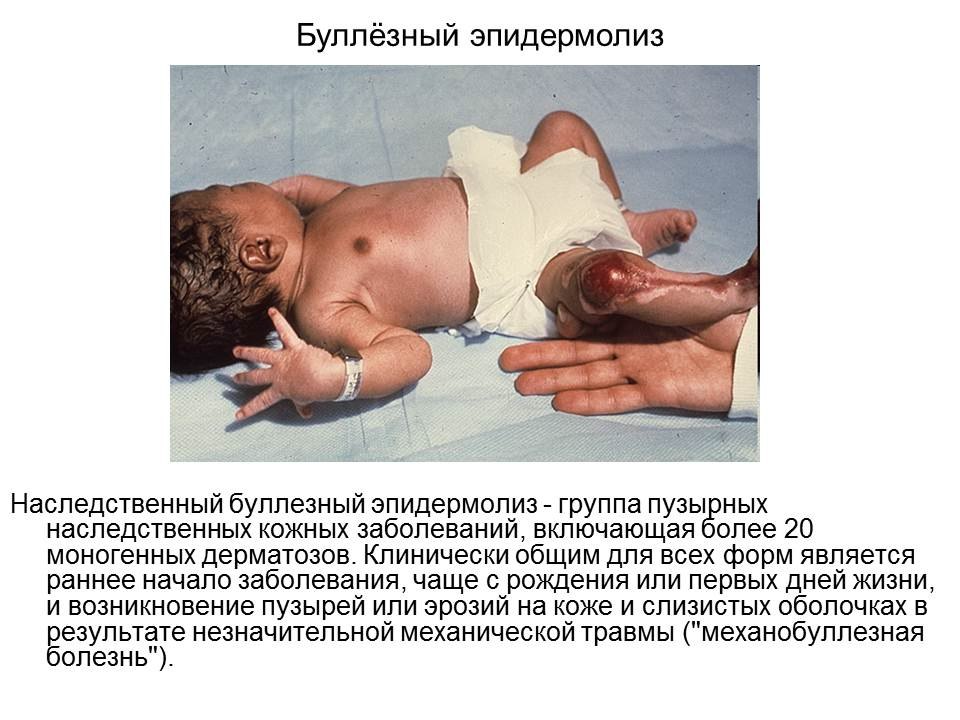
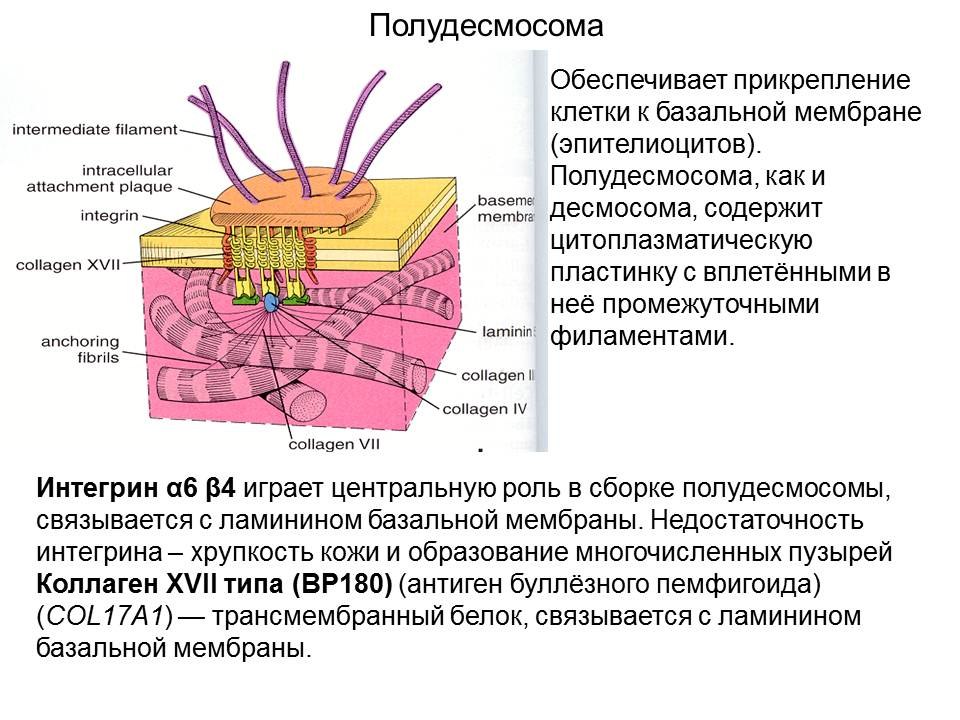
Что такое буллёзный пемфигоид? Каковы причины буллёзного эпидермолиза?
Буллёзный пемфигоид — доброкачественное хроническое заболевание кожи; первичный элемент — пузырь, формирующийся субэпидермально без признаков акантолиза. — Аутоиммунное заболевание, часто встречающееся у пожилых людей, представленное пузырьками или пузырями.
Характеризуется аутосомно-доминантным типом наследования
Синдром Элерса-Данло
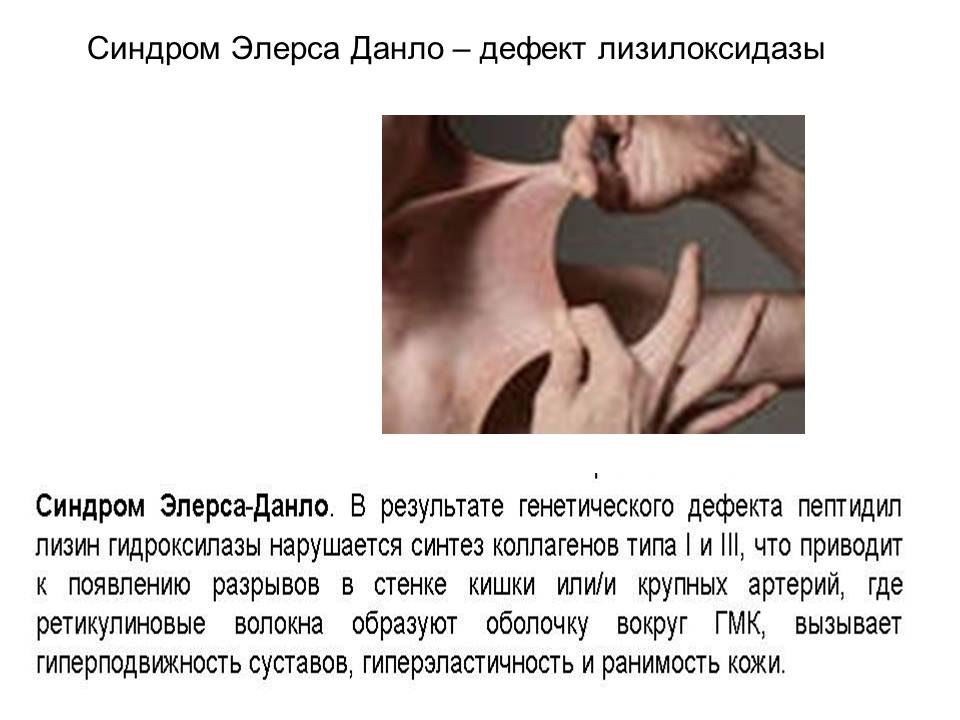
Синдром Марфана
Дефект фибриллина

Синдром Вильямса

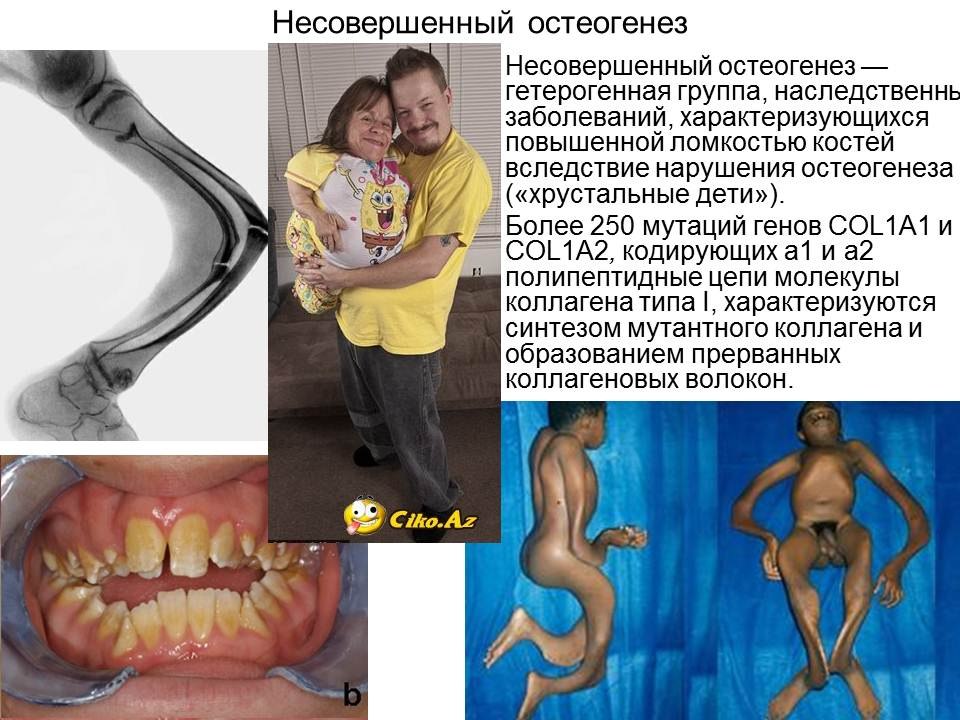
Несовершенный остеогенез
Ахондроплазия

56.Черепно-ключичный дизостоз

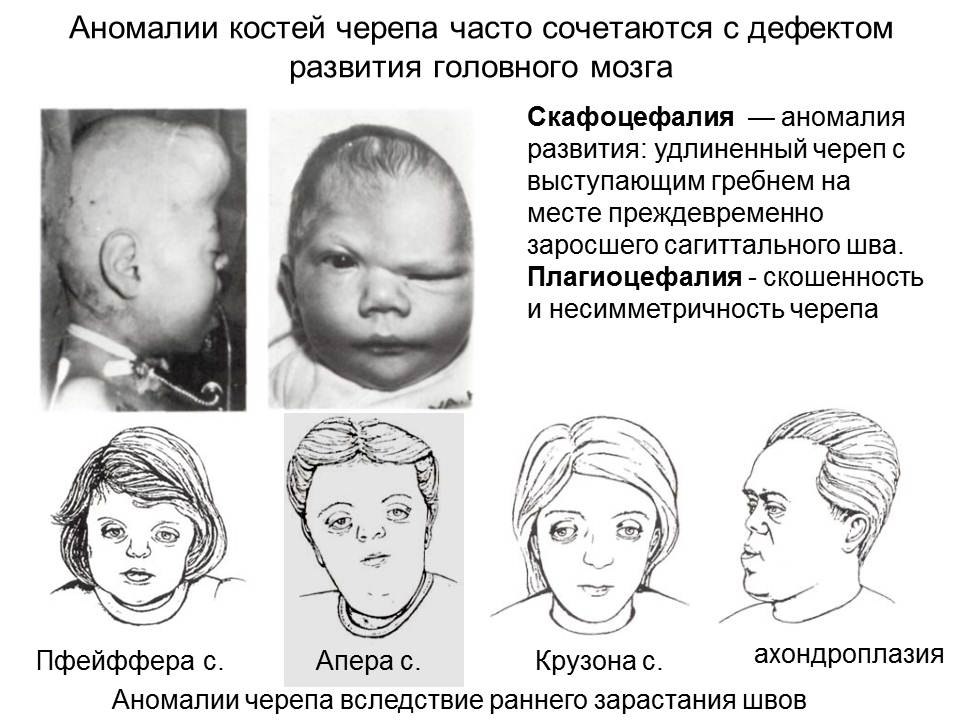
Аномалии формирования черепа (скафоцефалия, плагиоцефалия) и примеры синдромов с дефектами черепа
Примеры: Черепно-ключичный дизостоз
Анемия при недостаточности глюкозо-6-фосфатдегидрогеназы

Микросфероцитарная гемолитическая анемия (болезнь Минковского–Шоффара)
Как известно, для нормального функционирования эритроцита необходимо поддержание его нормальной формы, иначе эритроцит просто не пройдёт через просвет сосуда. В поддержании двояковогнутой формы эритроцита участвует белок спектрин. При анемии Минковского-Шоффара имеется дефект синтеза данного белка. К тому же происходит нарушение работы АТФ-азы, ввиду чего возникает повышенное поступление натрия и воды в клетку. Это приводит к тому, что эритроцит приобретает форму сферы и имеет маленький размер. Из-за этого резко падает способность эритроцита восстанавливать свою структуру после прохождения сосудов, поэтому когда эти клетки крови проходят межсинусные пространства селезёнки, те из них, которые имеют патологическую форму, разрушаются. Из-за этого вместо 120 дней при наследственном микросфероцитозе эритроцит живет всего лишь 10.
Серповидноклеточная анемия
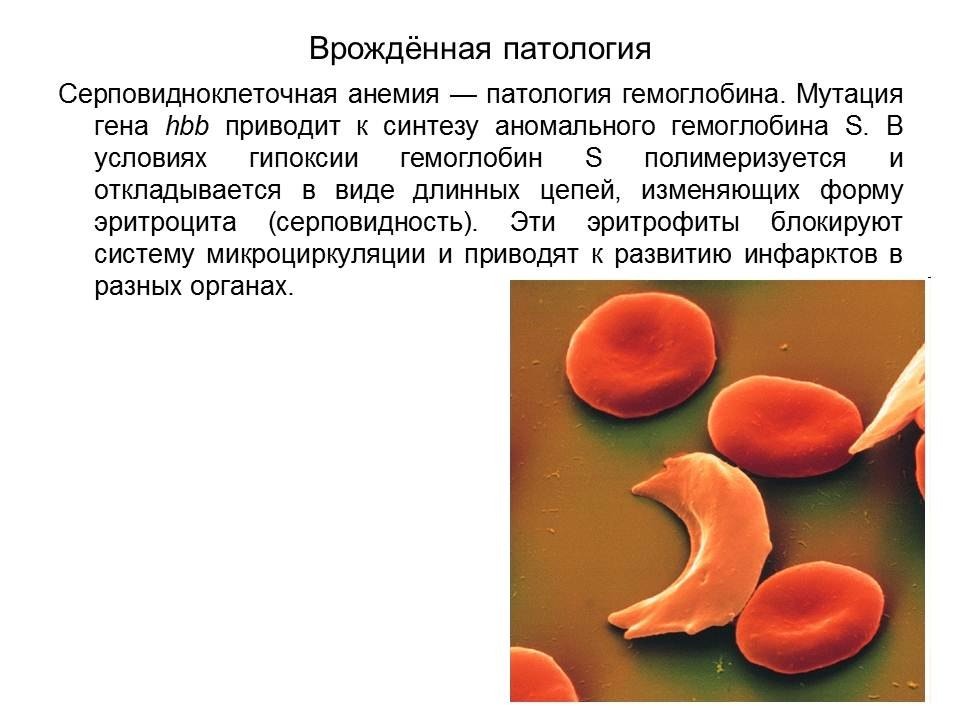
Талассемии

Агранулоцитоз наследственный (син. - врождённая нейтропения, наследуемый агранулоцитоз новорождённых, болезнь Костманна)

Дефекты функций нейтрофилов (синдромы Йова, Костманна, Шедьяка-Хигаши, ленивых лейкоцитов и др.)
- Иммунодефицит-Дефициты фагоцитоза-синдром Йова
- Первичный дефект фагоцитоза, частичный иммунодефицит, Патологические изменения гранул и ядер всех типов лейкоцитов, альбинизм, возможна гиперпигментация кожи, анемия, тромбоцитопения, изменения в костях, лёгких, сердце, а также психомоторные дефекты и предрасположенность к инфекциям.- синдром Шедьяка-Хигаши
- Ограниченная подвижность гранулоцитов и нарушение их выхода из костного мозга в кровь- синдром ленивых лейкоцитов
Тромбоцитопатии (тромбастения Глянцманна, синдром Бернара—Сулье, серых тромбоцитов синдром)
Тромбастения Гланцмана – наследственная болезнь из группы геморрагических диатезов, характеризующаяся недостаточностью ряда ферментов в тромбоцитах, вторичным нарушением ретракции кровяного сгустка и часто удлиненным временем кровотечения при нормальном или слегка пониженном количестве тромбоцитов, наличием гигантских тромбоцитов; выделяют формы с аутосомно-рецессивным и аутосомно-доминантным типом наследования.
Для тромбастении Гланцмана характерно возникновение гематом, кровотечений из слизистых и гиперменореи, а также тяжелых кровотечений в ответ на нормальные стимулы (например, хирургическое вмешательство или травму).
Синдром Бернара-Сулье- врожденная тромбоцитопатия, сопровождающаяся кровоизлияниями в кожу, слизистые оболочки, внутренние органы, развитием носовых кровотечений
Синдром серых тромбоцитов (недостаточность а-гранул): увеличение размеров тромбоцитов, серая окраска. снижение количества а-гранул и тромбоцитспецифических белков а-гранул.
Наследственные коагулопатии
Коагулопатии
собирательное обозначение болезненных состояний, обусловленных нарушениями физиологических механизмов свертывания крови, приводящие обычно к развитию геморрагического синдрома.
Наследственные коагулопатии вызываются генетически обусловленным снижением или извращением плазменных компонентов гемостаза. Наиболее распространенными формами являются гемофилия А, В, С, афибриногенемия.
Наследственные коагулопатии делятся на:
- Гемофилии: А-дефицит VIII фактора, В-дефицит IX фактора, С-дефицит XI фактора, Д-дефицит XII;
- Парагемофилия: дефицит II, V, VII, X факторов;
- Нарушение образования фибрина, дефицит фибриногена (I фактора).
Болезнь фон Виллебранда
наследственное заболевание крови, характеризующееся возникновением эпизодических спонтанных кровотечений, которые схожи с кровотечениями при гемофилии. Заболевание наследуется по принципу аутосомного доминирования.
Причина кровотечений — нарушение свертываемости крови из-за недостаточной активности фактора Виллебранда.
Наиболее характерным и специфическим симптомом при болезни Виллебранда являются кровотечения из
слизистых полости рта, носа, внутренних органов.
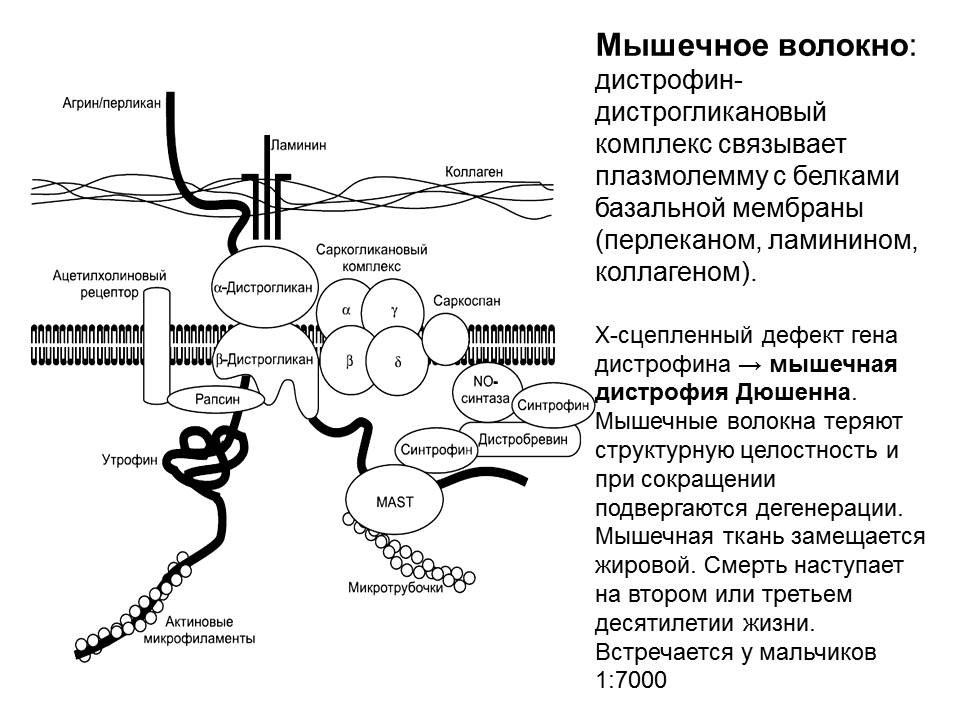
Мышечная дистрофия Дюшенна, Миотоническая дистрофия, Дистрофия мышечная Фукуяма



Миотонические миопатии (миотонии) врожденные (Томсена миотония, врождённая парамиотония и гиперкалиемический пароксизмальный паралич)


Синдром вялого ребенка (болезнь Оппенхайма)

Craniorachischisis totalis, spina bifida и ее виды, Анэнцефалия, Менингоцеле, Миеломенингоцеле, Менингогидроэнцефалоцеле, Энцефалоцеле, Гидроцефалия
Craniorachischisis totalis (краниорахисхйзис) - редко встречающийся дефект - незаращение нервной трубки. АФП - гликопротеин, синтезирующийся в желточном мешке эмбриона, желудочно-кишечном тракте и печени. Его уровень в амниотической жидкости возрастает в три-пять раз вследствие поступления из открытой нервной трубки. АФП проходит через плаценту и появляется в сыворотке крови беременной в более низких концентрациях, чем в околоплодных водах. Концентрация в сыворотке крови плода, в сыворотке беременной женщины и околоплодных водах максимальна в 15-16 недель беременности, затем происходит её снижение.
Спина Бифида (Spina bifida, расщепленный позвоночник) – порок развития позвоночника, представляющий собой неполное закрытие нервной трубки в не полностью сформированном спинном мозге. Кроме того, позвонки над открытой частью спинного мозга сформированы не полностью. Возникает порок на 3 – 4-й неделе беременности.
- spina bifida occulta - скрытое незаращение позвоночника;
- spina bifida cystica uverta - открытое расщепление позвоночника с формированием кистозной спинномозговой грыжи;
- rhachischiasis posterior (totalis et partialis) - расщепления позвоночника и мягких тканей с распластыванием спинного мозга, которые возникают на всем протяжении позвоночника или только в какой-то его части.
1. Оболочечные формы (менингоцеле) - расщепление позвоночника с выпячиванием в дефект твердой мозговой оболочки, но без вовлечения в процесс нервных структур. Твердая мозговая оболочка после выхода из костного дефекта истончается и исчезает. Купол грыжевого мешка представлен тонкой пиальной оболочкой. Кожа грыжевого выпячивания истончена, а на вершине нередко отсутствует. Содержимое грыжевого мешка - мозговые оболочки и ликвор (спинномозговая жидкость), форма его - обычно стебельчатая с суженной ножкой. Костный дефект захватывает обычно два-три позвонка. Каких-либо клинических проявлений при данной форме спинномозговых грыж не отмечается и только угроза разрыва грыжевого мешка, увеличивающиеся его размеры служат основанием для хирургической пластики дефекта.
2. Корешковая форма (менингорадикулоцеле) - расщепление позвоночника с выпячиванием в дефект оболочек спинного мозга и его корешков, которые частично могут заканчиваться в стенке мешка или входить в него, создавая петлю, но в дальнейшем, распространяясь в межпозвонковые отверстия, формируют нормальные нервы. Костный дефект захватывает 3-5 позвонков. Неврологический дефект при этой форме спинномозговых грыж зависит от количества вовлеченных в патологический процесс корешков, слепо заканчивающихся в стенке грыжевого мешка. В зависимости от этого дефекты могут проявляться от легкой слабости в конечностях и тазовых нарушений до грубых парезов и недержания мочи.
3. Мозговая форма (менингомиелоцеле или менингомиелорадикулоцеле) - расщепление позвоночника с вовлечением в грыжевой мешок оболочек, спинного мозга и его корешков. Пиальная оболочка выстилает грыжевой мешок, твердая мозговая оболочка заканчивается в зоне расщепления позвоночника, спинной мозг и корешки часто слепо заканчиваются в грыжевом мешке. Костный дефект обычно широкий и протяженный, захватывает от 3 до 6-8 позвонков. Шейки, как таковой, грыжевой мешок не имеет и из спинномозгового канала непосредственно переходит в грыжевое выпячивание. Кожа на вершине выпячивания отсутствует, грыжа покрыта тонким просвечивающимся листком пиальной оболочки. Степень неврологического дефекта всегда тяжелая - отсутствие движений в конечностях, их недоразвитие, деформации, недержание мочи и кала. Именно эта мозговая форма спинномозговых грыж встречается наиболее часто, и она нередко приводит к разрыву грыжевого мешка с истечением спинномозговой жидкости - к ликворее.
4. Кистозная форма (миелоцистоцеле) - достаточно редкая форма спинномозговых грыж, при которых конечный отдел спинного мозга резко расширен за счет центрального канала спинного мозга. Поэтому грыжевой мешок выстлан изнутри цилиндрическим эпителием, как и центральный канал. Нервные корешки отходят от наружной поверхности грыжевого выпячивания и направляются к межпозвонковым отверстиям. Степень неврологического дефекта, как и при мозговой форме, тяжелая - отсутствие движений в конечностях, грубые тазовые нарушения.
5. Осложненная форма (spina bifida complicata) характеризуется сочетанием одной из вышеперечисленных форм спинномозговых грыж с доброкачественными опухолями (липомами, фибромами), которые фиксированы к оболочкам, спинному мозгу или его корешкам.
Анэнцефалия -полное или частичное отсутствие больших полушарий головного мозга, костей свода черепа и мягких тканей.
Энцефалоцеле (энцефало- + греч. kēlē выбухание, грыжа; син.: гидроцефалоцеле — нрк, гидроэнцефалоцеле — нрк) — черепно-мозговая грыжа, содержащая оболочки и вещество головного мозга, но не включающая его желудочки.
Гидроцефалия расширение желудочковых систем мозга и субарахноидальных пространств за счет избыточного количества цереброспинальной жидкости.
Амиотрофии (Синдром Дежерина–Сотта, Спинальная амиотрофия Верднига–Хоффманна)


Нейрокристопатии

Дисплазия эктодермальная гидротическая (синдром Клустона)
В отличие от классического варианта ангидротической эктодермальной дисплазии синдром Клустона наследуется по аутосомно-доминантному типу, чем и объясняется одинаковая заболеваемость обоих полов.
Ведущими признаками этого аутосомнодоминантного заболевания служат дистрофия, гипоплазия или отсутствие ногтей, редкие волосы и гиперкератоз ладонных и подошвенных поверхностей.
Зубы обычно не изменены, хотя иногда они мелкие и с кариесом. Потоотделение не нарушается. У некоторых больных кожа в областях коленных, локтевых и голеностопных суставов гиперпигментирована.
Псориаз
Хроническое неинфекционное заболевание, дерматоз, поражающий в основном кожу. В настоящее время предполагается аутоиммунная природа этого заболевания. Обычно псориаз вызывает образование чрезмерно сухих, красных, приподнятых над поверхностью кожи пятен. Однако некоторые больные псориазом не имеют никаких видимых поражений кожи. Вызванные псориазом пятна называются псориатическими бляшками. Эти пятна являются по своей природе участками хронического воспаления и избыточной пролиферации лимфоцитов, макрофагов и кератиноцитов кожи, а также избыточного ангиогенеза (образования новых мелких капилляров) в подлежащем слое кожи.
Избыточная пролиферация кератиноцитов в псориатических бляшках и инфильтрация кожи лимфоцитами и макрофагами быстро приводит к утолщению кожи в местах поражения, её приподнятию над поверхностью здоровой кожи и к формированию характерных бледных, серых или серебристых пятен, напоминающих застывший воск или парафин («парафиновые озёрца»). Псориатические бляшки чаще всего впервые появляются на подвергающихся трению и давлению местах — поверхностях локтевых и коленных сгибов, на ягодицах
Ихтиозы
Представляют собой группу наследственных заболеваний, при которых нарушен процесс ороговения кожи. На коже появляются чешуйки по виду напоминающие рыбью чешую. На сегодняшний день дерматологи выделяют как минимум двадцать восемь разнообразных форм. Большинство форм ихтиоза провоцируются мутациями или нарушениями экспрессии генов, отвечающих за кодирование различных форм кератина.Достоверно причины возникновения заболевания до сегодняшнего дня не изучены. Считается, что ихтиоз обуславливается генетически и возникает на этапе формирования тканей зародыша. Существует точка зрения, что ихтиоз – заболевание, связанное с нарушениями в работе вегетативной нервной системы. Также в группу риска попадают люди с гиповитаминозом A.
Альбинизм
Врождённое отсутствие пигмента кожи, волос, радужной и пигментной оболочек глаза.Различают полный и частичный альбинизм.
В настоящее время считается, что причиной альбинизма является отсутствие (или блокада) фермента тирозиназы, необходимой для нормального синтеза меланина — особого вещества, от которого зависит окраска тканей.
В генах, ответственных за образование тирозиназы, могут возникать самые различные нарушения. От характера нарушения зависит степень недостатка пигмента у людей с альбинизмом.
Гиперкератозы
Это чрезмерное утолщение рогового слоя эпидермиса. Понятие гиперкератоз происходит от двух греческих слов hyper – много и keratosis – образование кератина. Клетки рогового слоя начинают усиленно делиться, что в сочетании с нарушениями слущивания эпидермиса и приводит к утолщению, которое может быть от нескольких миллиметров до нескольких сантиметров.
Гиперкератоз не является самостоятельным заболеванием. Утолщение рогового слоя и нарушение процесса ороговения наблюдаются при ихтиозе, лишаях, эритродермиях и других заболеваниях. Даже у здоровых людей гиперкератоз проявляется в той или иной мере на локтях, стопах, иногда на коленях. Экзогенные причины гиперкератозов, то есть причины, возникающие извне – это длительное и избыточное давление на кожу стоп, иногда на кожу тела из-за тесной или грубой одежды.
1, Фолликулярный гиперкератоз –В результате избыточного ороговения и нарушения отслойки верхних слоев эпидермиса происходит закупорка протока фолликула чешуйками кожи.
2, При диссеминированном гиперкератозе на коже появляются полиморфные элементы, напоминающие короткие и толстые волосы, которые располагаются изолированно без тенденции к слиянию на коже туловища и конечностей. Иногда имеются скопления группами в виде кисточки из 3-6 пораженных фолликулов.
3, диссеминированный гиперкератоз
4, Гиперкератоз стоп- Основными причинами гиперкератоза стоп является тесная и неудобная обувь, нерегулярный уход за ногами, наследственные и приобретенные патологии стопы, избыточная масса тела
Недержания пигмента синдром
Данный синдром представляет собой достаточно редкое заболевание, характеризующееся поражением кожи, нервной системы, глаз, скелета, реже других органов. В большинстве случаев наследуется Х-сцепленно доминантно. Плоды мужского пола погибают еще внутриутробно, в связи с чем заболевание встречается почти исключительно у женщин.
Характерным для данного заболевания является то, что изменения со стороны кожи протекают по стадиям, хотя такая стадийность может быть далеко не во всех случаях. В начале заболевания на коже появляются очаги воспаления, затем они трансформируются в очаги гиперпигментации (более темного цвета), затем наступает стадия депигментации (исчезновение пигмента), истончение и уплотнение кожи. Поражение кожи располагается преимущественно на боковой поверхности туловища и внутренней поверхности конечностей. Кожные изменения могут иметь как линейную форму, так и в виде причудливых фигур.
Нейрофиброматоз
Тяжелое врожденное заболевание, для которого характерно развитие в подкожной жировой клетчатке множественных нейрофибром, неврином черепных и спинномозговых нервов. Характеризуется образованием множественных нейрофибром и пигментных пятен главным образом на коже и слизистых оболочках, сопровождающееся неврологическими, психическими и гормональными нарушениями, а также изменениями в костях. При нейрофиброматозе могут поражаться все ткани и органы, но чаще кожа, подкожная клетчатка, нервные сплетения, в т.ч. межмышечное (мейсснерово) и подслизистое (ауэрбахово), нервные стволы и корешки.
ЭТИОЛОГИЯ не выяснена. Большинство авторов придерживаются теории дизонтогенетического происхождения нейрофиброматозы, другие связывают его с нарушением функции щитовидной железы, надпочечников.
Менкеса синдром (болезнь курчавых волос)
Клинически: слабо пигментированные, редкие курчавые волосы, судороги, физическое и отставание в умственном развитии, прогрессирующее поражение мозга, гипоплазия гонад. тяжелая умственная отсталость.
Тип наследования — Х-сцепленный рецессивный.
Гипертрихоз
Заболевание, проявляющееся в избыточном росте волос, не свойственному данному участку кожи, не соответствующему полу и/или возрасту.
Клинически различают врождённую (общую и ограниченную) и приобретённую формы гипертрихоза.
Причины гипертрихоза могут быть различными: наследственная предрасположенность, заболевания различных органов или желез, вырабатывающих мужские половые гормоны. Избыточный рост андрогензависимых волос у женщин называется гиперсутизмом.
Варденбурга синдром
Наследственное заболевание. Имеет следующие клинические признаки: телекант (латеральное смещение внутреннего угла глаза), гетерохромия радужки, седая прядь надо лбом и врождённая глухота.
Патология конечностей включает такие аномалии, как гипоплазия кистей и мышц, ограничение подвижности локтевых, лучезапястных и межфаланговых суставов, слияние отдельных костей запястья и плюсны. Снижение слуха при этом заболевании врождённое, воспринимающего типа, связанное с атрофией преддверно-улиткового органа (кортиев орган). Глухота вызвана нарушениями спирального (кортиева) органа с атрофическими изменениями в спинальном узле и слуховом нерве.
Ашера (Чарлза) синдром
группа синдромов слепоглухоты. Из 16000 слепоглухих людей в США более половины, как полагают, имеют синдром Ашера, - комбинация прогрессирующей пигментной ретинопатии и врождённой нейросенсорной тугоухость. Идентифицировано несколько локусов на 3, 10, 11, 14 и 21 хромосомах (см. также Наследственные болезни: картированные фенотипы).
Клинически: пигментный ретинит с началом в возрасте 10 лет, потеря зрения, катаракта, глубокая врождённая нейросенсорная тугоухость, умственная отсталость, психозы, атаксия, нарушения речи.
Гиперплазия коры надпочечников (адреногенитальный синдром)
группа наследственных болезней, в основе которых лежит недостаточность ферментов на различных уровнях синтеза стероидных гормонов коры надпочечников — кортизона и альдостерона. Тип наследования аутосомно-рецессивный. Частота 1 : 5000-1 : 6500.
Патогенез . Наследственный дефект в ферментативных системах (в большинстве случаев дефицит или недостаточность 21-гидроксилазы и дефицит 11-гидро-ксилазы) приводит к Снижению содержания в крови кортизона и альдостерона. Синтез половых гормонов при этом в коре надпочечников не нарушается. Низкий уровень кортизола в крови по принципу обратной связи стимулирует гипоталамо-гипофизарную систему и повышение секреции АКТГ. В свою очередь высокий уровень АКТГ способствует гиперплазии коры надпочечников именно той зоны, в которой не,нарушен синтез гормонов — преимущественно андрогенов. Одновременно с андрогенами образуются промежуточные продукты синтеза кортизона. В зависимости от характера ферментативного дефекта выделяют следующие формы адреногенитального синдрома: вирильную (простую, компенсированную) и сольтеряющую.
Гипертиреоидизм плода
Гипертиреоз - синдром, обусловленный избытком тиреоид-ных гормонов в крови.
При гипертиреозе повышена функция щитовидной железы. Она увеличивается в размерах и формируется зоб.
Гипертиреоз может быть генетически обусловленным. Эта патология встречается при аутоиммунном заболевании Грейвcа (они же – диффузный токсический зоб, Базедова болезнь, синдром Перри или Флаяни). Аутоиммунная этиология гипертиреоза щитовидной железы встречается в 70% случаев заболевания.
Возможной причиной гипертиреоза могут послужить узелковые новообразования на щитовидной железе. Одиночные узлы или так называемый многоузловой зоб стимулируют активность щитовидной железы и приводят к повышенной концентрации тиреоидных гормонов в организме.
Гипертиреоз может также спровоцировать воспаление щитовидной железы (острый или подострый тиреоидит).
Гиперпаратиреоз неонатальный семейный
Минимальные диагностические признаки: гиперкальциемия, повышение в сыворотке уровня иммунореактивного паратгормона.
На первых неделях жизни появляются запоры, затруднения дыхания, гепатоспленомегалия, полидипсия, полиурия, гипотония, судороги и анемия. Сухожильные рефлексы повышены. Отмечаются выраженная гиперкальциемия, гипофосфатемия, гиперкальциурия, гиперфосфатурия, аминоацидурия.
Рентгенологически выявляются деминерализация, субпери остальная резорбция костей и патологические переломы. Частым признаком заболевания является нефролитиаз. Без лечения дети погибают в первые месяцы жизни. Популяционная частота неизвестна.
Тип наследования — аутосомно-рецессивный и аутосомно-доминантный.
Врождённый гипотиреоидизм
Врожденный гипотиреоз - гетерогенная группа заболеваний, проявляющаяся врожденным дефицитом тиреоидных гормонов, развивающимся вследствие дисгенезии щитовидной железы или гипофизарной системы, а также вследствие врожденных дефектов синтеза тиреоидных гормонов и различных экзогенных воздействий (медикаменты, материнские блокирующие антитела и прочее). Другими словами, термином "врожденный гипотиреоз" обозначается гипотиреоз любого генеза, который манифестирует и диагностируется при рождении.
ВРОЖДЕННЫЙ ГИПОТИРЕОЗ встречается у новорожденных детей все чаще. Он возникает при недостатке йода в пище, а также может быть связан с недоразвитием щитовидной железы, неспособностью ее к нормальному образованию гормонов. Иногда болезнь возникает из-за приема некоторых препаратов мамой во время беременности.
Признаки врожденного гипотиреоза у детей
Диагноз «врожденный гипотиреоз» ставят как мальчикам, так и девочкам. В большинстве случаев новорожденный с подобной проблемой не отличается от других детей. Однако внимательная мама может уже с первых дней заметить признаки низкой активности щитовидной железы: малыш вяло сосет, у него большой живот, сухая кожа, низкая температура тела, выраженная желтуха. Да и крик у малыша особый, хриплый. Личико отечно, особенно веки. Часто у таких детишек выявляется пупочная грыжа.
Эктопия сердца. Пентада Кантрелла
Эктопия сердца - врожденное заболевание характеризующееся ненормальным положением сердца - вне грудной клетки: чаще всего в расколе грудной клетки, реже в брюшной полости или шее.
Пентада Кантрелла
Данное врожденное заболевание характеризуется двумя основными дефектами: эктопией сердца и дефектом передней брюшной стенки (чаще всего наблюдается омфалоцеле, но может встречаться и гастрошизис) в сочетании с нарушением развития трех, связанных между собою структур: дистального отдела грудины, передней части диафрагмы и диафрагмального отдела перикарда. Имеются сообщения о вариантах классической формы, которые характеризуются неполным проявлением данного синдрома.
Этиология. Неизвестна. Иногда может сочетаться с хромосомными аномалиями.
Патогенез. Остановка развития сегмента латеральной мезенхимы в период от 14-го до 18-го дня после зачатия приводит к незакрытию брюшной стенки и неполному слиянию наружных первичных тяжей.
Врождённые пороки сердца с дефектами перегородок (межпредсердных, межжелудочковых)
Причиной врождённого порока сердца могут быть генетические или экологические факторы, но, как правило, сочетание того и другого
Дефект межпредсердной перегородки (ДМПП)
Иногда дети рождаются с отверстием в межпредсердной перегородке.
Когда имеется широкое сообщение между предсердиями, большое количество артериальной «алой» крови из левой половины сердца сбрасывается в правую где кровь венозная «синяя». Все это количество крови протекается через правые отделы сердца и вызывает значительную перегрузку их (правого предсердия и правого желудочка).
Процесс этот продолжается непрерывно и со временем приводить к серьезным нарушения в работе сердца. Большинство детей не имеет каких-либо заметных симптомов в раннем детстве.
Хирургическое лечение закрытие в детстве дефекта межпредсердной перегородки в условиях искуственого кровообращения ( иногда с помошью заплаты) предотвратит серьезные осложнения. И не только это, отдаленные перспективы жизни также становятся весьма неплохими.
Дефект межжелудочковой перегородки (ДМЖП)
При наличии крупного отверстия между желудочками большое количество артериальной крови из левых отделов сердца сбрасывается через него в правые. Затем кровь опять перекачивается в легкие. Такое кровообращение, при котором кровь, уже побывавшая в легких, возвращается туда вновь является неэффективным. Сердце, которому необходимо перекачивать дополнительную кровь, перегружено и увеличивается в размерах.
Симптомы обычно появляются через несколько недель после рождения. Некоторые дети с крупным дефектом межжелудочковой перегородки отстают в физическом развитии и имеют сниженный вес. В сосудах легких в связи с большим количеством притекающей крови давление может быть повышенным. Некоторое время спустя повышенное давление может вызвать стойкое повреждение сосудов легких. Поэтому часто необходима как можно более ранняя операция.
Если отверстие мало, сердце не испытывает больших перегрузок. В этом случае единственным признаком, указывающим на болезнь, является громкий шум, выявляемый при выслушивании сердца. Операция в этом случае может и не понадобиться, отверстие постепенно может закрыться само.
Если ребенок себя чувствует очень плохо даже в раннем детском возрасте только операция может облегчить его страдания и устранить повышенное давление крови в сосудах легких. С помощью операции суживания легочной артерии (операция Мюллера) уменьшается просвет легочной артерии (ЛА), и кровоток в легких снижается. Эта паллиативная (облегчающая) процедура позволяет ребенку нормально развиваться. Когда ребенок подрастет, врачи смогут снять тесьму, суживающую легочную артерию, и произвести операцию на открытом сердце.
ствола и аорты. Транспозиция магистральных сосудов. Дефекты дуги аорты
Транспозиция магистральных сосудов (ТМС) — ВПС, характеризующийся дискордантностью желудочково-артериального соединения при конкордантности соединения остальных сегментов сердца. Иными словами, аорта отходит от морфологически правого желудочка, а лёгочный ствол — от морфологически левого.
Клиника: Общий цианоз. Дифференцированный цианоз, когда верхняя половина тела более синюшна, чем нижняя, патогномоничен для ТМС в сочетании с открытым артериальным протоком. Тахикардия. Одышка. Увеличение размеров сердца и печени.
Этиология :причины, вызывающие ВПС (см. Тетрада Фалло).
Дефекты дуги аорты
Коарктация аорты - это врожденное сегментарное сужение грудной аорты, создающее два режима кровообращения в большом круге и вызывающее определенные клинические симптомы.
Перерыв дуги аорты может быть полным или (реже) сегментарным, когда между дугой и нисходящей аортой выявляется атрезированный сегмент в виде фиброзного тяжа. Данная форма порока может рассматриваться как крайняя степень коарктации.
Аневри́зма — выпячивание стенки артерии (реже — вены) вследствие её истончения или растяжения.
Дуга аорты двойная – дуга аорты представлена двумя стволами: один располагается впереди трахеи, другой – позади пищевода. Слева от них оба ствола соединяются вновь. Передняя дуга обычно тоньше задней или представлена плотной связкой. Образуется двойная дуга в результате персистирования правой IV аортальной дуги.
Дуга аорты правосторонняя – развивается из эмбриональной правой дуги при редукции левой. Располагается позади пищевода. Из остатков левой дуги в таких случаях нередко образуется дивертикул.
Дуга аорты шейная – в случае инволюции 4 жаберных дуг дуга аорты может развиться из артерии III жаберной дуги. В этом случае дуга аорты располагается на шее над вырезкой грудины. Встречается чрезвычайно редко.
Комбинированные пороки сердца (триада, тетрада, пентада Фалло, синдром висцеральной гетеротаксии, Миллера–Уайта–Лева синдром)
Болезнь Фалло (триада, тетрада, пентада)
Это наиболее часто встречающийся порок, протекающий с цианозом
Характеризуются поступлением недостаточного количества крови в малый круг кровообращения (из-за стеноза легочного ствола) и сбрасыванием венозной крови в большой круг через транспозированную аорту и дефект межжелудочковой перегородки, причем в большей степени эти нарушения определяются размерами и степенью выраженности стеноза легочной артерии. Возможно сочетание тетрады Фалло с дефектом межпредсердной перегородки (пентада) или отсутствие в составе порока декстропозиции аорты (триада Фалло). У детей с такими аномалиями сердца, как правило, наблюдается отставание в физическом и умственном развитии, одышка и тотальный цианоз при беспокойстве, крике, кормлении. У них снижена толерантность к физическим нагрузкам, поэтому при утомлении они присаживаются на корточки.
Гетеротаксия висцеральная - заболевание, характеризующееся различными врождёнными аномалиями (сочетайные пороки сердца и транспозиция внутренних органов; например, гетеротаксия висцеральная Х-сцепленная
Миллера — Уайта — Лёва с.— врожденные аномалии сердца и крупных сосудов (возможно, наследственного характера): дефект межжелудочковой перегородки, декстропозиция аорты, гипертрофия правого желудочка, в отличие от тетрады Фалло (Fallot) — расширение легочной артерии, систолически-диастолический шум на всех основных местах выслушивания сердца.
Холт–Орама синдром (синдром «рука–сердце»)
Наследственное сочетание аномалий развития сердца и верхних конечностей. Наследование аутосомно-доминантное. В классической форме характеризуется дефектом межпредсердной перегородки второго типа и аномалией развития больших пальцев кистей.
Пороки развития руки варьируют от недоразвития или отсутствия 1-го пальца кисти и трёхфалангового 1-го пальца кисти до недоразвития или полного отсутствия лучевой кости с формированием лучевой косорукости. Чаще поражается левая рука. Наблюдаются и другие скелетные изменения: гипоплазия лопаток и ключиц, сколиоз, воронкообразная деформация грудины, искривление мизинца, сросшиеся пальцы (синдактилия), гипоплазия других пальцев кисти. В 85 % случаев у больных обнаруживаются различные формы врожденных пороков сердца: дефекты межпредсердной и межжелудочковой перегородок, открытый аортальный проток, коарктация аорты, стеноз легочной артерии, пролапс митрального клапана и др
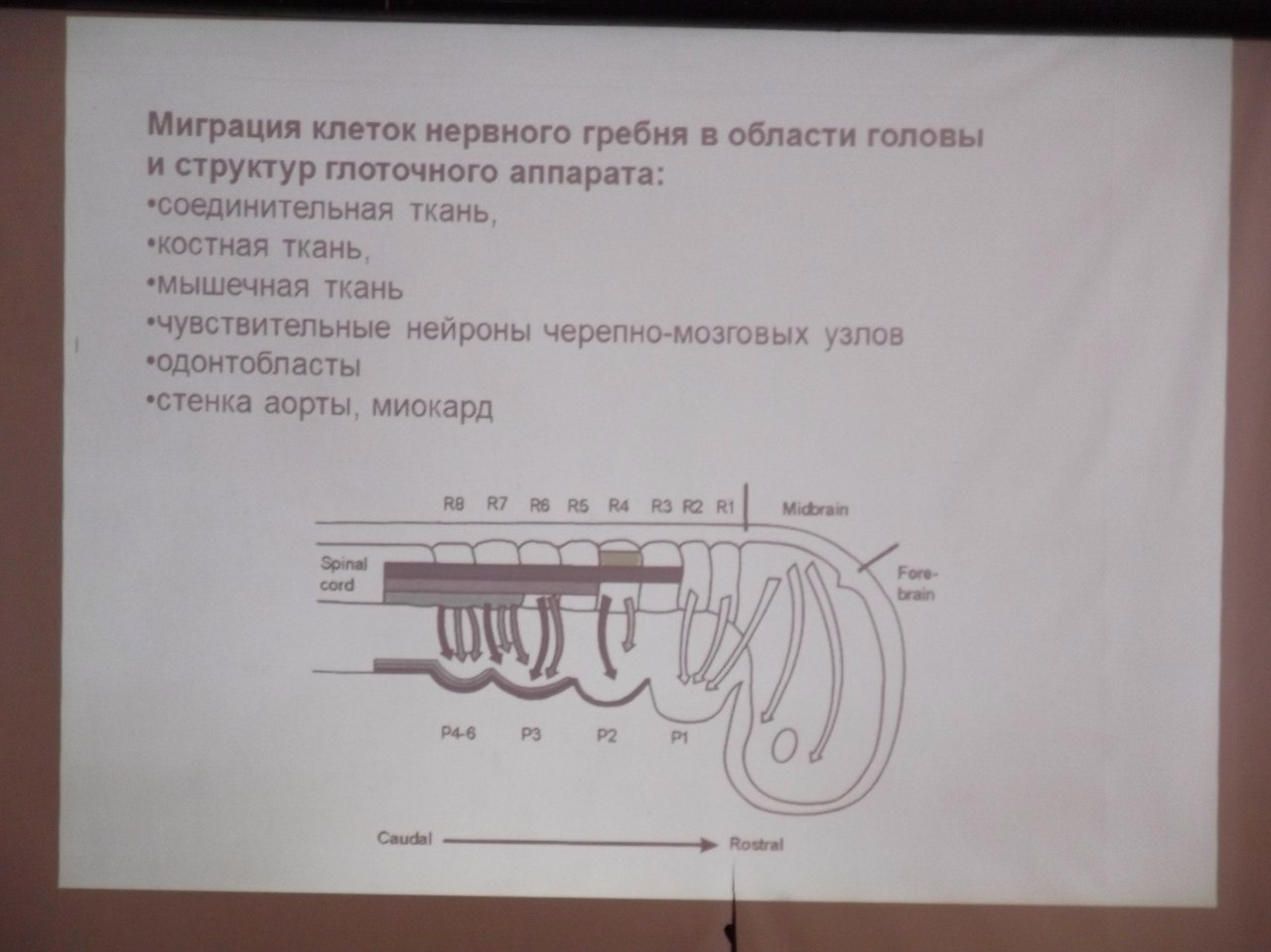
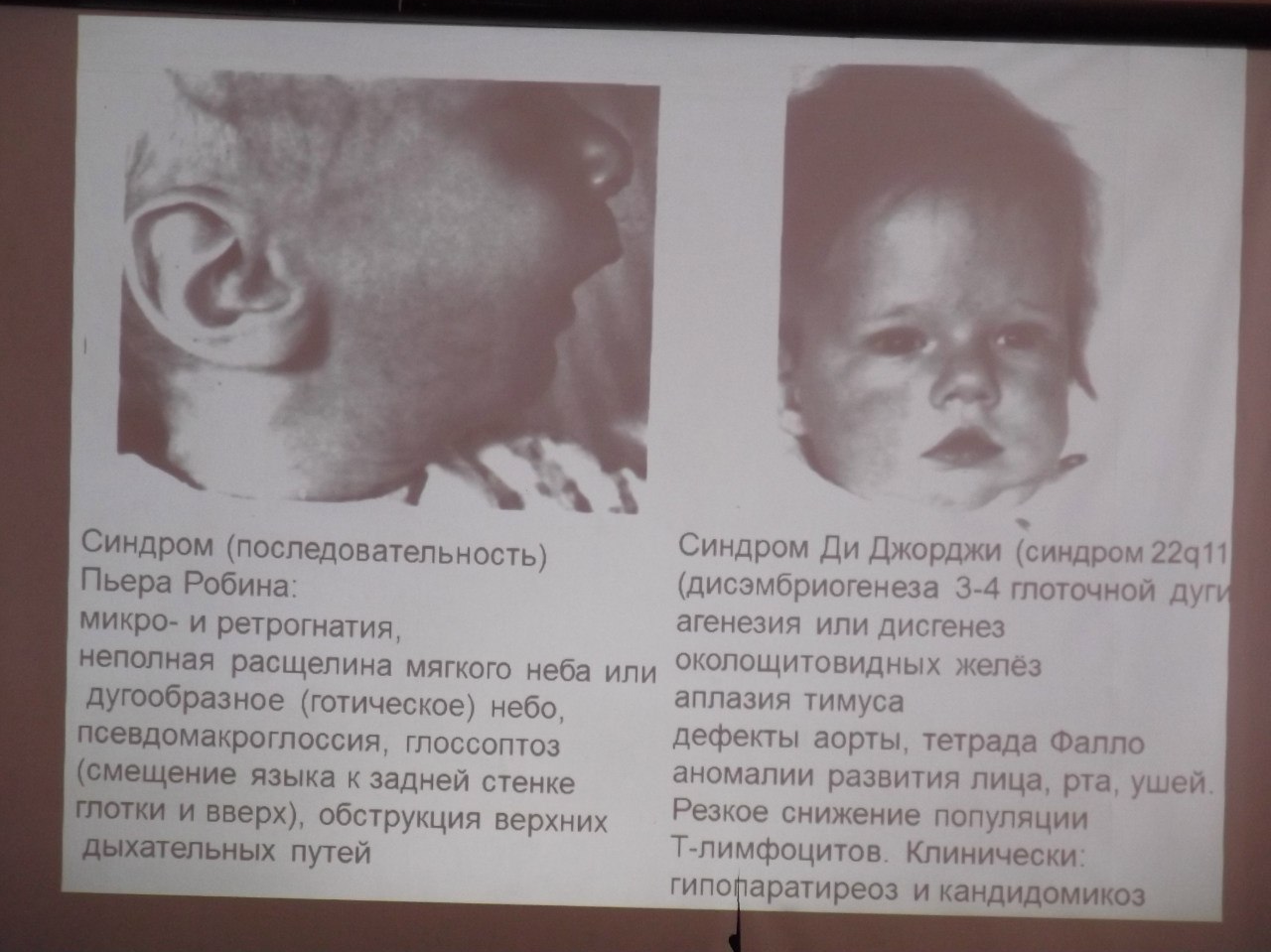
Дефекты миграции нервного гребня (Ди Джорджи синдром, Синдром Пьера Робина, Рото-лице-пальцевой синдром)
Синдром рото-лице-пальцевой - группа наследственных заболеваний, проявляющихся множественными врождёнными пороками развития (как правило, лица и пальцев). Различают несколько типов синдрома. Клинически: умственная отсталость, расщелина верхней челюсти, широкий корень носа, маленькие ноздри, гипоплазия хрящей носа, срединная расщелина верхней губы, добавочная гиперпластичная уздечка губы, дольчатый язык с гамартомами, асимметричная расщелина нёба, синдактилия, брахидактилия, полидактилия, дизартрия, неуклюжая походка, гнёздная алопецйя, поликистоз почек, почечная недостаточность, аге-незия мозолистого тела на КТ, неравномерная минерализация костей кистей и стоп. МКБ.
Дефекты развития лица и органов ротовой полости
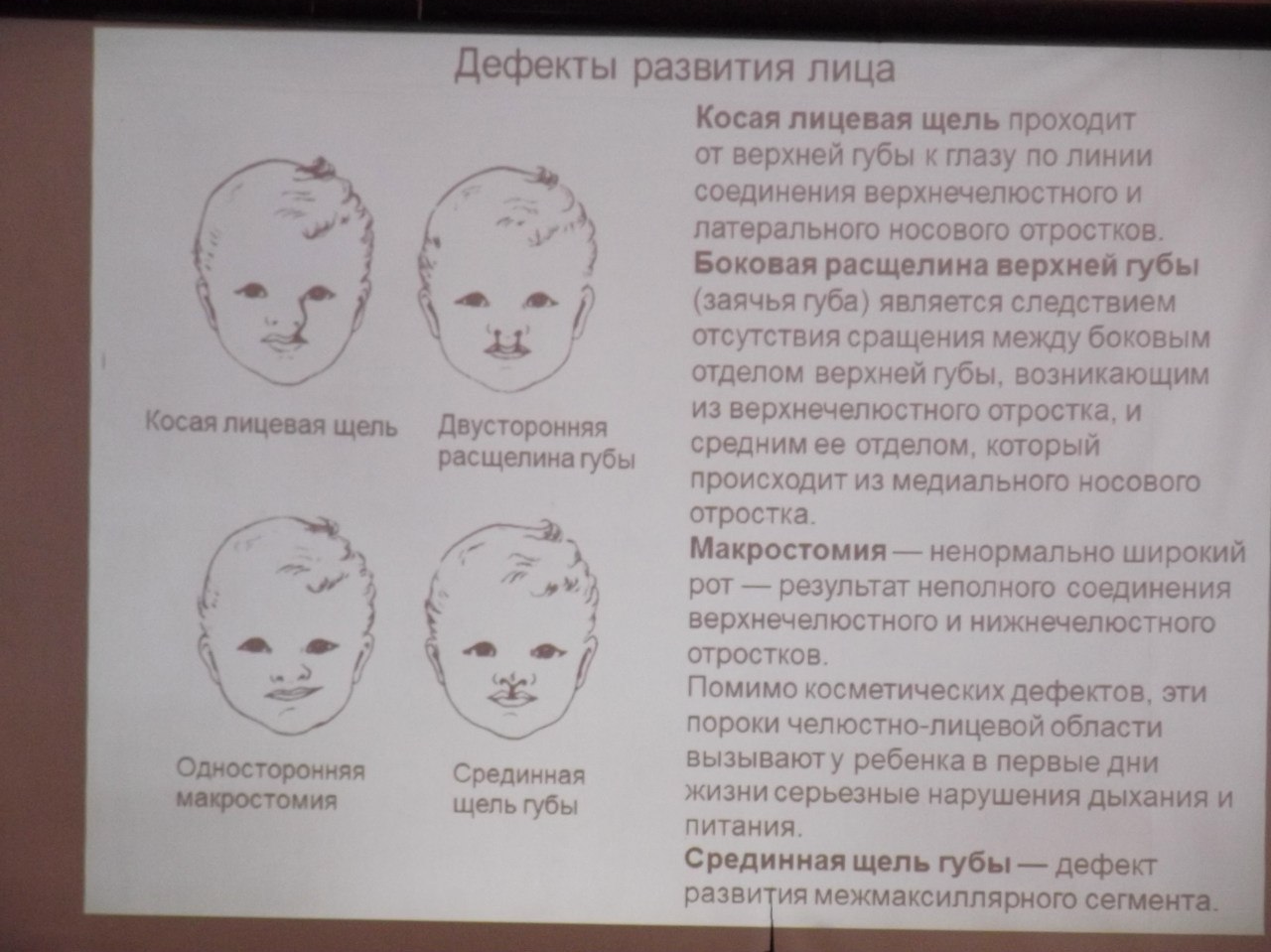
Аномалии развития лица
Апросомия – отсутствие лица как результат остановки в развитии закладок лица. На поверхности лица отмечаются только отдельные узлы.
Ариния – полное отсутствие наружного носа.
Ацефалия – врожденное полное отсутствие головы. Может сочетаться с отсутствием верхних конечностей (ацефалобрахия), желудка (ацефалогастрия), сердца (ацефалокардия), нижних конечностей (ацефалоподия), позвоночного столба (ацефалорахия), грудной клетки (ацефалоторация).
Искривление перегородки носа – частый порок, развивается при отставании в росте свода и дна полости рта.
Киста лица – опухолеподобное образование врожденного происхождения, встречающееся в местах костных швов на лице. Происхождение ее связывают с ростом в глубине тканей эктодермы, отшнуровавшейся в эмбриональном периоде. Различают дермоидные и эпидермальные кисты. Наиболее типичной локализацией является переносица, граница костного и хрящевого отделов носа, наружный край глазницы.
Киста носа дермоидная – располагается на спинке носа, образуется в результате незаращения эмбриональных щелей. Преимущественно локализуется под кожей у места соединения носовых костей с хрящами.
Колобома крыла носа – поперечная, неглубокая одно- или дву- сторонняя щель свободного края крыла носа. Чаще сопутствует сложным порокам лица.
«Лицо птичье» – лицо со скошенным и западающим назад подбородком при недоразвитии нижней челюсти и анкилозах височно-нижнечелюстного сустава. Наблюдается при синдроме Франческетти – Цвалена.
«Лицо рыбье» – лицо с резко суженным ротовым отверстием. Наблюдается при синдроме Франческетти – Цвалена.
Мелосхиз – расщелина щеки с увеличением размеров рта.
Микроформы расщелин верхней губы и неба – помимо выраженных форм расщелин, упомянутых выше, встречаются и небольшие признаки, получившие название микроформ. Сюда относятся скрытая или явная расщелина только языка, диастема, скрытая и начальная расщелины красной каймы губ, деформация крыла носа без наличия расщелины губы.
Нос добавочный (син.: хоботок, proboscis) – в легких случаях представляет собой вырост в виде трубки, располагающейся у корня носа. В тяжелых случаях вместо носа имеется трубчатое кожистое образование с одним слепо заканчивающимся отверстием.
Отсутствие перегородки носа – бывает полное или частичное. Встречается редко. Отсутствие половины носа врожденное – аплазия крыла и боковой поверхности носа в пределах хрящевой части, обычно сопровождается атрезией костного отверстия, ведущего в полость носа с той же стороны. Сохранившаяся половина носа гипоплазирована.
Перфорация перегородки носа врожденная – отверстие в костной или хрящевой части носовой перегородки.
Разрез глаз антимонголоидный – опущены наружные углы глазных щелей.
Расщелина верхней губы (син.: незаращение верхней губы, хейлосхиз, «губа заячья») – щель в мягких тканях губы, проходящая сбоку от фильтрума. Может быть одно- и двухсторонней, полной или частичной, подкожной или подслизистой.
Расщелина верхней губы и неба сквозная (син.: хейлогнатопала- тосхиз) – щель губы, альвеолярного отростка и неба. Может быть одно- и двусторонней. При сквозных расщелинах име- ется широкое сообщение между полостями носа и рта. Может сочетаться с полидактилией и аномалиями мочеполового аппарата.
Расщелина верхней губы срединная (син.: расщелина верхней губы пренебная) – щель в мягких тканях верхней губы, располагающаяся по средней линии. Сопровождается уздечкой и диастемой; может сочетаться с расщелиной альвеолярного отростка и удвоенной уздечкой. Аномалия очень редкая.
Расщелина лица косая (син.: расщелина параназальная, расщелина боковая, колобома косая) – редко встречающийся обычно односторонний порок развития. Различаю носоглазную и ротоглазную формы. Обе формы в ряде случаев распространяются на лоб и височную область, могут быть полными и неполными. Ротоглазничные расщелины встречаются в 2 раза чаще носоглазничных и нередко сочетаются с другими пороками: расщелинами губы и неба, мозговыми грыжами, гидроцефалией, микрофтальмом, деформацией пальцев кисти и стопы.
Расщелина нижней губы и нижней челюсти срединная – очень редкий порок. Встречаются частичные и полные формы. При полных формах альвеолярный отросток и тело нижней челюсти соединяются соединительнотканной перемычкой. Обе половины челюсти умеренно подвижны относительно друг друга. Язык концевым отделом может быть сращен с нижней челюстью. Известны случаи одновременной срединной расщелины верхней, нижней губы и нижней челюсти.
Свищ носа дермоидный – располагается на спинке носа, образуется в результате незаращения эмбриональных щелей.
Синофриз – сросшиеся брови.
Телекант – смещение внутренних углов глазных щелей латерально при нормально расположенных орбитах.
Трицефалия – наличие на одной голове трех лицевых поверхностей при общем туловище. *Цебоцефалия – недоразвитие наружного носа вплоть до его отсутствия, сочетающееся с уменьшенным расстоянием между глазами, вследствие чего лицо больного напоминает морду обезьяны. Объем черепа, как правило, уменьшен. Характерно слияние обоих полушарий головного мозга с наличием одного общего желудочка. Обонятельные нервы, мозолистое тело и прозрачная перегородка не развиты.
Аномалии развития органов полости рта
Аглоссия – отсутствие языка, как изолированная аномалия не описана. Наблюдается крайне редко при тяжелых гипоплазиях лица и челюстей.
Анкилоглоссия (син.: уздечка языка короткая) – прикрепление уздечки в области кончика языка или ее укорочение, приводящее к ограничению подвижности языка. Крайняя степень такой аномалии – приращение языка.
Ахейлия – отсутствие одной или обеих губ. Брахихейлия – врожденное укорочение средней части верхней губы, при котором она не перекрывает верхние зубы.
Гипсистафилия – высокое узкое небо.
Глоссоптоз – сочетание врожденного недоразвития и западения языка.
Губа двойная (син.: удвоение губ) – складка слизистой оболочки, располагающаяся параллельно красной кайме верхней губы и напоминающая дополнительную губу. Встречается довольно часто, преимущественно у мужчин.
Макроглоссия – чрезмерное увеличение языка с выраженной складчатостью слизистой оболочки. Часто сочетается с макрогенией. Встречается сравнительно часто.
акростомия – чрезмерно увеличенная ротовая щель. Обусловлена несращением тканей верхней и нижней частей щеки и краев губ между собой. Бывает одно- и двусторонней, является признаком аномаладов I и II жаберных дуг.
Макрохейлия – чрезмерно увеличенные губы вследствие разрастания в их толще соединительной ткани.
Микроглоссия – малые размеры языка, как изолированная аномалия не описана. Односторонняя микроглоссия является одним из признаков сочетанных пороков I и II жаберных дуг, при срединной расщелине нижней челюсти, аномаладе Робена.
Микростомия (син.: рот малый, микростома) – чрезмерно уменьшенная ротовая щель. Как самостоятельный порок наблюдается редко. Обычно сочетается с тяжелыми пороками производных I жаберной дуги.
Микрохейлия – очень малые размеры губ.
Небо арковидное – небо с острым углом у вершины.
Платистафилия – широкое и плоское небо.
реддверие полости рта мелкое – аномалия мягких тканей переднего отдела альвеолярного отростка нижней челюсти, состоящая в резком сужении или полном отсутствии зоны прикрепленной слизистой оболочки ниже десневого края. В норме эта зона в среднем составляет 5-6 мм. Прикрепленная слизистая оболочка играет защитную, буферную функцию для зубных сосочков, подвергающихся постоянной травме при откусывании, во время речи и при других движениях губ. Если зона прикрепленной десны узкая, то травмирующие движения передаются на сосочки, постоянно оттягивают и отслаивают их от корней зубов, появляется воспаление, постепенно формируются патологические зубодесневые карманы. В результате неуклонного прогрессирования воспалительно-дистрофического процесса в мягких тканях и кости обнажаются лунки и корни зубов, зубы расшатываются и выпадают в молодом возрасте. Частота аномалии среди детей – 6.9%, среди взрослых – 5.3%.
Расщелина неба (син.: палатосхиз, «пасть волчья», uranoschisis) – бывает полной (щель в мягком и твердом небе), частичной (только в мягком или только в твердом небе), срединной, одно- и двусторонней, сквозной или подслизистой.
Рот двойной – крайне редкий порок, проявляющийся добавочной ротовой щелью, открывающейся в добавочную ротовую полость меньших размеров, чем основная ротовая полость. Обе полости не сообщаются.
Свищ нижней губы – обычно парный и располагается на красной кайме губы по обе стороны от средней линии. Представляет собой проток добавочных слизистых желез. Встречается очень редко. *Синхейлия – срастание верхней и нижней губ.
Уздечка верхней губы – низкое прикрепление уздечки верхней губы, достигающей основания межзубного сосочка центральных резцов. В таких случаях уздечка оказывается более широкой, иногда представлена тяжем, ограничивающим подвижность губы. Нередко сочетается с центральной диастемой. Встречается очень часто.
Язык двойной – расщепление языка по средней линии.
Язык добавочный – наличие у корня языка добавочного слизисто-мышечного выступа. Внешним видом и подвижностью напоминает язык, только гораздо меньших размеров. Крайне редко встречающийся порок.
Хиршпрунга болезнь (мегаколон)
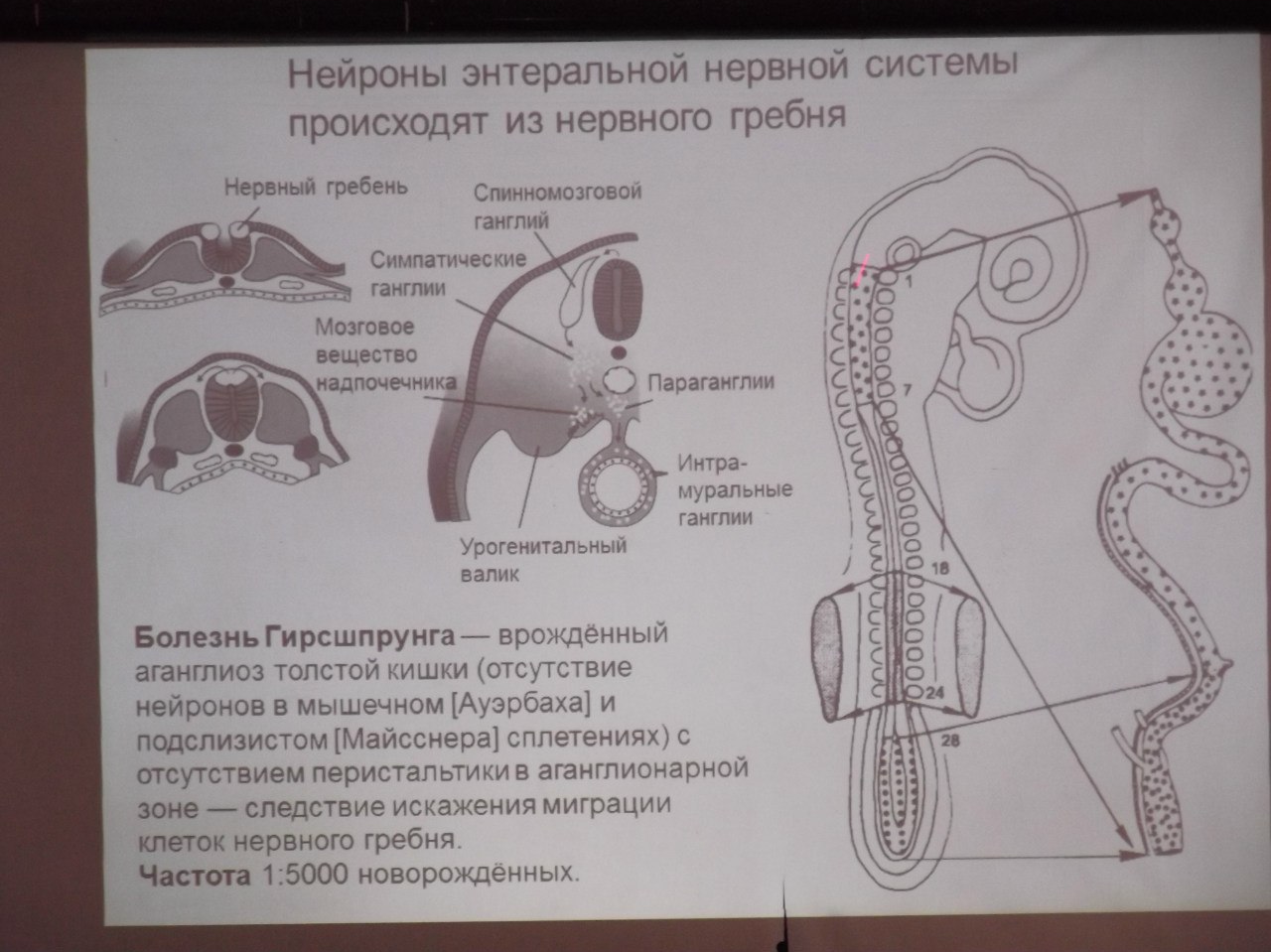
Полное отсутствие или дефицит периферических нервных рецепторов и нарушение проводимости в нервных путях стенки толстого кишечника приводят к затруднению продвижения каловых масс по гладким мышцам или суженной части толстой кишки, что ведет к резкому расширению и увеличению стенки вышерасположенных ее отделов. Увеличение расширенной части толстой кишки формируется вследствие активизации перистальтики для продвижения содержимого через аганглионарную зону.
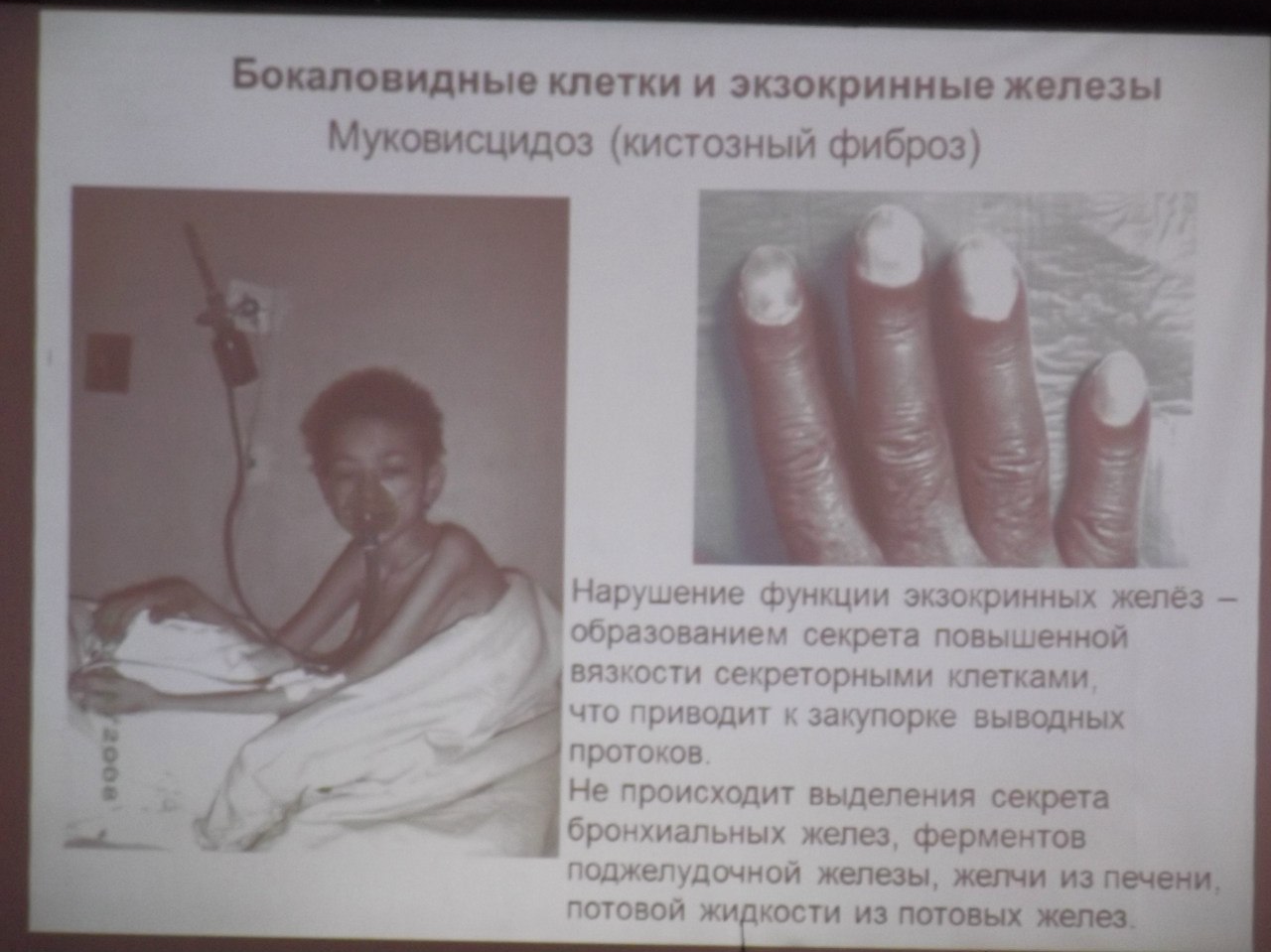
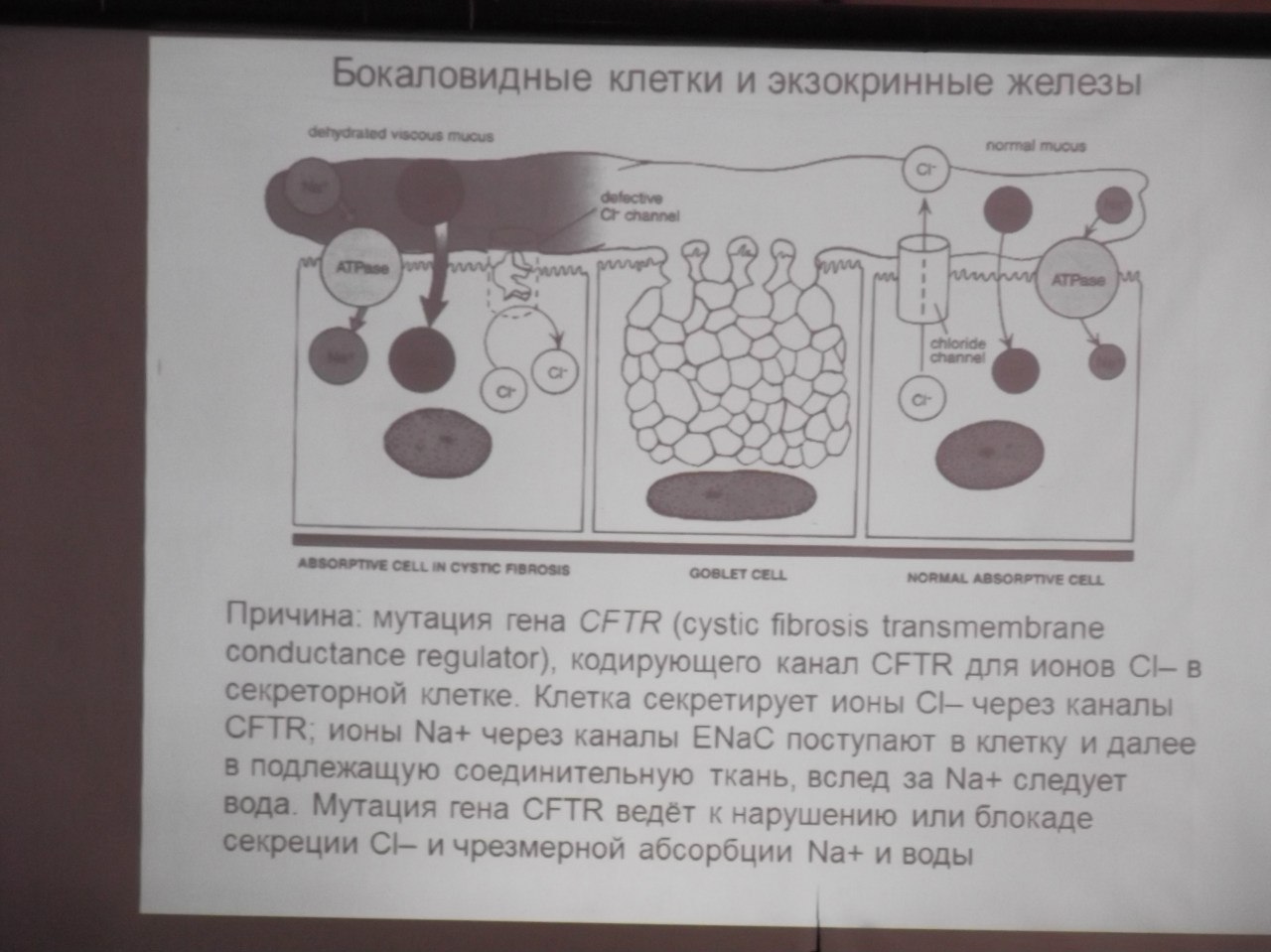
Муковисцидоз

Атрезия и стеноз желчных протоков
Атрезия желчных путей (особенно внутрипеченочных) в большинстве случаев связана с перенесенным внутриутробно гепатитом, чаще вызванным одним из реовирусов. У некоторых детей возникновение этого порока развития обусловлено неблагоприятными факторами, действовавшими на 4–8-й нед внутриутробной жизни.
В этиологии атрезии желчных протоков большинство отечественных и зарубежных авторов придают значение таким факторам, как продуктивное воспаление, вызывающее дегенерацию эпителия протоков, облитерацию их просвета и околопротоковый склероз. Прогрессирование процессов альтерации, пролиферации и фибро-зирования во внутриутробный период и после рождения ведет к полной обтурации просвета желчных протоков .
Патогенез болезни связан с нарушением выделения желчи вследствие непроходимости желчных протоков. Развиваются желтуха, билиарный цирроз печени, портальная гипертензия, печеночная недостаточность. При отсутствии хирургического лечения дети умирают вскоре после рождения.
Патологическая анатомия. Желчные протоки в месте атрезии представлены тонким фиброзным тяжем.
стеноз желчных протоков
Заболевание представляет собой комплекс различных нарушений, характеризующихся общим признаком — затруднением оттока желчи из желчных протоков в двенадцатиперстную кишку. Стеноз возникает обычно в нижнем отделе общего желчного протока на почве желчнокаменной болезни, воспалительного или опухолевого процесса в протоке, в сфинктере Одди, ампуле и папилле Фатера, а также вследствие фиброза, рубцевания и спаек.
Трахео-пищеводные свищи
Развивающиеся из одного зачатка пищеводная и дыхательная трубки при различных нарушениях в течение внутриутробного периода имеют патологические сообщения между собой — свищи. Наиболее часто свищ возникает между трахеей и пищеводом. Во время кормления у ребенка возникает кашель, он синеет (цианоз). Особенно яркая картина развивается при больших размерах свища. На фоне попадания пищи в дыхательные пути может развиться пневмония.
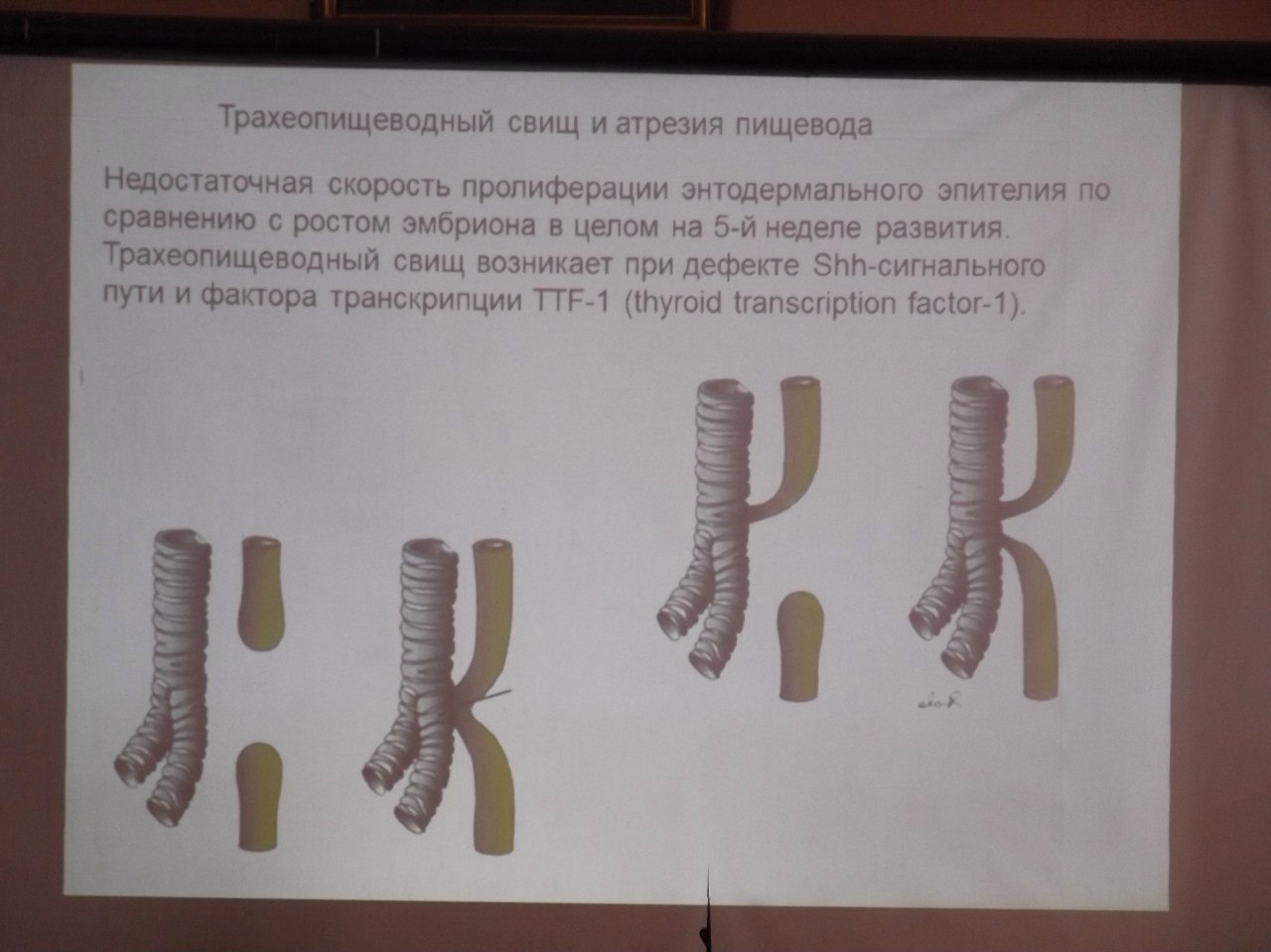
Синдром Картагенера
комбинированный порок развития, характеризующийся образованием бронхоэктазов в сочетании с полным или частичным обратным расположением внутренних органов и полипозом слизистой оболочки носа. Часто сочетается с другими врожденными аномалиями: полидактилией, агенезией или гипогенезией лобных пазух, пороками развития позвонков и ребер, мочевыводящих путей, сердца, гипофункцией некоторых эндокринных желез (щитовидной, гипофиза, надпочечников) с задержкой роста, поражением сетчатки (пигментный ретинит, расширение сосудов сетчатки) и др.
Этиология и патогенез. Не изучены. Имеет значение генетическая предрасположенность, передаваемая по аутосомно-рецессивному типу. Не исключена возможность врожденной предрасположенности бронхиального дерева к хроническому воспалению с последующим развитием бронхоэктазов.

Респираторный дистресс-синдром новорожденных

Врождённый альвеолярный протеиноз (ЛАП):

102.Первичная (врождённая) эмфизема (недостаточность α1антитрипсина)

Протеолиз — процесс ферментативного гидролиза белков, катализирующийся протеолитическими ферментами (протеазами)
Поликистоз почек
Поликистоз почек — врожденное заболевание, при котором в обеих почках появляются и постепенно увеличиваются кисты, что приводит к атрофии функционирующей паренхимы. Относится к наследственным аномалиям развития и часто встречается у членов одной семьи.
Этиология, патогенез. Причина возникновения аномалии неизвестна. Патогенез обусловлен пороком эмбрионального развития канальцев, часть которых трансформируется в кисты. Почки у большинства больных увеличены, содержат множество кист различных размеров, между которыми расположены участки сохранившейся паренхимы, местами замещенной соединительной тканью. Чашечки и лоханки сдавлены и деформированы. Кисты могут нагнаиваться.
Аутосомно-доминантная и аутосомно-рецессивная поликистозная болезнь почек относится к цилиопатиям — группе заболеваний, для которых характерно нарушение нормальной работы ресничек на поверхности ряда клеток, за счет которых обеспечивается «прием» сигналов из внеклеточной среды. Белки полицистин-1, полицистин-2 и фиброцистин входят в состав первичных ресничек на поверхности клеток млекопитающих. В клетках эпителия почечных канальцев первичные реснички располагаются со стороны просвета почечных канальцев, и предполагается, что это обеспечивает их сенсорную функцию току мочи. В результате неправильного восприятия сигналов из-за нарушенной работы первичных ресничек в клетках эпителия почечных канальцев происходит накопление циклического аденозинмонофосфата, на снижение уровня которого направлено действие ряда экспериментальных методик лечения поликистоза почек.
На макроуровне для поликистоза характерно наличие множественных кист (отсюда название: поли- + киста + -оз) в обеих почках. Кисты образуются из-за повышенной пролиферации и дедифференциации фильтрующего эпителия нефрона. В результате вместо нормальных почечных канальцев образуются наполненные жидкостью пузырьки — кисты, что приводит к значительному увеличению объема почек (вес почки больного может достигать 35 кг). Кисты в почке больного возникают фокально не более чем в 2-5 % нефронов, но из-за увеличения объёма кист происходит сдавление соседних здоровых нефронов, и постепенно почка теряет фильтрующую функцию.
Экстрофия мочевого пузыря
это врождённый порок развития мочевого пузыря, при котором мочевой пузырь оказывается не внутри, а снаружи. Передняя стенка мочевого пузыря отсутствует, как и соответствующий ей участок брюшной стенки, которая расщеплена, и таким образом мочевой пузырь оказывается вовне. Моча льётся наружу через отверстия мочеточников.
Состояние экстрофии образуется очень рано во время развития эмбриона, примерно на 4-5 неделях его развития.
Экстрофия мочевого пузыря часто сочетается с пороками развития верхних мочевых путей, а также с аномалиями других органов и систем.
Экстрофированная слизистая стремительно, уже к концу первых суток жизни, подвергается изменениям в связи с постоянным раздражением и присоединением воспаления. Быстро развивается восходящая инфекция мочевых путей. Причины неправильного развития пока не до конца ясны. Именно в это время начинают формироваться органы и ткани организма из разъединяющихся, делящихся и соединяющихся клеток. Одна из теорий предполагает, что именно из-за какого-то неправильного механизма такого деления и соединения клеток, что-то происходит с клоакальной мембраной, что не даёт ей закрыться, и это приводит к тому, что мочевой пузырь оказывается вне брюшной полости. Другая теория предполагает, что на этом этапе развития эмбриона слой клеток над мочевым пузырем очень тонок, он не может удержать мочевой пузырь внутри и расщепляется, опять же мочевой пузырь оказывается снаружи.
Дивертикул Меккеля
локальное мешковидное
выпячивание стенки подвздовшной кишки,
образовавшеяся вследствие неполного
заращения желточного протока, который
участвует в питании плода(в норме
подвергается обратному развитию после
6—8 нед. внутриутробного периода)/ Может
никак себя не проявлять, но может
воспаляться и симулировать клиническую
картину острого аппендицита.
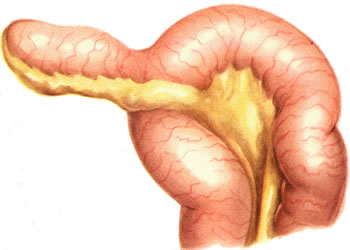
Наиболее частыми клиническими проявлениями М.д, служат его воспаление, непроходимость кишечника и кишечное кровотечение. Воспаление (дивертикулит) могут спровоцировать инородные тела, гельминты, попавшие в просвет дивертикула, а также застой кишечного содержимого
Урахус
Урахус— трубчатое образование у эмбриона, соединяющее передний отдел верхушки мочевого пузыря и пупок между брюшиной и поперечной фасцией живота, образуется из верхнего отдела аллантоиса. По данному протоку моча плода выводится в околоплодные воды. С 5 месяцев внутриутробной жизни начинается облитерация протока, которая завершается к моменту рождения, с превращением его в срединную пупочную связку. Однако при определенных условиях проток перекрывается не полностью, в результате чего формируются его аномалии:
-Пупочный свищ — незаращение части урахуса, находящейся в области пупка. Проявляется такая аномалия постоянным мокнутием ранки пупка.
-Пузырно — пупочный свищ — полное незаращение урахуса. Данный вид характеризуется постоянным выделением из ранки пупка мочи.
-Дивертикул мочевого пузыря — незаращение части урахуса, отходящей от мочевого пузыря.
-Киста урахуса — незаращение средней части урахуса.
Пузырный занос (полный и неполный) и его причины
Пузырный занос - заболевание плодного яйца, отличительными признаками которого являются перерождение ворсин хориона в пузырьки с жидкостью, разрастание эпителия ворсин, особенно синцития.
Причиной возникновения пузырного заноса является наличие у эмбриона двойного набора хромосом отца при недостаточном количестве или же вообще отсутствии хромосом матери. Такая аномалия случается, когда одновременно 2 сперматозоида оплодотворяют «неполноценную» яйцеклетку - с задержкой набора хромосом или безъядерную. При этом в первом случае развивается неполный пузырный занос, а во втором - полный.
Полный пузырный занос возникает при однородительской дисомии, когда по неизвестным причинам происходит потеря материнских генов и дублирование отцовского гаплоидного генома (зигота имеет кариотип 46,ХХ). Иногда (5%) полный пузырный занос вызван оплодотворением пустой (безъядерной) яйцеклетки двумя сперматозоидами, приводящим к кариотипу 46,XY или 46,XX. Эмбрион погибает на ранних стадиях развития, до установления плацентарного кровообращения
Неполный пузырный занос вызван триплоидией в результате оплодотворения яйцеклетки двумя сперматозоидами (диспермия) с задержкой гаплоидного набора материнских хромосом. Клетки концептуса содержат один гаплоидный набор материнских хромосом и диплоидный набор отцовских хромосом - кариотип может быть 69.XXY, 69.ХХХ или 69.XYY. Плод погибает на 10 нед внутриутробного развития.
Патогенез:
При значительном накоплении жидкости в ворсинах сосуды трофобласта атрофируются. Синцитий, покрывающий пузырьки, способен пролиферировать и ферментативно расплавлять децидуальную оболочку, прорастать и внедряться в мышечный слой матки, разрушая мышечные элементы и сосуды. Иногда инвазивная способность покровного эпителия пузырьков столь значительна, что они разрушают стенку матки, проникают в брюшную полость и могут послужить причиной внутреннего кровотечения. Это деструирующая форма пузырного заноса, которая по характеру роста напоминает опухоль. Обычно она связана с опасным для жизни кровотечением.
Омфалоцеле
Омфалоцеле (грыжа пупочного канатика, пуповинная грыжа, эмбриональная грыжа) — вид врождённого дефекта передней брюшной стенки, при котором петли кишечника, печень и, иногда, другие органы выходят за пределы брюшной полости в грыжевом мешке. Омфалоцеле обусловлено дефектом развития мышц передней брюшной стенки
В некоторых случаях, предположительно, омфалоцеле может являться следствием генетического рассройства (синдром Эдвардса, синдром Патау).
В норме в ходе эмбриогенеза петли кишечника выходят за пределы брюшной полости, выпячиваясь в пупочный канатик, однако на сроке десяти недель беременности возвращаются обратно; при омфалоцеле же органы остаются в пупочном канатике. Грыжевой мешок при омфалоцеле в лёгких случаях может содержать единичные петли кишечника, в тяжёлых — практически все органы живота.
Сочетается с другими аномалиями и хромосомными дефектами в 70% случаев. Хромосомные аномалии встречаются в 20% случаев (синдром Эдвардса, синдром Патау).. Из многочисленных сочетанных аномалий, встречающихся при омфалоцеле, особое внимание занимает синдром Беквита- Видемана (пуповинная грыжа, макроглоссия, органомегалия). Наблюдается у 12% детей с омфалоцеле.
Многоводие и маловодие
Околоплодные воды, или амниотическая жидкость (АЖ) — среда обитания плода, выполняющая одновременно несколько функций: создание пространства для свободных движений растущего плода, защита от механической травмы, поддержание температурного баланса, предотвращение компрессии пуповины в родах, осуществление транспортной функции и участие в обмене веществ.
Многоводие – это патология беременности, при которой происходит избыточное накопление околоплодных вод - более 1,5- 2 литра жидкости. Соответственно, маловодие - это недостаток околоплодных вод, при котором количество околоплодной жидкости составляет менее 500 мл.
Если говорить о многоводии, то в большинстве случаев оно возникает в результате инфекционных и вирусных заболеваний у беременной, таких как хламидиоз, микоплазмоз, цитомегаловирусная инфекция и так далее.Также многоводие может вызвать сахарным диабет у матери, резус-конфликт между кровью матери и плода, врожденные пороки развития плода. Многоводие почти всегда встречается при многоплодной беременности.
Маловодие, также как и многоводие, может быть обусловлено инфекционно-воспалительными заболеваниями матери. К другим, немаловажным факторам, провоцирующим маловодие, относят повышенное артериальное давление беременной, поздний гестоз, ожирение у матери, перенашивание беременности, пороки развития плода, нарушения мочевыделительной системы плода, нарушение кровоснабжения в плаценте.
Многоводие является частой причиной различного рода пороков в развитии плода, всевозможных патологий, вплоть до его перинатальной смерти. Кроме того, многоводие осложняет роды, которые, как известно, являются тяжелым испытанием не только для мамы, но и для ребенка. В результате ослабления родовой деятельности, которое часто наблюдается при многоводии, у ребенка может наступить асфиксия(удушье), негативно влияющая на общее состояние малыша, деятельность его центральной нервной системы.
При маловодии стенки матки расположены вблизи от поверхности тела плода, таким образом, ребенку недостаточно места, необходимого для нормального развития. Вследствие этого у плода могут наблюдаться отклонения в развитии костной системы, конечностей. Ребенок в той или иной степени начинает отставать в росте и массе, кожа его становится сухой, покрытой многочисленными морщинами.
Патологии в развитии плода могут наблюдаться как при умеренном маловодии, хотя степень такой гипотрофии, как правило, более легкая, больше шансов на рождение здорового малыша, так и при выраженном, когда у плода наблюдаются явные отставания в развитии, гипоксия, нарушения мозговой и нервной деятельности, снижение тонуса конечностей, вялость.
при данных состояниях можно родить нормального ребенка.
Синдром (последовательность) Поттера
Синдром Поттера представляет собой сочетание двусторонней агенезии почек и характерных аномалий лица (приплюснутый нос, гипертелоризм, эпикант, узкие глазные щели, борозда под нижними веками, микрогнатия и мягкие, большие, деформированные, низко расположенные ушные раковины), 40% детей рождаются недоношенными, большинство погибают в первые часы жизни. При этом синдроме могут встречаться следующие пороки развития: двусторонняя гипоплазия легких, аномалии гениталий, атрезия ануса, отсутствие сигмовидной и прямой кишки, атрезия пищевода и двенадцатиперстной кишки, единственная пупочная артерия и деформация нижней челюсти туловища и нижних конечностей. Типичное лицо, гипоплазия легких и аномалии конечностей развиваются вследствие характерного для данного синдрома маловодия. Описаны случаи односторонней агенезии почек у родственников.
Placenta accrete
Отделение зрелой плаценты от стенки матки в III периоде родов в нормальных условиях происходит довольно быстро и легко во время первого сокращения. Отделение происходит в спонгиозном слое основной децидуальной оболочки.
Затруднения в отделении зрелой плаценты, удлиняющие третий период родов, могут быть вызваны резко выраженным соединением плаценты со стенкой матки. Такое соединение обычно является результатом состояния, при котором ворсинки развивающейся плаценты не задерживаются в спонгиозном слое, а проникают глубже вплоть до поверхности, отделяющей децидуальную оболочку от миометрия и даже врастают в него. Приросшие плаценты охватывают группу плацент, ворсины которых прилегают к мышце матки, врастают в неё или прорастают её.
Приращения плаценты делится в зависимости от глубины проникновения ворсин в мышцу матки на:
1,истинное приращение плаценты (placentae accretae cerae) – соединение плаценты со стенкой матки таково, что её ворсины достигают миометрия, слприкосаются с его поверхностью, однако не повреждают его структуры и не углубляются между мышечными между мышечными волокнами.
2. плаценты врастающие (placentae incretae) связаны со стенкой матки сильнее, так как их ворсины внедряются на различную глубину в миометрий, нарушая его непрерывность в зоне плацентарной площадки.
3,плаценты прирастающие (placentae percretae) разрушает структуру миометрия на всю глубину, проникая сквозь этот слой вплоть до параметрия или брюшины.
Амниотические перетяжки
волокнистые нити (амниотические тяжи), возникающие в плодном пузыре (амнионе). Проходя через его полость, они могут опутывать, связывать или прорезать части тела плода или пуповину, вызывая различные пороки развития. Возникающие в результате поражения называют синдромом амниотических перетяжек.
Из-за опутывания тяжами может нарушиться кровоток частей плода. В результате есть риск возникновения отёка, застоя лимфы, вызывающего припухлость, некроза (омертвение ткани, требующее хирургической ампутации после рождения) или внутриутробной ампутации. Происходит это на ранних сроках беременности, чаще всего с одной стороны тела, затрагивая выступающие части плода − пальцы, руки. Другие возможные последствия:
- Рука новорождённого с амниотической перетяжкой
-кольцевые перетяжки (вдавления) конечностей и пальцев;
-сращение пальцев рук и/или ног;
-расщелины губы или нёба;
-гемангиома;
-различные черепно-лицевые дефекты, дефекты позвоночника, пуповины и/или тела.
Есть две теории появления амниотических перетяжек. Одна из основных теорий объясняет их возникновение частичным прорывом амниотического пузыря на ранних сроках беременности, при котором хорион остается нетронутым.
Волокнистые нити, возникшие из-за прорыва, плавают в амниотической жидкости и могут «поймать» и опутать те или иные части плода. Так как по мере роста плода размер нитей не меняется, возникают вдавления, нарушающие кровоток, со всеми возможными последствиями.
Существует также теория сосудистых нарушений. Так как предыдущая теория не объясняет частые сочетания амниотических перетяжек и расщелин (губы, нёба), возникло предположение о внутренних сосудистых нарушениях или нарушении кровообращения как основном или дополнительном факторе возникновения тяжей
Синдром Дауна
Синдром Дауна, трисомия 21, - одна из форм геномной патологии, при которой чаще всего кариотип представлен 47 хромосомами вместо нормальных 46, поскольку хромосомы 21-й пары, вместо нормальных двух, представлены тремя копиями (трисомия, см. также плоидность).
Синдром Дауна представляет собой врожденное нарушение развития, которое дает о себе знать в виде значительного нарушения роста костей, а также других аномалий физического развития ребенка. У детей, рожденных с синдромом Дауна, отмечаются следующие особенности строения тела: узкие раскосые глаза, маленькая округлая голова, сухие тонкие волосы, маленькие уши и нос, влажная отечная кожа, толстые губы. У этих детей также короткие и толстые пальцы, длинный язык, маленький рост, а также не до конца развитые половые органы. Больные синдромом Дауна значительно отстают в умственном развитии. Уже, будучи взрослыми, люди с данным синдромом размышляют как семилетние дети. Следует отметить еще и то, что все больные синдромом Дауна чаще всего очень ласковые и покорные.
синдром Дауна не является наследственной патологией
Синдром Дауна — хромосомная патология, характеризующаяся наличием дополнительных копий генетического материала по 21-й хромосоме, либо полностью (трисомия), либо частично (например, за счёт транслокации). Последствия от наличия дополнительной копии сильно различаются в зависимости от степени копии, генетической истории и чистой случайности. Синдром Дауна встречается как у людей, так и у других видов (например был обнаружен у обезьян и мышей). Совсем недавно исследователи вывели трансгенных мышей с наличием 21-й человеческой хромосомы (в дополнение к стандартному набору мышей). Добавление генетического материала может проводиться в разных направлениях. Типичный человеческий кариотип обозначается как 46,XY (мужской) или 46,XX (женский) (различие в поле несёт Y-хромосома).
Синдром Патау
хромосомное заболевание, которое характеризуется наличием в клетках дополнительной хромосомы 13.
Причины
В большинстве случаев причиной появления синдрома Патау является трисомия 13, которая означает, что каждая клетка тела имеет три копии 13 хромосомы вместо обычных двух. Кроме того, бывают случаи (их доля очень незначительна), когда лишь некоторые клетки организма имеют дополнительную копию хромосомы в результате смешанной популяции клеток с различным числом хромосом; такие случаи называют мозаичным синдромом Патау.
Данное заболевание может также возникать, когда часть 13 хромосомы привязывается к другой хромосоме (транслокуеться) до или в момент зачатия. Больные люди имеют две копии 13 хромосомы, плюс дополнительный материал из нее, подключенный к другой хромосоме. С транслокацией, человек имеет частичную трисомия 13 хромосомы, в связи с чем физические признаки синдрома часто отличаются от типичного случая.
В большинстве случаев синдром Патау не наследуется, а возникает как случайное событие в процессе формирования половых клеток (яйцеклеток и сперматозоидов). Ошибка в делении клеток, которая называется нерасхождением, может привести к появлению репродуктивных клеток с аномальным числом хромосом. Например, яйцеклетка или сперматозоид могут получить дополнительную копию хромосомы. Если одна из этих атипичных половых клеток будет вовлечена в генетической структуре ребенка, ребенок будет иметь дополнительную 13 хромосому в каждой из клеток организма. Мозаицизм синдрома Патау также не наследуется, а возникает как случайное нарушение при делении клеток в начале развития плода.
Синдром Патау может быть унаследован в связи с транслокацией. Здоровый человек может нести измененный генетический материал между 13 и другими хромосомами. Эта перестройка называется сбалансированной транслокацией, поскольку не было получено дополнительного материала с 13 хромосомы. Люди, являющиеся носителями данного типа, находятся в зоне повышенного риска рождения детей с этим заболеванием, хотя они и не имеют признаков синдрома Патау.
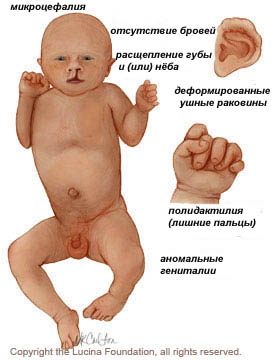
Проявления и физические признаки
Плод, который выживает в течение беременности и рождения, имеет следующие отклонения:
Нервная система:
- отклонения психического и моторного развития;
- микроцефалия;
- голопрозэнцефалия (нарушение формирования полушарий мозга);
- структурные дефекты глаз, в том числе микрофтальмия, аномалия Питерса, катаракта, колоб, дисплазия или отслоение сетчатки, сенсорный нистагм, пробковая потерю зрения и гипоплазия зрительного нерва;
- менингомиелоцеле (спинномозговой дефект)
Костно-мышечные и кожные:
- полидактилия («лишние пальцы»)
- низко посаженные и деформированные ушные раковины;
- выступающая пятка;
- деформация ноги, стопа выглядит как качеля;
- омфалоцеле (брюшной дефект, пупочная грыжа);
- аномальный вид кисти;
- перекрытие пальцами большого пальца;
- врожденное отсутствие кожи (отсутствуют участки кожи / волос);
- волчья пасть, заячья губа (расщепление неба).
Урогенитальные:
- аномальные гениталии, - дефекты почек.
Другие:
- пороки сердца (дефект межжелудочковой перегородки);
- одна пуповинная артерия.
Риск повторенияЕсли один из родителей является носителем транслокации, то шансы пары зачать ребенка из трисомией 13 составляют менее 1% (меньше, чем при синдроме Дауна).
Синдром Эдвардса
Синдром Шерешевского-Тернера
Синдром Вольфа-Хиршхорна
Синдром Кляйнфельдтера
Синдром «кошачьего крика»