

Релаксація. Ширина ліній
При виконані умов резонансу електронні переходи між двома підрівнями відбуваються в обох напрямках з однаковою ймовірністю і відповідно супроводжуються поглинанням чи випромінюванням енергії. Можливість спостерігати саме спектри поглинання пов’язана з більшою заселеністю нижнього енергетичного рівня у відповідності до закону Больцмана, а значить і з більшою кількістю переходів з нижнього рівня на верхній. В магнітному полі при кімнатній температурі на нижньому рівні є невеликий надлишок заселеності n1 – n2, проте він становить лише соті відсотка, оскільки величина розщеплення ΔЕ, як вказувалося вище, є незначною 0,2 – 1 см-1.
Під впливом електромагнітного випромінювання заселеність рівнів дуже швидко вирівнюється і поглинання енергії припиняється. Таке явище називають насиченням. Без наявності процесів, які б дозволили відновити початкову заселеність енергетичних рівнів і дали б можливість безперервно спостерігати поглинання енергії, зареєструвати спектри ЕПР було б неможливо. Такі процеси називаються процесами релаксації. Один із них – це процес передачі енергії від системи спінів оточуючому середовищу (гратці). Він відбувається в результаті взаємодії між спінами парамагнітних часток і коливаннями гратки і називається спін-гратковою релаксацією. Спін-граткова релаксація відбувається не лише в твердих тілах, а й в інших середовищах, оскільки “коливання гратки” в загальному випадку являє собою молекулярно тепловий рух всередині середовища: газу, рідини чи твердого тіла. Швидкість спін-граткової релаксації, як правило позначають 1/Т1, де Т1 час спін-граткової релаксації. Це час, за який система із збудженого стану повертається в основний стан, тобто час, за який встановлюється початкова заселеність рівнів. Час Т1 є дуже важливою характеристикою зразка, оскільки визначає ширину лінії ЕПР, а часто і принципову можливість реєстрації ЕПР спектрів. Ширина лінії ЕПР є обернено пропорційною часу спін-граткової релаксації.
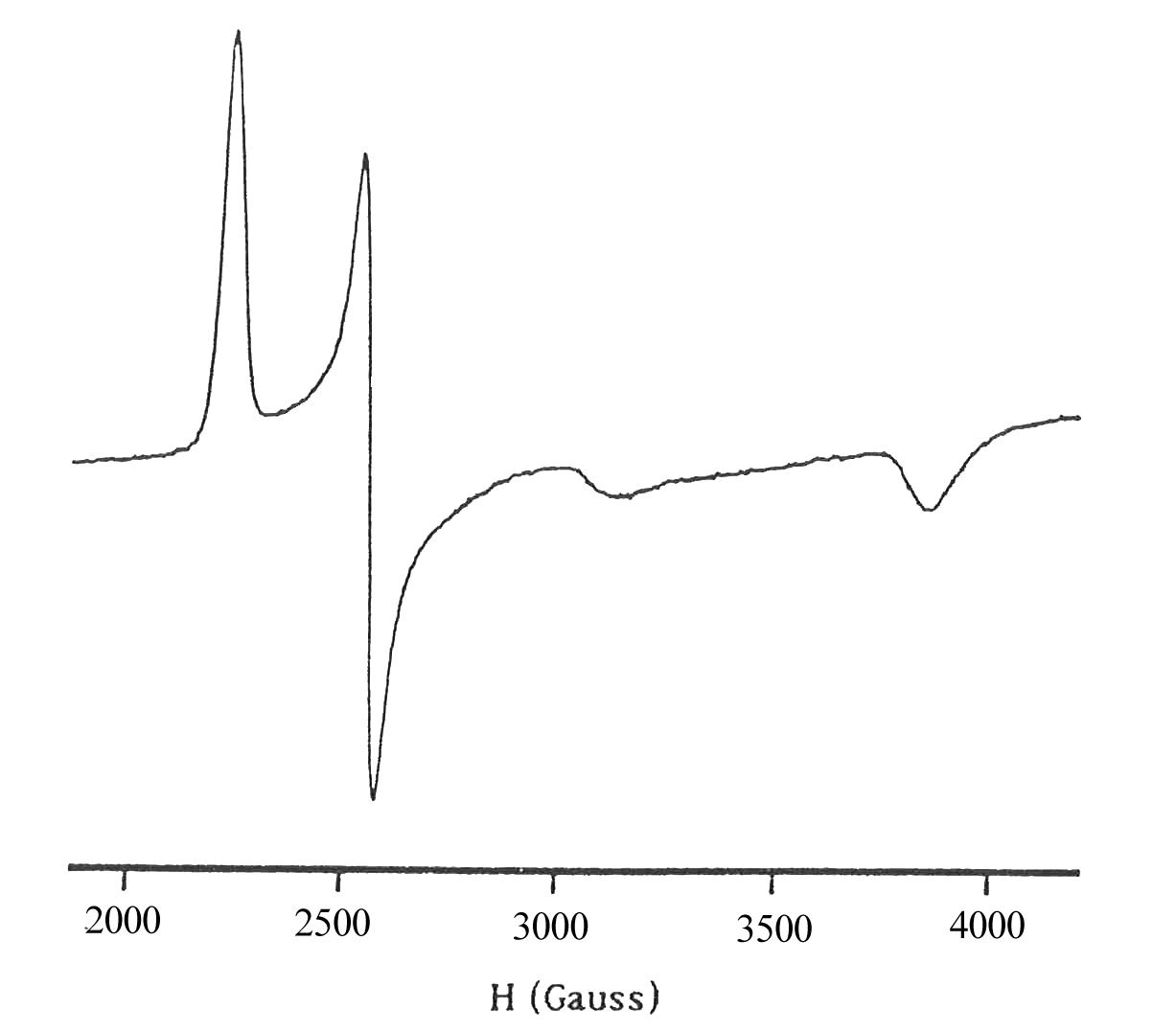
Для вільних радикалів значення Т1 дуже велике і лінії ЕПР дуже вузькі та добре розділені. Для комплексів перехідних металів час спін-граткової релаксації завжди набагато менший, тому ширина ліній в їх спектрах ЕПР майже завжди більша, ніж для вільних радикалів, а іноді лінії є настільки широкими, що їх взагалі не вдається зареєструвати. На рис. 3 для прикладу зображено спектри ЕПР комплексу феруму і вільного радикалу. Навіть наближена оцінка показує, що ширина ліній у спектрі комплексу більша щонайменше у тисячу разів, ніж ліній радикалу (в першу чергу зверніть увагу на масштаб).
Температура зразка дуже сильно впливає на час релаксації і на ширину лінії: чим менша температура, тим більший час Т1 і тим вужчі лінії. У деяких випадках при температурах рідкого азоту чи гелію можна одержати спектри ЕПР, які не можна зареєструвати при кімнатній температурі із-за їх дуже великої ширини.
|
|
б |
|
Рис.3. Спектри ЕПР комплексу феруму(ІІІ) (a) і аніон-радикалу n-бензосеміхінону (б). |
|
 а
а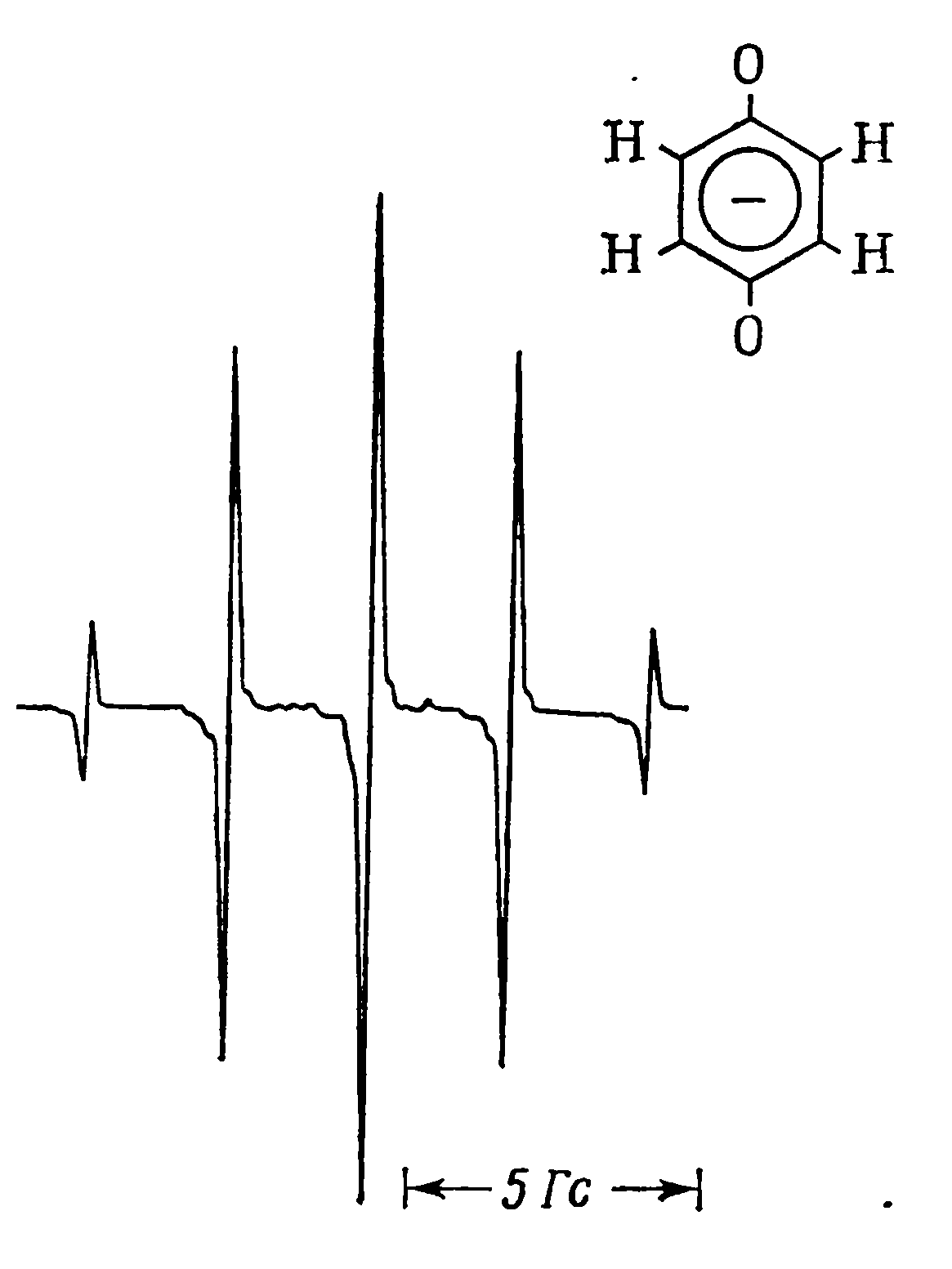
Час Т1 значно залежить також від розщеплення орбітальних рівнів енергії: чим більше розщеплення, тим більший час Т1 і тим вужчими є лінії . У тих випадках, коли розщеплення є незначним, як у іонів лантаноїдів, час релаксації великий, лінії поглинання мають дуже велику ширину і спектри ЕПР можуть не спостерігатись навіть при температурі рідкого гелію. Незначним є значення орбітального розщеплення і для d-металів з парною кількістю електронів. Тому для електронних конфігурацій іонів металів d2, d4, d6, d8 спектри ЕПР при температурах 77 К і вище також, як правило, не спостерігаються. У комплексних сполуках перехідних елементів з непарною кількістю d електронів орбітальні розщеплення є значно більшими, і лінії ЕПР будуть достатньо вузькими. Дійсно, як показала практика, реєструвати спектри ЕПР при температурах в інтервалі 77–300 К вдається лише для комплексів металів з непарною кількістю d-електронів, перелік яких наведено в табл.1. Для інших парамагнітних сполук металів ширина ліній при даних умовах занадто велика, хоч при температурі рідкого гелію спостерігати переходи ЕПР для них іноді й можливо.
Таблиця 1. Іони металів, для яких спостерігається явище ЕПР при температурах 77-300 К
|
Конфігурація |
Парамагнітні іони |
|
d1 |
Ti3+, V4+, Cr5+ , Mo5+, W5+ , Nb4+, Mn6+, Re6+, Os7+ |
|
d3 |
V2+, Cr3+ |
|
d5 |
Mn2+, Fe3+, Mo1+ , Ru3+, V0, Cr1+, Os3+ |
|
d7 |
Co2+, Ni3+, Rh2+, Pd3+, Pt3+, Fe1+ |
|
d9 |
Cu2+, Ag2+, Ni1+ |
|
f7 |
Gd3+, Eu2+ |
Серед іонів металів, які наведено в табл. 1, більшість мають нехарактерні ступені окиснення. Метод ЕПР дає можливість просто і надійно зафіксувати їх утворення в розчинах, чи в твердих фазах і в цьому відношенні є назамінним аналітичним методом. У багатьох випадках довести утворення сполук металів із незвичними ступенями окиснення за допомогою інших методів значно важче, а іноді і неможливо, особливо коли сполуки існують лише в розчині або утворюються в незначній кількості.
Таким чином, наявність неспарених електронів (тобто парамагнетизм) є необхідною, але недостатньою умовою для реєстрації ЕПР спектрів. Так, наприклад, методом ЕПР не можна досліджувати парамагнітні комплекси нікелю(ІІ) (конфігурація d8) і навіть високоспінові комплекси кобальту(ІІ) (конфігурація d7), оскільки час релаксації для них є досить малим. У той же час низькоспінові комплекси кобальту(ІІ) дають якісні спектри ЕПР навіть при кімнатній температурі.
У парамагнітній речовині на кожний електрон, крім зовнішнього магнітного поля, діють магнітні поля всіх інших неспарених електронів даного зразка. Ця взаємодія між електронами, називається спін-спіновою взаємодією. Електрони знаходяться в постійному русі, причому кожний електрон у кожний момент часу має своє специфічне оточення. Унаслідок цього величина внутрішніх локальних полів, які діють на електрони, відрізняється одна від одної. А значить умови резонансу для різних електронів будуть виконуватися не при однакових значеннях зовнішнього магнітного поля. Наслідком цього є уширення ліній ЕПР. Оскільки в магнітноконцентрованих зразках (індивідуальні парамагнітні сполуки) відстань між сусідніми парамагнітними частинками невелика, то в результаті досить значний спін-спінових взаємодій лінії мають значну ширину і тонка структура в спектрах ЕПР таких зразків є не розділеною.
Оскільки величина спін-спінової взаємодії є обернено пропорційною кубу відстані між сусідніми парамагнітними атомами чи іонами, то зменшити її, а значить і одержати більш вузькі лінії ЕПР можна шляхом діамагнітного розведення парамагнітних сполук (зразків). Основними способами діамагнітного розведення є використання рідких чи твердих розчинів парамагнітних речовин. Тому, якщо необхідно одержати добре розділені спектри ЕПР, проводять дослідження розчинів координаційних сполук (див. нижче). Звичайно при цьому використовують розчинники, які практично не взаємодіють з парамагнітною речовиною, тобто при розчиненні не повинні відбуватись суттєві зміни у складі чи будові речовини.
Швидкі хімічні процеси також впливають на ширину спектральних ліній. Так, наприклад, ацетилацетонат купруму(ІІ) має високу стійкість і не дисоціює у розчинах толуену, тому при кімнатній температурі лінії в спектрах ЕПР толуенових розчинів є відносно вузькими і добре розділеними (рис. 4а). У розчинах ацетилацетонату купруму в донорних розчинниках, наприклад, в ДМФА відбуваються процеси хімічного обміну лігандів:
![]()
Це приводить до значного уширення ліній, внаслідок чого вони часто є нерозділеними, що робить неможливим проводити інтерпретацію спектрів (рис. 4б). Тому спектри ЕПР розчинів комплексів у донорних розчинниках чи комплексів, термодинамічна стійкість яких є недостатньо високою, досліджують у заморожених склоподібних розчинах при температурі кипіння рідкого азоту (77 К), оскільки при таких умовах процеси хімічного обміну значно уповільнюються.
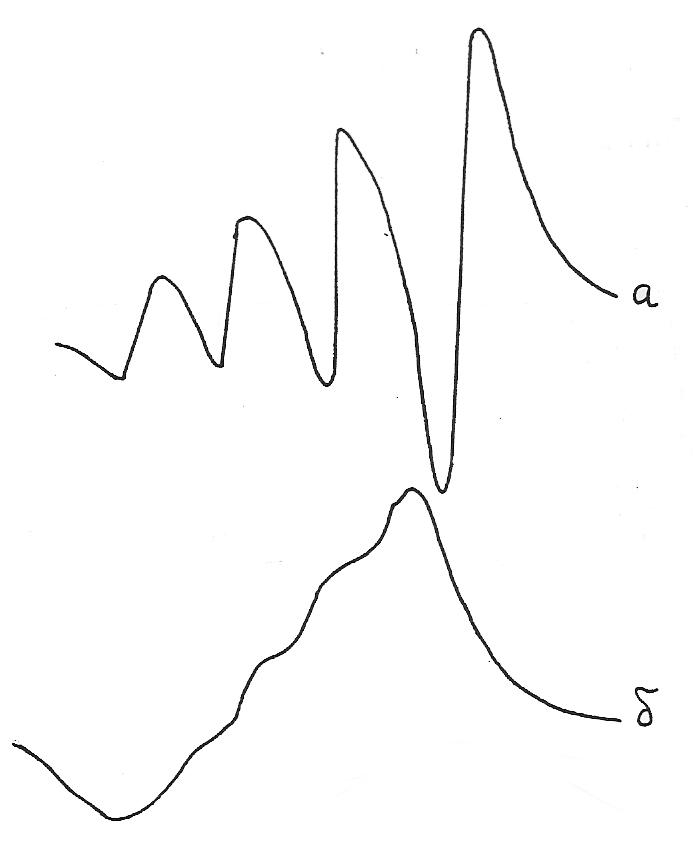
Рис. 4. спектри ЕПР ацетилацетонату купруму в толуолі (а) і в ДМФА (б) при 273 К
Все сказане відноситься лише до моноядерних комплексів, або до біядерних з великою відстанню металметал. Якщо в бі- чи в поліядерних координаційних сполуках внутрішньомолекулярна відстань між парамагнітними іонами є невеликою, тоді можливість реєстрації спектрів ЕПР і їх форма визначаються процесами спінового обміну, кількісною мірою яких є величина обмінного інтегралу.
g-фактор. Анізотропія g-фактора
Для того, щоб зі спектрів ЕПР одержати інформацію про будову речовини, потрібно провести аналіз параметрів спектру. Одним із таких параметрів є g-фактор, який є мірою ефективного магнітного моменту електрона і визначає положення смуги поглинання в магнітному полі. g-Фактор “вільного” електрона дорівнює 2,0023. Спектри ЕПР радикалів мають значення g-фактора, яке відрізняється від g-фактора вільного електрона лише у третьому знаці після коми. Так, наприклад, для вільного радикалу дифенілпікрилгідразиду (ДФПГ), який використовується в ЕПР як зовнішній стандарт, g =2,0036.
У випадку комплексів перехідних металів g-фактор може суттєво відрізнятися від чисто спінового значення. Так, наприклад, для NbCl4 в етанолі g = 1,80, а для хлориду гадолінію в етанолі одне із значень g–фактора дорівнює7,2. Це є наслідком спін-орбітальної взаємодії, яка для іонів перехідних металів має значно більшу величину, ніж для вільних радикалів. Магнітні властивості неспарених електронів перехідних металів залежать не тільки від спінового моменту(1), але і від орбітального моменту. Тому спектри ЕПР комплексів перехідних металів характеризуються ефективним g-фактором (далі просто g-фактор), який враховує вклади від спін-орбітальної взаємодії і є мірою ефективного магнітного моменту електрона.
Відхилення g-фактора від 2,0023 є пропорційним різниці енергій орбітальних енергетичних рівнів, що змішуються в результаті спін-орбітальної взаємодії. Для випадку одного неспареного електрона значення ефективного g-фактора в межах простої теорії кристалічного поля визначається за рівнянням:
![]() ,
(7)
,
(7)
де g0 = 2,0023 (g-фактор вільного електрона); λ – константа спін-орбітальної взаємодії; n – коефіцієнт, який залежить від того, які орбіталі змішуються у результаті спін-орбітальної взаємодії; ΔЕ – різниця в енергії між орбіталлю, що містить неспарений електрон та орбіталлю, з якою відбувається змішування. Знак “+” чи “-“ визначається ступенем заповнення d-орбіталі: “+” для металів з d1-4 електронними конфігураціями; “-“ – для металів з d6-9 електронними конфігураціями ; для d5 конфігурацій g ≈ g0 ≈ 2,0. Тому для іонів металів з d1-4 оболонками, як правило, g < 2.0, а для іонів металів з d69 оболонками g 2.0.
Спін-орбітальна взаємодія відповідає також за анізотропію магнітних властивостей, яка характерна для полікристалічних зразків координаційних сполук, що мають низьку симетрію. Спіновий кутовий момент орієнтується в залежності від напрямку зовнішнього поля, а орбітальний кутовий момент, що пов'язаний з електронами, які рухаються по молекулярним орбіталям, прив'язаний до орбітальної хвильової функції. На рис.5 показано дві різні орієнтації магнітної орбіталі (тобто орбіталі, на якій знаходиться неспарений електрон) відносно напрямку магнітного поля.
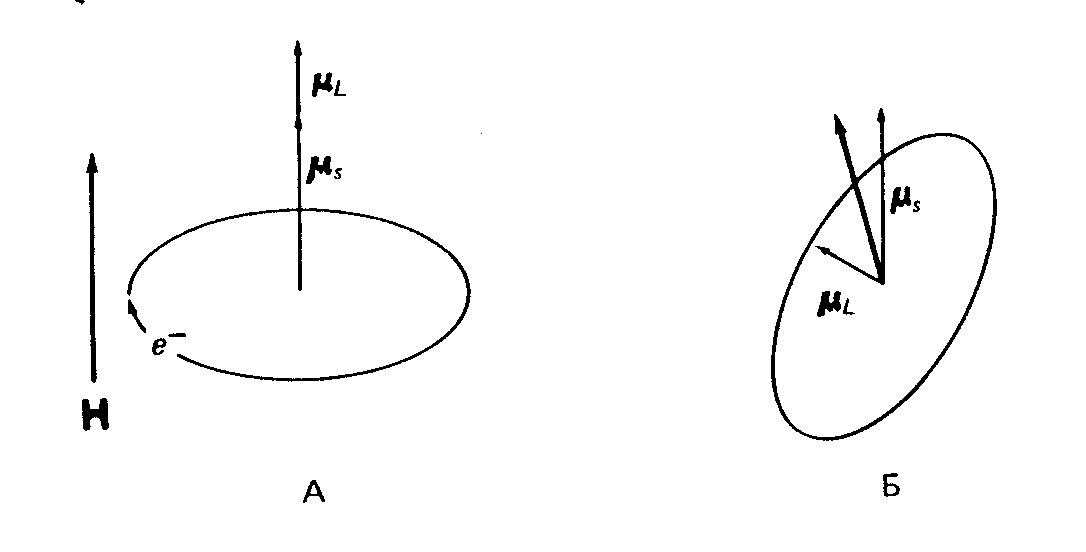
Рис.5.
Як видно з рис. 5, при різній орієнтації молекули відносно зовнішнього магнітного поля векторна сума моментів μL і μS буде різною, тобто різною буде ефективний магнітний момент електрона. Тому в залежності від орієнтації кристала в магнітному полі g-фактор буде мати різні значення.
Звичайно, якщо комплекс має кубічну симетрію (наприклад, октаедричну), то її магнітні властивості не залежать від орієнтації молекули у зовнішньому магнітному полі. Спектр ЕПР у цьому випадку буде представляти собою одиночну (синглетну) лінію, тобто буде ізотропним (рис. 6а).
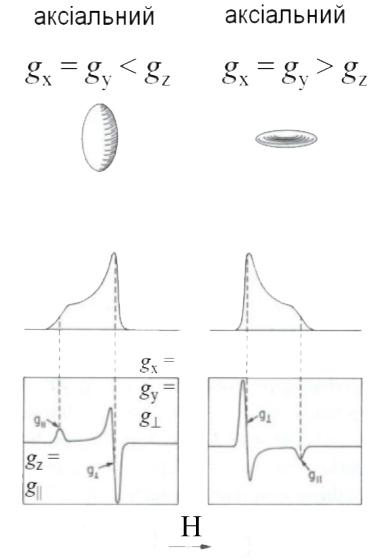
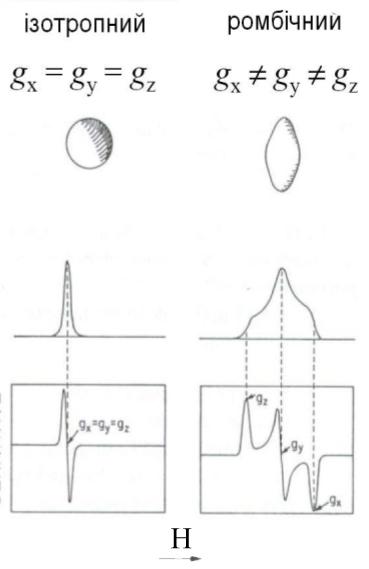
Рис. 6. Форма спектрів ЕПР для полікристалічних комплексів металів різної симетрії
У випадку ромбічної симетрії лігандного оточення іона металу g-фактор буде мати три значення: gx, gy і gz у залежності від орієнтації кристала відносно зовнішнього магнітного поля. У спектрі ЕПР порошкоподібних зразків при цьому спостерігається три смуги поглинання (рис. 6). Відповідні анізотропні значення g-фактора знаходять із рівнянь:
![]()
![]()
![]() (8)
(8)
Анізотропні значення g-фактора сполук ромбічної симетрії зв’язані з ізотропним значенням gо, яке можна визначити зі спектрів ЕПР рідких розчинів цих сполук виразом:
![]() . (9)
. (9)
При аксіальній симетрії (точкові групи D3h, D4h, D5h, C4v, C3v) у спектрах ЕПР полікристалічних речовин спостерігаються дві смуги поглинання, яким відповідають два значення g-фактора: g=gx=gy і gII = gz (рис. 6). Вони зв’язані з ізотропним g-фактором залежністю
![]() . (10)
. (10)
_______________________
Сказане є справедливим лише для випадків, коли осі симетрії кристала співпадають з молекулярними осями симетрії. Якщо ні, то для характеристики анізотропних систем потрібен g-тензор, який враховує спін-орбітальні вклади в g-фактор при різних орієнтаціях кристала.
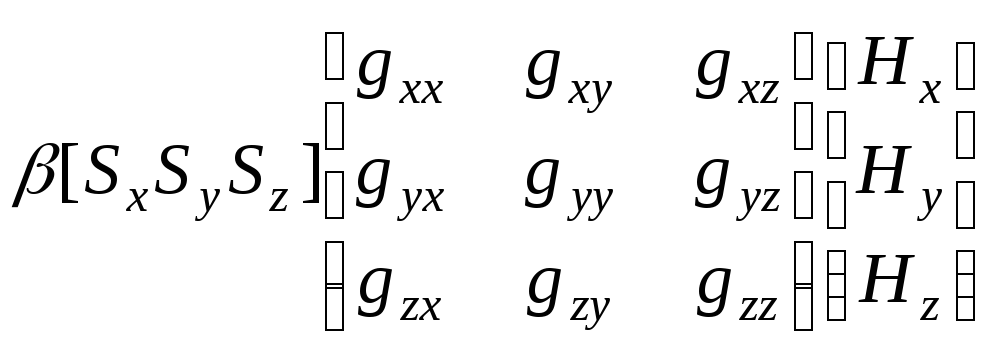
Лінії, які відповідають перпендикулярній орієнтації кристалів відносно магнітного поля, легко відрізнити в спектрах ЕПР від ліній паралельної орієнтації, оскільки останні мають значно меншу інтенсивність.
Як видно з рис. 6, за спектрами ЕПР полікристалічних зразків можна легко розрізнити ромбічну, кубічну і аксіальну симетрію. У випадку аксіальної симетрії форма спектрів ЕПР залежить від основного стану центрального іона металу (тобто від того, на якій орбіталі знаходиться неспарений електрон).
Наприклад,
якщо комплекс купруму(ІІ) має основний
стан
![]() ,
тобто має форму квадрата, витягнутого
октаедра чи тетрагональної піраміди
(див. розділ 2, рис. 2.7), анізотропні
значення g-факторів
знаходяться у співвідношенні:
,
тобто має форму квадрата, витягнутого
октаедра чи тетрагональної піраміди
(див. розділ 2, рис. 2.7), анізотропні
значення g-факторів
знаходяться у співвідношенні:![]() ,
і спектр ЕПР має форму зображену на
рис. 6б. У
випадку основного стану
,
і спектр ЕПР має форму зображену на
рис. 6б. У
випадку основного стану
![]() (трикутна біпіраміда, сплюснутий октаедр
чи чотирикутна піраміда) взаємоспіввідношення
параметрів будуть іншими, а саме:
(трикутна біпіраміда, сплюснутий октаедр
чи чотирикутна піраміда) взаємоспіввідношення
параметрів будуть іншими, а саме:![]() .
Форма спектра ЕПР для комплексів
купруму в цьому випадку зображена на
рис. 6в.
.
Форма спектра ЕПР для комплексів
купруму в цьому випадку зображена на
рис. 6в.
Проте слід особливо зауважити, що висновки щодо симетрії молекул одержані при дослідженні спектрів ЕПР полікристалічних сполук не є однозначними. Вони справедливі лише у тому випадку, якщо відповідні осі симетрії молекул, що знаходяться у межах однієї елементарної комірки, розташовані паралельно. Якщо існує розупорядкування осей (а це можна визначити лише з результатів рентгеноструктурного анлізу), то висновки, зроблені при ЕПР спектроскопічних дослідженнях полікристалічних зразків, можуть бути некоректними або повністю невірними.
Як
приклад можна навести ЕПР спектри
порошкоподібних зразків солей Міллона,
![]() і Бектона,
і Бектона,
![]() (рис. 7). Як слідує із спектра, неспарений
електрон у іона
(рис. 7). Як слідує із спектра, неспарений
електрон у іона
![]() повинен знаходитись на
повинен знаходитись на
![]() орбіталі і комплекс повинен мати
відповідну цьому основному стану
геометрію. Насправді цей іон має
плоско-квадратну будову, але молекули
в елементарній комірці розташовані
перпендикулярно одна до одної, що
приводить до “аномальної”
форми
спектра ЕПР.
орбіталі і комплекс повинен мати
відповідну цьому основному стану
геометрію. Насправді цей іон має
плоско-квадратну будову, але молекули
в елементарній комірці розташовані
перпендикулярно одна до одної, що
приводить до “аномальної”
форми
спектра ЕПР.
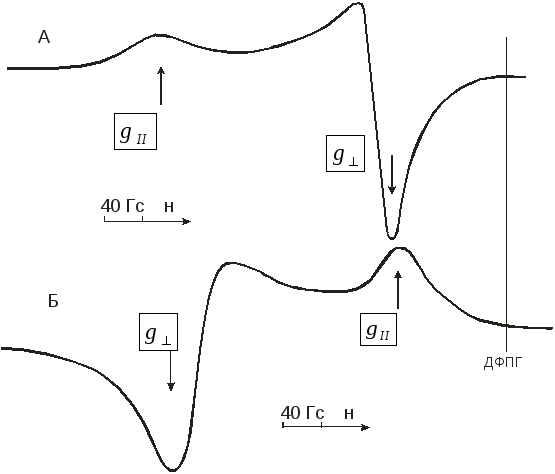
Рис. 7. Спектри ЕПР порошків солей Міллона (а) і Бектона (б)
Така залежність спектрів ЕПР полікристалічних комплексів не лише від симетрії молекул, але і від особливостей кристалічної будови сполук є причиною того, що спектри ЕПР координаційних сполук перехідних металів переважно досліджують у розчинах. Ще однією значною перевагою досліджень у розчинах є можливість спостерігати розділену надтонку і додаткову надтонку структуру спектрів ЕПР. У магнітноконцентрованих полікристалічних зразках комплексів тонка структура спектрів, як правило, не розділяється, із-за значних спін-спінових взаємодій між близько розташованими парамагнітними іонами.