

7. Вычисление энтропии.
В качестве отправной точки необходимо воспользоваться соотношением (4.21).
Тогда для изотермического процесса (Т = const):
![]() ,
,
где dq = dU + pdV, а dU = 0,
поэтому
![]() .
(4.39)
.
(4.39)
Из уравнения состояния идеального газа:
![]() ,
(4.40)
,
(4.40)
если n = 1 моль, следовательно:
![]() .
(4.41)
.
(4.41)
После интегрирования:
![]() .
(4.42)
.
(4.42)
Для случая P = const:
![]() ,
,
где dqP = CPdT, поэтому
![]()
и после интегрирования:
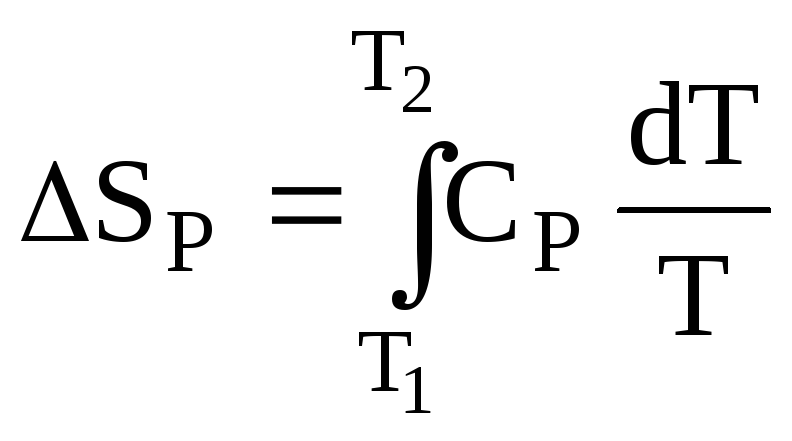 .
(4.43)
.
(4.43)
Если СР = const, то
![]() .
(4.44)
.
(4.44)
По аналогии для случая V = const:
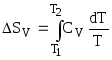 .
(4.45)
.
(4.45)
и если СV = const, то
![]() .
(4.46)
.
(4.46)
Если изотермическое превращение является фазовым - плавление, испарение, сублимация или полиморфный переход, то:
![]() ,
(4.47)
,
(4.47)
где q - энергетический эффект фазового перехода;
Т - температура фазового перехода.
Таким образом, рассмотрены некоторые уравнения, позволяющие вычислять изменение энтропии, если известны изменения параметров системы и ее теплоемкость. Существуют и другие методы определения изменения энтропии, например по измерениям электродвижущих сил гальванических элементов, о чем речь пойдет в главе “Электрохимия”.
8. Направление протекания процессов в изолированных системах и термодинамические условия равновесия.
Из уравнения второго закона термодинамики (4.39) следует, что если dq = 0, то справедливо неравенство:
![]() ,
(4.48)
,
(4.48)
т. е. в изолированной системе самопроизвольно реализуются процессы, сопровождающиеся увеличением энтропии и, как крайний случай, сохранение постоянства энтропии при обратимых процессах.
Самопроизвольный процесс протекает до тех пор, пока система не перейдет в равновесное состояние, для которого энтропия максимальна для заданных условий.
Условие максимума возрастающей функции:
dS = 0 и d2S < 0, (4.49)
которое является справедливым применительно к изолированным системам, т.е. для таких, у которых U и V = const, а также Н и Р.
Тогда (4.48) и (4.49) в более строгой форме записи:
![]() ;
;
![]() и
и![]() .
(4.50)
.
(4.50)
9. Энергия Гиббса. Энергия Гельмгольца.
Энтропия - основная
функция, позволяющая судить о направлении
самопроизвольных процессов в изолированных
системах. Чтобы использовать ее в
практических целях нужно исследовать
как самое систему, так и ее окружение.
Хотя рассчитать изменение энтропии
окружения или окружающей среды довольно
просто
![]() ,
можно предложить метод, автоматически
учитывающий наличие окружающей среды.
,
можно предложить метод, автоматически
учитывающий наличие окружающей среды.
Из первого закона термодинамики следует:
dq = dU +dA.
Из второго закона:
dq
![]() TdS,
TdS,
поэтому
TdS
![]() dU + dA.
(4.51)
dU + dA.
(4.51)
Неревенство (4.51) представляет собой обобщенную форму записи первого и второго начал термодинамики.
Из (4.51) следует, что:
dA
![]() TdS - dU.
(4.52)
TdS - dU.
(4.52)
При постоянных Т и V (4.52) интегрируется непосредственно:
AV
![]() T(S2
- S1)
- (U2
- U1)
T(S2
- S1)
- (U2
- U1)
или
AV
![]() TS2
- TS1
- U2
+ U1,
TS2
- TS1
- U2
+ U1,
откуда
AV
![]() -[( U2
-
TS2)
- (U1
- TS1)],
(4.53)
-[( U2
-
TS2)
- (U1
- TS1)],
(4.53)
где U - TS = F, поэтому:
AV
![]() -( F2 -
F1)
или
-( F2 -
F1)
или
![]() .
(4.54)
.
(4.54)
F - новая функция состояния, называемая энергией Гельмгольца (свободная энергия при постоянном объеме системы или изохорно-изотермический термодинамический потенциал).
Соотношение (4.54) читается: “... работа неизолированной системы в условиях V, T = const не более убыли энергии Гельмгольца”.
Величина АР
= AV
- p![]() есть величина максимальной полезной
работы, где АV
- максимальная полная работа.
есть величина максимальной полезной
работы, где АV
- максимальная полная работа.
По аналогии АР может быть определена в виде разности двух значений некоторой функции G - энергии Гиббса:
AР
![]() -( G2 -
G1)
или
-( G2 -
G1)
или
![]() .
(4.55)
.
(4.55)
Так же как энтальпия отличается от внутренней энергии на величину pV, так и G отличается от F на величину pV:
G = F + pV = U - TS + pV = H - TS. (4.56)
Итак, окончательно:
F = U - TS и G = H - TS, (4.57)
где F и G - новые функции состояния.