

Лекциця ВМС 11
.docЛекция 11.
Релаксационные свойства аморфных полимеров.
Согласно законам термодинамики всякий самопроизвольный процесс, происходящий в той или иной системе, приближает её к состоянию равновесия. Однако если сам переход имеет конечную скорость, то в процессе перехода можно зафиксировать ряд промежуточных неравновесных состояний. Процессы, связанные с переходом системы из неравновесных состояний в равновесное, называются релаксационными.
Механические свойства любых реальных твёрдых тел, в том числе и полимерных, носят релаксационный характер. Основные случаи проявления релаксационных свойств полимеров в высокоэластическом состоянии следующие.
1. К образцу полимера приложено постоянное напряжение σ0. Если при этом течение отсутствует, то напряжению σ0 соответствует определённая равновесная высокоэластическая деформация εравн. Однако равновесная высокоэластическая деформация, соответствующая напряжению σ0, достигается не мгновенно. Как видно из рис. 1, равновесная деформация является верхним пределом, к которому во времени стремится реальная деформация.
ε







 σ0
σ0
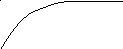 εравн
1
2
εравн
1
2
 σравн
σравн

t t
2. Образец полимера подвергают очень быстрой (практически мгновенной) деформации ε и закрепляют в деформированном состоянии. При этом в образце возникает напряжение σ0, значительно превышающее равновесное напряжение σравн. Со временем в результате перегруппировки звеньев и изменения конформаций макромолекул напряжение в образце уменьшается (рис. 2). Этот процесс называется релаксацией напряжения.
 σ
σ





3
σ = σ0 sin ωt
 π
2π 3π 4π t
π
2π 3π 4π t



 0
0
ε
 ε
= ε0
sin
(ωt-δ)
ε
= ε0
sin
(ωt-δ)
 0
0
π 2π 3π 4π t
3. К образцу полимера приложено переменное напряжение, действующее, например, по синусоидальному закону σ = σ0 sin ωt. В этом случае приложенное напряжение σ характеризуется двумя величинами – амплитудой σ0 и частотой ω (или периодом Т = 2π/ω). Под действием периодических напряжений в образце возникают периодические деформации, также изменяющиеся по синусоидальному закону. Однако синусоида деформации сдвинута по фазе относительно синусоиды напряжения на угол δ: ε = ε0 sin (ωt - δ). Возникновение разности фаз между напряжением и деформацией обусловлено релаксационными явлениями, вызывающими запаздывание изменений деформации по сравнению с соответствующими изменениями напряжения.
В случае простых релаксирующих систем отклонение какой-либо измеряемой величины от равновесных значений уменьшается с течением времени по экспоненциальному закону:
Δx = Δx0е-t/τ,
где Δх0 и Δх – отклонения значений измеряемой величины от равновесного значения для начального и данного моментов времени соответственно; τ – постоянная для данной системы, называемая временем релаксации.
Время релаксации характеризует скорость перехода системы в равновесное состояние. Зависимость деформации полимера от времени при приложении к нему постоянной нагрузки можно описать уравнением ε = εравн(1 – е-t/τ), а процесс релаксации напряжения – уравнением σ - σравн = (σ0 – σравн)е-t/τ.
В разных системах время достижения состояния равновесия различно. Например, скорости релаксационных процессов в жидкостях зависят, подобно вязкости жидкостей, от соотношения энергий межмолекулярного взаимодействия и теплового движения. Чем выше вязкость жидкости, тем медленнее протекают релаксационные процессы, т.е. тем больше времена релаксации. При комнатной температуре время релаксации обычных низкомолекулярных жидкостей мало и составляет 10-8 – 10-10 с. Однако при понижении температуры скорость молекулярных перегруппировок быстро уменьшается и при отсутствии кристаллизации жидкости при дальнейшем охлаждении превращаются в стеклообразные тела, обладающие бесконечно большим временем релаксации.
Очевидно, что время релаксации при прочих равных условиях должно зависеть от размеров молекул жидкости, возрастая при переходе от низших гомологов к высшим. Естественно было бы ожидать, что у полимеров, обладающих очень длинными молекулами, времена релаксации должны быть очень велики. Однако вследствие гибкости макромолекул релаксационные процессы в полимерах на самом деле характеризуются широким набором (спектром) времён релаксации, содержащим как очень малые времена, соответствующие перегруппировкам малых частей макромолекул, так и очень большие времена, соответствующие передвижению целых макромолекул или их больших участков.
Релаксационные процессы обусловливают так называемый гистерезис, проявляющийся в несовпадении деформационных кривых ε = f(σ), получаемых при постепенном увеличении напряжения и при постепенном его уменьшении. При нагружении образца полимера деформация его за конечный промежуток времени не успевает развиться полностью. Следовательно, значения деформации оказываются меньше равновесных. При разгрузке образца он не успевает полностью сократиться и в каждый момент времени значение деформации оказывается больше его равновесного значения. Поэтому при неравновесной деформации кривые нагрузка – удлинение не совпадают. График, отражающий эти зависимости, имеет вид петли, которая называется петлёй гистерезиса.
σ


 4
ε 5
4
ε 5


 ω1
> ω2
> ω3
ω1
> ω2
> ω3




ω3 ω2 ω1
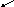



 ε1
ε Т
ε1
ε Т
Площадь петли гистерезиса может быть представлена в виде суммы двух интегралов: ε1 0
Sn = ∫σdε + ∫σdε
-
ε1
Площадь петли гистерезиса – это разность между удельной работой, затраченной при нагружении и полученной при разгрузке образца. Чем больше площадь петли гистерезиса, тем больше потери механической энергии. Эта энергия превращается в тепло и частично может затрачиваться на активацию сопутствующих химических процессов.
При переменном нагружении образца, когда напряжение изменяется по синусоидальному закону, необратимо затраченная за один цикл удельная работа деформации А = πσ0ε0 sin δ тем больше, чем больше разность фаз напряжения и деформации.
Явление гистерезиса необходимо учитывать при создании изделий из полимерных материалов, работающих в режиме циклического нагружения. Очевидно, что при изготовлении, например, автомобильной шины механические потери следует максимально уменьшить, так как в противном случае при эксплуатации шины будет происходить разогрев материала, способствующий его разрушению.
Для более детального анализа релаксационных явлений при деформации полимеров в широком интервале температур рассмотрим процесс развития деформации под действием синусоидально изменяющегося напряжения с постоянной амплитудой.
На рис. 5 показана зависимость амплитуды деформации от температуры при различных частотах (или периодах) действия силы. При низких температурах (в области стеклообразного состояния) амплитуда деформации очень мала и практически не зависит от частоты действия силы. В области стеклообразного состояния время релаксации намного больше времени деформации, поэтому практически сколь угодно длительный промежуток времени оказывается недостаточным для перегруппировки звеньев макромолекул. С повышением температуры время релаксации уменьшается, так как вследствие увеличения интенсивности теплового движения звеньев их перегруппировки происходят чаще. При высоких температурах в области высокоэластического состояния время релаксации звеньев очень мало и в образце практически при любом значении времени действии силы высокоэластическая деформация успевает развиться до значений, близких к равновесному. Поэтому в этой области температур амплитуда деформации также практически не зависит от частоты действия силы.
Однако для каждого полимера существует такой интервал температур, в котором время релаксации и время развития деформации соизмеримы. В этой промежуточной области температур (переходной области из стеклообразного в высокоэластическое состояние) наблюдается резкая зависимость амплитуды деформации от частоты действия силы. Если время действия силы больше времени релаксации, деформация успевает развиться. Если время действия силы меньше времени релаксации, высокоэластическая деформация не успевает развиться. Так, если при некоторой температуре и частоте действия силы ω3 в материале развивается деформация, близкая к равновесной, то при этой же температуре и частоте действия силы ω1 амплитуда деформации может быть очень мала и материал ведёт себя как стеклообразное тело.
t

 g
δ
6
g
δ
6

 ω3
ω2
ω1
ω1
> ω2
> ω3
ω3
ω2
ω1
ω1
> ω2
> ω3

 T
T
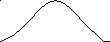
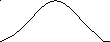
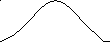
На рис. 6 показана зависимость тангенса угла сдвига фаз между напряжением и деформацией от температуры при различных частотах действия силы. Как видно, угол сдвига фаз невелик при очень малых и больших значениях частоты и температуры: при малой частоте и большой температуре потому, что релаксационные процессы успевают пройти полностью за время действия силы, а при большой частоте и низкой температуре потому, что релаксационные процессы вообще не успевают пройти и материал ведёт себя как упругое стеклообразное тело. Это означает, что сдвиг фаз проходит через максимум в переходной области, в которой время деформации сравнимо со временем релаксации полимера. Следует отметить, что наличие у полимеров спектра времён релаксации приводит к наложению сдвигов фаз, вызываемых различными релаксационными процессами. В результате этого наложения получается один пологий максимум сдвига фаз, охватывающий значительный интервал температур.
Принцип температурно-временной суперпозиции. Сравнение температурных зависимостей амплитуды деформации и tg δ при различных частотах действия силы показывает, что увеличение частоты (т.е. уменьшение времени действия силы) и понижение температуры одинаково влияют на деформацию или угол сдвига фаз. Одно и то же значение деформации или угла сдвига фаз можно получить, изменяя либо частоту, либо температуру. Это свидетельствует об эквивалентности температуры и времени воздействия силы. В этой эквивалентности и заключается суть принципа температурно-временной суперпозиции. Исходя из этого принципа, можно рассчитать зависимость механических свойств от частоты действия силы в столь широком диапазоне частот, который иногда вообще не может быть реализован в лабораторных условиях. Это достигается проведением опыта в относительно узком диапазоне частот при разных температурах. Затем на основе принципа эквивалентности времени действия силы и температуры строится единая обобщённая кривая при заданном значении температуры, охватывающая широкую область частот действия силы.





 σ
7
σ
7


 T1
T1

 T2
T2


 T3
T3

 T4
T4


 T5
T5
 T6
T6
lg t
На рис. 7 приведены кривые релаксации напряжения полимера при различных температурах. Согласно принципу температурно-временной суперпозиции кривые релаксации напряжения, снятые при разных температурах, можно наложить на один обобщённый график путём простого их смещения вдоль оси логарифма времени на величину, зависящую от температуры. Выбрав в качестве температуры приведения какую-либо произвольную температуру, например Т5, станем сдвигать остальные кривые вдоль оси логарифма времени по отношению к стандартной кривой до тех пор, пока участки кривых не совместятся и не образуют одну обобщённую кривую. Отрезок, на который следует сдвинуть каждую исходную кривую вдоль оси логарифма времени для получения обобщённой кривой, называется фактором сдвига или параметром приведения аТ. Фактор сдвига представляет собой отношение времени релаксации полимера при температуре Т к времени его релаксации при температуре приведения Т0, т.е.
lg aT = lg τT/τ T0
В методе приведения очень важным является правильный выбор температуры приведения Т0. Очень удобной была бы комнатная температура, однако в этом случае характер зависимости параметра приведения от температуры будет неодинаков. В то же время установлено, что все полимеры при температурах, равноудалённых от температуры стеклования, при одинаковом молекулярно-массовом распределении имеют одинаковые времена релаксации. Самой лучшей температурой приведения могла бы быть температура стеклования каждого полимера, но это связано с рядом затруднений, поскольку измерения вязкости и других механических свойств очень сложно проводить вблизи температуры стеклования. Поэтому в качестве температуры приведения была предложена температура Т0 = Тс + 50, лежащая на 50 градусов выше Тс любого полимера.
Зависимость коэффициента аТ от температуры определяется уравнением Вильямса-Лэндела-Ферри (ВЛФ):
lg aT = lg η(T)/η(T0) = - C1(T-T0)/(C2+T-T0),
где С1 и С2 – эмпирические константы, η – коэффициент вязкости полимера.
При Т0 = Тс+50 коэффициенты в уравнении ВЛФ для многих полимеров принимают значения: С1=8,86; С2=101,6, что свидетельствует об универсальности этого уравнения.
Принцип температурно-временной суперпозиции имеет большое практическое значение. С его помощью удаётся построить обобщённые кривые, простирающиеся на многие десятичные порядки по времени, что позволяет прогнозировать вязкоупругие характеристики полимеров на длительное время их эксплуатации.