

Лекциця ВМС 12
.docЛекция 12.
Вязкотекучее состояние аморфных полимеров.
В вязкотекучем состоянии под действием внешних сил в полимерных телах развиваются необратимые деформации. Вместе с тем вязкому течению полимеров всегда сопутствуют и обратимые (высокоэластические) деформации, развитие которых обусловлено отклонением в процессе течения конформаций макромолекулярных цепей от равновесных. Например, изменение деформации образца полимера в вязкотекучем состоянии под действием постоянного напряжения имеет сначала нестационарный характер, а затем скорость деформации перестаёт зависеть от времени (рис. 1).
ε



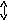
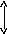




 lg η
lg η
 1
2
1
2
необратимая
деформация
 ηо
ηо
 высоко-
η∞
высоко-
η∞

 эластическая
эластическая
t lg σc
Установление стационарности указывает на завершение релаксационных процессов развития высокоэластической деформации. Дальнейшее возрастание деформации обусловлено только вязким течением.
Для полимеров в вязкотекучем состоянии наиболее важной характеристикой является их поведение при сдвиге. Связь между скоростью вязкого течения γ и напряжением простого сдвига σс определяется законом Ньютона σс = ηγ, где η – коэффициент пропорциональности, называемый вязкостью. Вязкость характеризует сопротивление полимера сдвигу или его внутреннее трение. При постоянной температуре вязкость (т.е. отношение напряжения к скорости сдвига) может не зависеть от режима деформирования. Среды, удовлетворяющие такому условию, называются ньютоновскими. К ним относится большинство низкомолекулярных жидкостей. Непрерывная перестройка структуры таких жидкостей под действием теплового движения происходит настолько быстро, что внешнее воздействие практически не оказывает заметного влияния на этот процесс. В то же время влияние теплового движения на изменение структуры жидкости может быть соизмеримым или даже более слабым, чем влияние сдвига. Тогда
напряжение изменяется медленнее, чем скорость сдвига, и, следовательно, с повышением напряжения и скорости сдвига их отношение (вязкость) уменьшается. Вязкость, изменяющаяся в зависимости от напряжения и скорости сдвига, называется эффективной, а жидкости, у которых напряжение сдвига изменяется непропорционально скорости сдвига, называются неньютоновскими. Явление зависимости вязкости от напряжения и скорости сдвига называется аномалией вязкости.
Измерение зависимости вязкости полимеров η от напряжения сдвига σс показывает, что для полимерных систем характерен эффект аномалии вязкости, заключающийся в уменьшении вязкости по мере увеличения напряжения сдвига (рис. 2). Аномалия вязкости полимерных систем связана с комплексом структурных изменений, происходящих при деформировании. К таким изменениям при сдвиговых деформациях прежде всего относится механическое разрушение пространственной сетки, образованной межмолекулярными связями.
При небольших значениях σс вязкость полимеров асимптотически приближается к предельному значению, которое называется наибольшей (начальной) ньютоновской вязкостью η0. Увеличение напряжения, приводящее к распаду флуктуационной сетки и облегчающее взаимное перемещение сегментов, вызывает снижение вязкости, но только до такого состояния, при котором предельно разрушенная надмолекулярная структура не будет успевать восстанавливаться во время течения. После этого вязкость, упав до η∞ - величины, называемой наименьшей ньютоновской вязкостью, снова перестанет меняться.
Вязкость полимеров сильно зависит от температуры. Для области высоких температур, далёких от температуры стеклования полимера, выполняется экспоненциальная зависимость вязкости от температуры, характеризуемая энергией активации вязкого течения: η = A exp (Ea/RT).
По мере возрастания молекулярной массы полимера энергия активации вязкого течения перестаёт зависеть от молекулярной массы. Это отражает то обстоятельство, что движение макромолекул при течении осуществляется в результате диффузии их отдельных участков. Энергия активации вязкого течения линейных полимеров зависит от химического строения звена и увеличивается с возрастанием жёсткости цепи.
Хотя вероятность осуществления отдельного элементарного акта течения для достаточно длинных цепей не зависит от длины макромолекулы, от неё существенно зависят абсолютные значения вязкости, так как для необратимого передвижения макромолекул необходимо, чтобы путём независимых перемещений отдельных сегментов произошло относительное смещение их центров тяжести. Чем больше длина цепи (т.е. молекулярная масса полимера), тем большее число согласованных перемещений участков цепей для этого требуется.
Теоретические расчёты и экспериментальные данные показывают, что зависимости вязкости от молекулярной массы полимеров складываются из двух участков. В области низких значений молекулярных масс η ~ М. Однако по достижении некоторой критической молекулярной массы она начинает оказывать несравненно более сильное влияние на вязкость. В этой области выполняется соотношение η ~ М3,5.
Стеклообразное состояние аморфных полимеров.
В стеклообразном состоянии полимер обладает физико-механическими свойствами твёрдого тела (не способен к текучести и высокоэластической деформации, имеет малые значения коэффициента термического расширения и сжимаемости) при сохранении структурной неупорядоченности, присущей жидкому или высокоэластическому состоянию. Стеклование обычно рассматривается как кинетическое явление, когда при понижении температуры или повышении частоты воздействия характерные времена перестройки макромолекулы, ответственной за проявление высокоэластичности, оказываются сравнимыми с временем опыта или периодом воздействия. Физические свойства полимеров в стеклообразном состоянии определяются соотношением энергий межмолекулярных взаимодействий и энергии теплового движения, а также плотностью упаковки полимера, т.е. величиной его свободного объёма. Энергия межмолекулярного взаимодействия либо не зависит от температуры, либо зависит очень незначительно. Энергия же теплового движения резко уменьшается с понижением температуры и при определённых её значениях становится недостаточной для преодоления внутри- и межмолекулярных взаимодействий. Это приводит к повышению вязкости полимера и к уменьшению интенсивности теплового движения звеньев, т.е. к повышению жёсткости его цепей.
Процесс стеклования связан с уменьшением свободного объёма полимера при сжатии его в результате охлаждения. Сегменты макромолекул могут участвовать в тепловом движении лишь при наличии достаточных по размеру пустот по соседству с ними. Если же свободный объём становится менее 2,5% от общего объёма полимера, то тепловое перемещение сегментов прекращается. Температура, при которой это происходит, и есть температура стеклования Тс. Но и после этого множество звеньев остаются подвижными, хотя и не могут перемещаться свободно, будучи прочно связанными с цепью, фиксированной во многих точках. Чем медленнее проводится охлаждение, тем ниже температура стеклования полимера и меньше его свободный объём.
Свойства полимерных стёкол тесно связаны с гибкостью макромолекул. Сравнительно жёсткие цепи, которые перемещаются, перегруппировываются и укладываются с трудом, дают стёкла с пониженной плотностью упаковки макромолекулы, с бóльшим «свободным пространством», чем у гибких цепей. «Рыхлость» упаковки растёт с увеличением длины макромолекулы, её степени полимеризации.
Вследствие сохранения некоторой подвижности у звеньев полимерной молекулы и их возможности перемещаться в «свободном пространстве» стёкла с такой упаковкой обладают некоторой способностью деформироваться, а также относительно малой хрупкостью. Очень гибкие цепи, наоборот, легко принимают конформацию, способствующую хорошей укладке их, и плотность упаковки у них почти такая же, как у низкомолекулярных стёкол. Поэтому полимерные стёкла, состоящие из подобных макромолекул, отличаются пониженной способностью к деформации, повышенными хрупкостью и упругостью, приближаясь по своим свойствам к низкомолекулярным стёклам.
Важная особенность полимерных стёкол, отличающая их от низкомолекулярных, заключается в способности в определённом интервале температур ниже Тс при приложении больших напряжений деформироваться на десятки и даже на сотни процентов.
Так как времена высокоэластической релаксации полимеров в стеклообразном состоянии очень велики, то при приложении сравнительно небольших напряжений стеклообразный полимер не может деформироваться по механизму высокоэластичности. Для объяснения способности полимерных стёкол к большим деформациям при приложении высоких нагрузок необходимо учесть, что время релаксации, вообще говоря, является функцией не только температуры, но и напряжения и при больших напряжениях может заметно уменьшаться.
Время релаксации, характеризующее скорость перегруппировок участков макромолекул, определяется соотношением
τ = τ0еU/kT
где U – активационный барьер (зависящий от энергии межмолекулярного взаимодействия); τ0 – период собственных колебаний кинетической единицы (в данном случае сегмента) относительно положения равновесия; Т – температура.
Влияние напряжения σ на время релаксации сводится к эффективному уменьшению энергии активации молекулярных перегруппировок и в первом приближении описывается уравнением
τ = τ0е(U-аσ)/kT
где а – константа, имеющая размерность объёма.
Большие деформации, развивающиеся в стеклообразных полимерах под влиянием больших напряжений, были названы вынужденно-эластическими, а само явление – вынужденной эластичностью. При вынужденно-эластической деформации не происходит смещения центров тяжести макромолекул друг относительно друга. Как и при высокоэластической деформации, изменение формы образца происходит за счёт изменения конформаций макромолекул. Однако в отличие от высокоэластической деформации этот процесс при данной температуре практически необратим.
σ


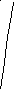 2
3
2
3
 1
1
I II III

ε
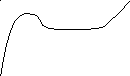
На рисунке 3 в координатах напряжение-деформация приведена типичная кривая растяжения стеклообразного полимера (кривая 1). Условно её можно разбить на несколько участков.
На начальном участке кривой растяжения (участок I) соблюдается закон Гука (напряжение пропорционально удлинению). Возникновение упругих сил при деформации обусловлено изменением внутренней энергии, как и при упругой деформации обычных твёрдых тел. Деформация на первом участке невелика (хотя и на порядок выше, чем у обычных твёрдых тел) и связана, главным образом, с изменением валентных углов и межмолекулярных расстояний.
Уменьшение наклона кривой σ = f(ε) по мере увеличения степени растяжения связано с началом развития в образце вынужденно-эластической деформации. С возрастанием напряжения скорость вынужденно-эластической деформации быстро увеличивается. В точке максимума на кривой σ = f(ε) скорость вынужденно-эластической деформации становится равной скорости растяжения, задаваемой прибором. Напряжение, при котором это наблюдается, называют пределом вынужденной эластичности (σв). По достижении σв происходит резкое сужение образца – образование так называемой «шейки». При переходе в шейку полимер ориентируется, и его свойства по сравнению со свойствами исходного материала существенно изменяются. Ориентированный материал обладает в стеклообразном состоянии более высокими значениями модуля упругости и предела вынужденной эластичности в направлении ориентации, чем изотропный материал. Когда при образовании шейки достигается степень вытяжки, обеспечивающая заметное возрастание σв, развитие вынужденно-эластической деформации в шейке резко замедляется. Процесс деформации продолжается у границ шейки, где сечение образца уменьшено, т.е. там, где напряжение повышено, а упрочнение ещё мало. На пологом участке кривой растяжения (участок II) напряжение при удлинении остаётся практически постоянным. Поперечное сечение шейки изменяется мало, и удлинение образца происходит, главным образом, за счёт вынужденной эластической деформации материала у границ шейки. Длина шейки при этом увеличивается. Растяжение с образованием шейки и дальнейшим её распространением является особенностью твёрдых полимеров.
После того, как границы распространяющейся шейки подойдут к концам рабочей части образца, полимер снова, как и на начальном участке кривой, деформируется однородно, как единое целое (участок III). Напряжение при этом снова начинает возрастать с увеличением удлинения.
Удлинения, возникающие на участке II кривой растяжения 1, после снятия нагрузки уменьшаются незначительно. Так как без приложения внешних напряжений тепловое движение в полимерном стекле не способно заметно изменять конформации макромолекул, фиксированные молекулярными взаимодействиями, то уже развившаяся вынужденно-эластическая деформация после снятия нагрузки оказывается фиксированной. Однако при нагревании полимера выше Тс, когда подвижность участков макромолекул возрастает, вынужденно-эластическая деформация полностью релаксирует.
Температурная зависимость предела вынужденной эластичности. Температура хрупкости. Зависимость σв от температуры при постоянной скорости деформирования представлена на рис. 4.
σ



4
 σп
σп


σв
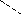
Тхр Тс Т
Прямая σв = f(T) пересекается с осью абсцисс в точке, соответствующей температуре стеклования полимера. При температуре стеклования значение σв близко к 0, и в образце уже при малых напряжениях развивается высокоэластическая деформация. По мере понижения температуры σв возрастает, поскольку для перегруппировки участков цепей требуются всё большие напряжения, и в конце концов становится выше прочности испытываемого полимера (σп). Иными словами, при достаточно низкой температуре разрыв макромолекул под действием приложенной силы, а следовательно, и нарушение целостности материала происходит раньше, чем успевают переместиться их отдельные участки. Эта температура называется температурой хрупкости полимера (Тхр). Дальнейшее понижение температуры несколько увеличивает напряжение, необходимое для разрыва (σп), но разрыву уже не предшествуют заметные вынужденно-эластические деформации материала. Кривая растяжения такого образца полимера соответствует кривой 2 на рисунке 3.
Температура хрупкости соответствует точке пересечения температурных зависимостей σв и σп. Ниже температуры хрупкости σп < σв, и образец хрупко разрушается прежде, чем достигается значение σв.