

Лекциця ВМС 14
.docЛекция 14.
Механическая прочность и долговечность полимеров.
Под прочностью понимают способность тел выдерживать без разрушения приложенную к ним нагрузку. Прочность обычно характеризуют величиной напряжения, вызывающего разрушение тела. Напряжение, вызывающее разрыв, называется разрывным напряжением или сопротивлением разрыву и выражается в МПа.
В течение длительного времени полагали, что потеря сплошности материала (разрыв или скол) наступает тогда, когда напряжение достигает некоторого предельного критического значения, и что при меньших напряжениях разрушения не происходит. Позднее было установлено, что такое представление о характере процесса разрушения неверно, так как все материалы при длительном воздействии внешних сил разрушаются при напряжениях, величина которых значительно меньше напряжений, возникающих при быстром воздействии силы. Это означает, что величина сопротивления разрыву зависит от времени действия силы. Время от момента приложения силы до момента разрыва называется долговечностью материала.
Временнáя зависимость прочности при статической нагрузке называется статической усталостью материала, временнáя зависимость прочности при динамической нагрузке – динамической усталостью материала.
Изучение одноосного растяжения полимеров позволило обнаружить связь между разрывным напряжением σ, долговечностью полимера под нагрузкой τд и абсолютной температурой Т. Эта зависимость выражается уравнением Журкова:
τд = τ0 exp [(U0 – γσ)/kT]
где k – константа Больцмана; U0 – энергия активации разрушения в отсутствие напряжения; τ0 – константа, равная 10-12-10-13 с, что отвечает периоду колебания атомов в твёрдых телах; γ – структурно-чувствительный коэффициент, отражающий влияние структуры материала на распределение в нём напряжений.
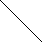
Уравнение Журкова предполагает линейную зависимость lg τд от σ или от обратной температуры 1/Т:
l
 g
τд
= lg
τ0
+ (U0
– γσ)/2,303kT.
g
τд
= lg
τ0
+ (U0
– γσ)/2,303kT.
 lg
τд
Т2
Т3
lg
τд
lg
τд
Т2
Т3
lg
τд

 Т1
σ1
σ2
Т1
σ1
σ2
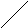
 σ3
σ3
0 - 0 -


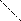




 - 13
- 13
- 13
- 13


σ 1/Т
На графиках видно, что прямые, соответствующие разным напряжениям или разным температурам, сходятся в одной точке, называемой полюсом. По наклону прямой в координатах lg τд – 1/Т можно экспериментально найти значение U0 – γσ = U, а по линейной зависимости U от σ определить U0 и γ.
Расчёты, проведённые для предельно ориентированных полимеров достаточно большой молекулярной массы, показали, что значения U0 близки к энергии химической связи между атомами главной цепи макромолекулы и совпадают со значениями энергии активации термической деструкции соответствующего высокомолекулярного соединения. Следовательно, в этих условиях, когда в максимальной степени проявляется совместное действие межмолекулярных сил и суммарный эффект их превосходит прочность валентных связей, разрушение образца происходит вследствие разрыва валентных связей макромолекулы, оно может быть рассмотрено как процесс термической деструкции, локализованной в небольшой части полимерного тела и ускоренной механическими напряжениями. Другими словами, механическое напряжение «помогает» тепловому движению, которое стремится разорвать связи между атомами. При случайном концентрировании избытка тепловой энергии на определённой связи происходит её разрыв. Вероятность разрыва определяется множителем е(Uo-γσ)/kT, величина которого зависит от приложенного напряжения. Растягивающее напряжение снижает величину энергии активации на γσ, тем самым увеличивается вероятность разрыва связей. Со временем число разрывов накапливается и образующийся дефект разрастается настолько, что происходит зарождение магистральной трещины, приводящей к разрушению всего образца.
В неориентированных полимерах процесс разрушения под действием флуктуаций тепловой энергии захватывает как химические, так, в значительной степени, и межмолекулярные связи. В результате также может образоваться дефект, дающий начало роста трещины.
Согласно статистической теории хрупкой прочности разрыв происходит не одновременно по всей поверхности разрушения, а последовательно, постепенно. Процесс начинается у самой опасной трещины или неоднородности (чаще всего на поверхности образца), где перенапряжение близко к теоретической прочности. С ростом этой первичной трещины увеличивается среднее напряжение на оставшемся уменьшенном сечении, вследствие чего перенапряжение на других, уже внутренних дефектах тоже достигает критического значения, а это приводит к появлению и развитию новых вторичных трещин. Образец разрывается в результате постепенного слияния всех трещин.
Всё изложенное выше относится к полимерам, которые мало деформируются перед разрушением. Это полимеры, надмолекулярная структура которых в момент разрушения сохраняется такой же, как в исходном образце, а не меняется кардинально в результате ориентации, как в эластомерах. Изменение надмолекулярной структуры в эластомерах, сильно деформирующихся в момент разрушения, приводит к тому, что зависимость долговечности от напряжения в них описывается уравнением, отличающимся от уравнения Журкова. Для эластичных пространственно сшитых полимеров
τд = Вσ-b,
где В и b – константы, зависящие от природы полимера. Константа В отражает также температурную зависимость долговечности:
τд = В0σ-beU/kT.
Установление достоверных зависимостей долговечности от напряжения или температуры для разных полимеров помогает создать основу для прогнозирования работоспособности изделий и конструкций из полимеров. Линейная зависимость lg τд от σ или от lg σ позволяет вести интерполяцию, т.е определять долговечность в пределах того интервала напряжений, который исследован экспериментально, или вести экстраполяцию, т.е определять долговечность при напряжениях и температурах, которые находятся вне исследованного интервала напряжений и температур.
Физико-химические свойства растворов полимеров.
Изучение поведения макромолекул в растворе имеет особое значение в связи с тем, что высокомолекулярные соединения не существуют в газообразном состоянии и всю основную информацию о свойствах индивидуальных макромолекул, их конформациях и размерах, молекулярных массах и распределениях по молекулярным массам можно получить только при изучении растворов. Кроме того, знание свойств полимерных растворов необходимо для использования полимерных материалов в растворённом состоянии и для их переработки из растворов.
Природа растворов полимеров.
Размеры макромолекул соизмеримы с размерами коллоидных частиц. Поэтому растворы полимеров обнаруживают ряд свойств, характерных для коллоидных золей (эффект Тиндаля, замедленная диффузия, тиксотропия и др.). Однако в отличие от коллоидных золей растворы полимеров являются молекулярно-дисперсными системами и удовлетворяют основным критериям истинных растворов: 1) самопроизвольность образования, термо-динамическая устойчивость, равновесность и обратимость; 2) постоянство концентрации во времени; 3) однофазность и гомогенность.
Критерием термодинамической устойчивости системы служит, в частности, подчинение её правилу фаз Гиббса. Правило фаз для конденсированных систем, в которых давление пара одного из компонентов равно нулю, имеет вид Ф + С = К + 1, где Ф – число фаз, К – число компонентов, С – число степеней свободы, т.е. число переменных, полностью определяющих состояние системы, которые можно произвольно изменять без нарушения числа фаз. Выражением подчинения системы правилу фаз является диаграмма состояния, или фазовая диаграмма, которая для двухкомпонентных систем имеет вид кривой растворимости в координатах температура – состав. В любой точке диаграммы свойства системы не зависят от пути достижения равновесия: разбавления, концентрирования, охлаждения или нагревания.
Для систем полимер – растворитель известны фазовые диаграммы различных видов. Поскольку размеры молекул полимера значительно превышают размеры молекул растворителя, состав на этих диаграммах выражают обычно в весовых или объёмных, но не в мольных долях.




 Тфр
Тфр Тфр
Тфр
Тфр
Тфр Тфр
Тфр


 ВКТР
ВКТР


 ВКТР
НКТР
ВКТР
НКТР



 А·
НКТР
А·
НКТР

 НКТР
ВКТР
НКТР
ВКТР





φ2´ φ2´´ φ2 φ2 φ2 φ2
Тфр – температура фазового расслоения, φ2 – объёмная доля полимера
Известны системы с верхней критической температурой растворения (ВКТР), выше которой ни при какой концентрации полимера в системе не наблюдается расслоения. При этом область выше кривой соответствует однофазной гомогенной системе, область ниже кривой – двухфазной гетерогенной системе. Например, в точке А система расслаивается на две равновесные фазы составов φ2´ и φ2´´. Примерами систем, обладающих ВКТР, являются системы: ацетат целлюлозы – хлороформ, полиизобутилен – бензол, полистирол – циклогексан и др.
Существуют системы, характеризующиеся нижней критической температурой растворения (НКТР). НКТР – это температура, ниже которой ни при какой концентрации полимера в системе не происходит расслаивание. Например, системы полиэтиленоксид – вода, метилцеллюлоза – вода, нитрат целлюлозы – этанол обладают НКТР. Для некоторых систем, например, полипропиленоксид – вода, реализуются замкнутые кривые растворимости с НКТР и ВКТР, причём НКТР < ВКТР и НКТР лежит в области ниже температуры кипения растворителя.
Известен ещё один вид фазовых диаграмм, для которых НКТР находится выше ВКТР и выше температуры перехода жидкость – пар для растворителя. Такие диаграммы характерны для систем, состоящих из компонентов, идентичных по химическому строению, но сильно различающихся по размерам. НКТР повышается с увеличением размеров молекул растворителя. Расслоение системы в данном случае обусловлено большой разницей в термических коэффициентах расширения компонентов. Диаграммы состояния такого типа получены, в частности, для систем полиэтилен – алканы, поливинилацетат – этилацетат, поливиниловый спирт – вода и др.
Применимость правила фаз к системам полимер – растворитель однозначно доказывает, что это термодинамически устойчивые, равновесные системы. Однако из-за большого размера молекул полимера равновесие в таких системах обычно устанавливается значительно медленнее, чем в растворах низкомолекулярных веществ (иногда в течение недель и месяцев). Скорость установления равновесия определяется скоростью взаимной диффузии и сильно зависит от природы полимера и растворителя, молекулярной массы и концентрации полимера, а также от его исходного физического состояния и исходной степени дисперсности полимерных частиц.
Особенности процесса растворения полимеров. Первой стадией растворения любого полимера является его набухание. Набухание – это процесс поглощения полимером низкомолекулярной жидкости, сопровождающийся увеличением объёма полимера и изменением конформаций его молекул. Большие молекулы полимера характеризуются низкими значениями коэффициентов диффузии. Поэтому смешение осуществляется медленно, и его промежуточные стадии легко фиксируются. При этом благодаря способности макромолекул изменять свою форму растворитель на промежуточных стадиях растворения не только заполняет пустоты между отдельными звеньями (процесс, аналогичный капиллярной конденсации в твёрдых пористых телах), но и увеличивает эффективные радиусы полимерных клубков и расстояния между их центрами масс, не нарушая при этом сплошности полимерного тела. Последнее приводит к значительному увеличению объёма полимерной фазы по сравнению с исходным. Набухший полимер фактически представляет собой раствор низкомолекулярной жидкости в полимере.
Если процесс растворения самопроизвольно прекращается на стадии набухания, то говорят об ограниченном набухании. Так, полимеры пространственно-сетчатого строения не могут полностью раствориться без разрыва химических связей. Они способны лишь ограниченно набухать, образуя гели. Состояние набухания характеризуют степенью набухания q, которую определяют как количество поглощённой полимером жидкости, отнесённое к единице массы или объёма полимера:
q = (m – m0)/m0 или (V – V0)/V0 или (V – V0)/m0
где m0 , V0 – масса и объём исходного полимера; m, V – масса и объём набухшего полимера.
Максимальная или равновесная степень набухания определяется природой полимера и растворителя (сродством между ними) и густотой пространственной сетки полимера (при её наличии). По значению равновесного набухания можно оценить густоту пространственной сетки. Процесс растворения может прекратиться на стадии ограниченного набухания и в случае линейных полимеров, если состав системы и условия соответствуют какой-либо точке на фазовой диаграмме, лежащей в области расслаивания.
Скорость набухания лимитируется скоростью взаимной диффузии компонентов системы и для полимеров, находящихся в исходном высокоэластическом состоянии обычно удовлетворительно описывается кинетическим уравнением первого порядка:
dq/dt = k(q∞ - q)
где k – постоянная набухания; q∞ - максимальная степень набухания; q – степень набухания в момент времени t.
После интегрирования этого уравнения получим:
ln (q∞ - q) = ln q∞ - kt (1)
По тангенсу угла наклона прямолинейной зависимости ln (q∞ - q) от времени можно определить постоянную набухания.
В случае линейных полимеров при изменении условий (температуры, концентрации и др.) ограниченное набухание может перейти в неограниченное, т.е. произойти растворение полимера. При этом макромолекулы диффундируют в растворитель вплоть до образования гомогенного раствора.
Поскольку растворы полимеров образуются самопроизвольно, их образование, как и всякий самопроизвольный процесс, сопровождается уменьшением энергии Гиббса, т.е.
ΔGсм = (Gр-ра – ΣGкомп) < 0
где ΔGсм – изменение энергии Гиббса при растворении; Gр-ра – энергия Гиббса раствора; ΣGкомп – сумма энергий Гиббса компонентов раствора до растворения.
Энергия Гиббса связана с изменением энтальпии и энтропии процесса уравнением
ΔGсм = ΔНсм - Т ΔSсм
где ΔНсм = Нр-ра – ΣНкомп и ΔSсм = Sр-ра – ΣSкомп – изменения соответственно энтальпии и энтропии системы при растворении, т.е. энтальпия и энтропия смешения.
Из условия ΔG < 0 следует, что самопроизвольное растворение полимера реализуется в нескольких вариантах.
-
ΔНсм < 0 и ΔSсм > 0.
Растворение экзотермическое и сопровождается возрастанием энтропии. При этом энергия взаимодействия между разнородными молекулами больше, чем между однородными (в расчёте на 1 моль растворителя и 1 моль звеньев полимера).
-
ΔНсм < 0 и ΔSсм < 0 при условии |ΔНсм| > |TΔSсм|.
Растворение также экзотермическое, но сопровождается уменьшением энтропии. Это может происходить, например, при растворении полярных полимеров в полярных растворителях. Энтропия уменьшается за счёт иммобилизации растворителя в образующихся сольватных оболочках вокруг звеньев макромолекул. При повышении температуры такие системы расслаиваются, т.е. они обладают НКТР.
3. ΔНсм > 0 и ΔSсм > 0 при условии |ΔНсм| < |TΔSсм|.
Растворение эндотермическое и сопровождается возрастанием энтропии. Это наблюдается в неполярных растворителях. Расслаивание в таких системах происходит при понижении температуры, т.е. они обладают ВКТР.
-
ΔНсм = 0 и ΔSсм > 0.
Растворение атермическое и сопровождается возрастанием энтропии. Это наблюдается, например, при растворении полимера в соответствующем ему гидрированном мономере (полиизобутилена в изооктане, поливинилацетата в этилацетате и др.). Как известно, величина ΔHсм = ΔUсм + + p ΔVсм характеризует изменение внутренней энергии (ΔUсм) и изменение объёма (ΔVсм) системы при растворении. Поэтому условие ΔНсм = 0 обычно означает, что и энергия взаимодействия, и средняя плотность упаковки молекул при растворении полимера в низкомолекулярной жидкости не изменяются по сравнению с исходными компонентами.