

Medgenetika_glava1_2
.pdfГБОУ ВПО «КАЗАНСКИЙ ГОСУДАРСТВЕННЫЙ МЕДИЦИНСКИЙ УНИВЕРСИТЕТ» МИНИСТЕРСТВА ЗДРАВООХРАНЕНИЯ РФ
КАФЕДРА МЕДИЦИНСКОЙ БИОЛОГИИ И ГЕНЕТИКИ
МЕДИЦИНСКАЯ ГЕНЕТИКА
Глава 1. Клинико-генеалогический метод. Косвенная ДНК-диагностика
Казань 2013
УДК 611:018(075.8) ББК 28.05+28.06
Печатается по решению Центрального координационно-методического совета
Казанского государственного медицинского университета.
Авторы:
Иллариошкин С.Н., Волков Е.М., Блатт Н.Л., Салафутдинов И.И., Исламов Р.Р.
Под общей редакцией: проф. Исламова Р.Р. Рецензенты:
Заведующий кафедрой гистологии, цитологии и эмбриологии КГМУ проф. Челышев Ю.А.
Заведующий кафедрой гистологии, цитологии и эмбриологии РНИМУ им. Н.И. Пирогова проф. Глинкина В.В.
Медицинская генетика. Глава 1. Клинико-генеалогический метод. Косвенная ДНК-диагностика.Учебно-методическое пособие/ Иллариошкин С.Н., Волков Е.М., Блатт Н.Л., Салафутдинов И.И., Исламов Р.Р. – Казань: КГМУ, 2013. – 46 с.
Учебно-методическое пособие составлено в соответствии с Государственным образовательным стандартом высшего профессионального образования (ФГОС-3) и типовой Учебной программой по дисциплине «Биология». В 1 главе «Клинико-генеалогиченский метод. Косвенная ДНК-диагностика» Учебно-методического пособия «Медицинская генетика» изложены цели и задачи данной темы, указаны формируемые компетенции, приведён теоретический обзор изучаемого материала, изложенный в лаконичной, рубрицированной форме и имеющий медицинскую направленность. Практические навыки включают усвоение правил составления родословной и применения полиморфных маркёров в косвенной ДНК-диагностике, изучение блоттинга по Саузерну, решение ситуационных задач по теме. Пособие содвключает типовые тестовые вопросы, вопросы самоконтроля и справочник терминов.
Учебно-методическое пособие предназначено для студентов младших курсов медицинских факультетов и медицинских вузов.
© Казанский государственный медицинский университет, 2013
Глава 1 КЛИНИКО-ГЕНЕАЛОГИЧЕСКИЙ МЕТОД.
КОСВЕННАЯ ДНК-ДИАГНОСТИКА
Цель занятия:
Сформировать представление о клинико-генеалогическом методе изучения генетики человека, косвенной ДНК диагностике наследственных заболеваний и блоттинге по Саузерну.
Задачи занятия:
1.Познакомиться с основными методами антропогенетики.
2.Узнать возможности и области применения клинико-генеалогичес- кого метода.
3.Научиться составлять и анализировать родословные.
4.Овладеть клинико-генеалогическим методом.
5.Изучить метод косвенной ДНК диагностики наследственных заболеваний.
6.Изучить метод блоттинга по Саузерну и область применения.
Формируемые компетенции:
ОК-1, ПК-2, ПК-3, ПК-9, ПК-31, ПК-32
Студент должен знать:
1.Возможности применения клинико-генеалогического метода в ме- дико-генетической консультации.
2.Типы наследования и их основные характеристики.
3.О полиморфизме длин рестрикционных фрагментов и полиморфных маркёрах наследственных заболеваний.
4.Принцип косвенной ДНК диагностики. Достоинства и недостатки.
5.Основные этапы протокола Саузерн блота.
Студент должен уметь:
1.Составлять родословную.
2.Анализировать родословную и определять тип наследования признака.
3.Работать с обучающей компьютерной программой по теме.
4
4.С помощью полиморфного маркёра анализировать носительство мутантного аллеля гена.
Студент должен владеть:
1.Основными понятиями и терминами по теме.
2.Практическими навыками клинико-гнеалогического метода.
3.Навыками косвенной ДНК-диагностики.
Оснащение занятия:
1.Таблицы
2.Мультимедийный проектор
3.Компьютеры
4.Билеты с примерами родословных
Хронологическая карта занятия:
1.Организационная часть.
2.Письменный тестовый контроль базового уровня знаний.
3.Разбор теоретического материала.
4.Самостоятельная работа студентов и текущий контроль за выполнением заданий.
5.Проверка выполненных работ в тетрадях и альбомах.
6.Установка задания для подготовки к следующей теме.
5
ТЕОРЕТИЧЕСКИЙ ОБЗОР
Человек — специфический объект генетического анализа
•Для человека неприменим основной метод генетических исследований, а именно метод экспериментальной гибридизации, поскольку данный метод вступает в непреодолимое противоречие с общечеловеческими нормами морали и Декларации ООН о «Правах человека».
•Редкая смена поколений (в среднем через 25 лет) не дает исследователю возможность обследования более 2-3 поколений одной семьи одновременно.
•Малое количество детей в современной семье не представляет возможности проследить расщепление признаков по законам Менделя.
•Большое количество групп сцепления (23 у женщин и 24 у мужчин) так же затрудняет анализ.
•В то же время есть и положительные стороны. Прежде всего, человек относится к одному из хорошо изученных биологических видов. Человечество обладает материальной культурой (письменные и иные источники информации, в частности истории болезни, акты рождения и смерти). И наконец, врач может установить личный (речевой) контакт с пациентом при его обследовании.
КЛИНИКО-ГЕНЕАЛОГИЧЕСКИЙ МЕТОД
Генеалогический метод — метод изучения родословных, с помощью которого прослеживается распределение какого-либо признака в семье или роду с указанием типа родственных связей между членами родословной. В медицинской генетике метод называется клинико-генеалоги- ческим. Это один из самых распространенных методов изучения генетики. С его помощью были установлены закономерности наследования многих признаков человека, а так же выявлены заболевания, имеющие наследственную природу. Достоинством данного метода является его простота и доступность, как для врача, так и для пациента. Клиникогенеалогический метод считается универсальным и широко применяется при решении медицинских (практических) и фундаментальных (теоретических) задач, например:
•установлении наследственного характера признака;
•определении типа наследования признака или заболевания;
•оценки пенетрантности гена;
•анализе сцепления генов и картировании хромосом;
•при изучении интенсивности мутационного процесса;
6
•Установлении механизмов взаимодействия генов. Клинико-генеалогический метод включает в себя два основных эта-
па — (1) составление родословной и (2) анализ родословной.
Как правило, родословная составляется для изучения одного или нескольких признаков (однако, при изучении более чем двух признаков одновременно, становится трудно установить характер наследования). Существует правило: чем больше поколений вовлечено в составление родословной и чем более обширной будет информация о членах родословной, тем более точным будет результат. Обычно данные собираются в форме опроса и/или с изучением медицинских карт (истории болезни) семьи. Делать окончательные выводы только на основе опроса и анализа родословной будет не верным Для подтверждения сделанных выводов, необходимо проводить дополнительные. клинические и лабораторные исследования. Для составления родословной используют специальные символы (рис. 1-1).
Наследование моногенных признаков у человека
В 1966 году в книге Виктора Мак Кьюсика «Менделевское наследование у человека» впервые была собрана информация о нормальных и патологических признаках человека, подчиняющихся правилам менделевского наследования. Данные этой базы постоянно пополняются и доступны в Интернете (http://www.ncbi.nlm.nih.gov/Omim/). В настоящее время существует описание более 20000 фенотипов, у которых 5 тысяч имеют установленный тип наследования.
Тип наследования (характер сегрегации патологических признаков в рядупоколений) является важнейшей и постояннойхарактеристикой любого моногенного заболевания. Каждому из типов наследования свойственны свои строго определенные менделевские законы. В настоящее время выделяются следующие типы наследования моногенных болезней человека:
•аутосомно-доминантный и аутосомно-рецессивный — характерны для заболеваний, гены которых локализованы на аутосомах;
•Х-сцепленный доминантный и рецессивный — характерны для заболеваний, гены которых расположены на Х-хромосоме;
•Y-сцепленный (голандрический) — характерен для редких фенотипических признаков, определяемых генами Y-хромосомы;
•митохондриальный (материнский) — характерен для заболеваний, связанных с мутациями митохондриальной ДНК.
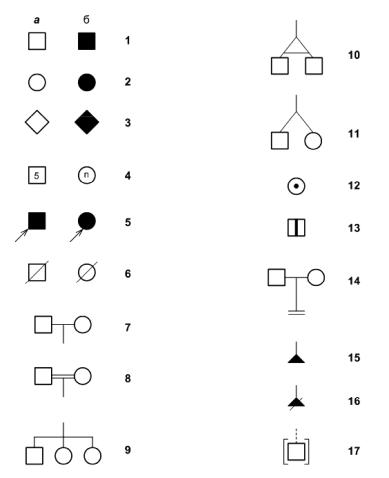
7
Рис. 1-1. Основные символы, рекомендуемые к использованию при составлении родословной. 1 – лица мужского пола; 2 – лица женского пола; 3 – пол неизвестен (а – здоровые, б – больные); 4 – несколько человек (а – число известно, б – число неизвестно); 5 – пробанд; 6 – умершие; 7 – брак; 8 – родственный брак; 9 – сибсы (дети одной родительской пары); 10 – монозиготные близнецы; 11 – дизиготные близнецы; 12 – облигатный гетерозиготный носитель мутантного гена (клинически здоров и не заболеет); 13 – носитель мутантного гена на пресимптоматической стадии (имеет высокий риск заболеть по достижении соответствующего возраста); 14 – бездетный брак; 15 – выкидыш; 16 – медицинский аборт; 17 – усыновление [из Иллариошкин С.Н., 2003].
8
Аутосомно-доминантный тип наследования
Аутосомно-доминантный тип наследования болезни встречается в случаях, когда патологический ген является доминирующим (по отношению к нормальномугенуиз той же пары) и определяет развитие симптоматики даже будучи в гетерозиготном состоянии — на одной из двух гомологичных неполовых хромосом (аутосом).
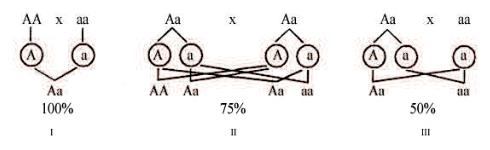
Обычно при аутосомно-доминантных заболеваниях в браке больного и здорового членов семьи распределение аллелей имеет вид Аа × аа (где А — доминантный мутантный ген, а — рецессивный нормальный ген) (рис. 1-2).
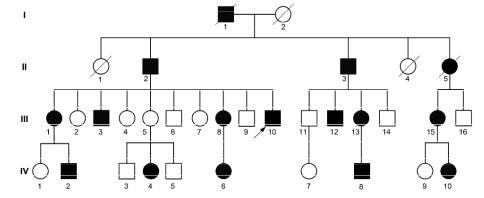
Родословная с аутосомно-доминантным типом наследования имеет характерные признаки (рис. 1-3):
1)прямая передача болезни от одного из родителей потомкам («вертикальная» передача болезни), в том числе передача заболевания детям от больного отца; нередко может прослеживаться манифестация заболевания в нескольких поколениях;
2)соотношение больных и здоровых лиц употомков больного индивидуума приближается к показателю 1:1 (50%);
3)для каждого из детей — потомков больного родителя риск унаследовать мутантный ген (т.е. риск возникновения заболевания) составляет 50%;
4)мужчины и женщины поражаются обычно в равной степени; в редких случаях может наблюдаться более высокая пенетрантность гена и более тяжелое течение заболевания у лиц одного пола — чаще у женщин.
Рис. 1-2. Распределение аллелей гена заболевания и вероятность проявления доминантного признака у потомства различных пар.
9
Рис. 1-3. Родословная семьи с аутосомно-доминантным заболеванием (например, болезнь Хантингтона, миотоническая дистрофия, синдром Марфана, большинство вариантов моторно-сенсорной невропатии Шарко-Мари-Тута) [из Иллариошкин С.Н., 2003].
Доминантные гены обладают различной пенетрантностью, т.е. вероятностью проявления мутантного гена у его носителей. Пенетрантность гена составляет 100%, если в семье заболевают все носители мутации. При неполной пенетрантности отдельные члены семьи — заведомые носители мутации — могут оставаться клинически здоровыми, но передать мутантный ген и заболевание своим детям; в таких случаях говорят о «пропуске поколения» в родословной (III-5 на рисунке 1-3). При данном типе наследования отсутствие симптоматики у родителей больного может объясняться: неполной пенетрантностью мутантного гена; ложным отцовством, или возникновением мутации de novo.
Доминантные гены обладают также различной экспрессивностью, т.е. выраженностью спектра клинических проявлений болезни у носителей мутации. Высокая экспрессивность мутантного гена приводит к появлению развернутой клинической картины болезни, низкая — к манифестации «стёртой» её формы («фрустрированная» форма, forme fruste). Правильная диагностика таких «стёртых» форм исключительно важна для медико-генетического консультирования, поскольку лица с тяжелой и «стёртой» клиникой равным образом являются носителями мутантного гена и могут передать заболевание потомкам.
Известны редкие примеры аутосомно-доминантных заболеваний, развивающихся в результате наследования рецессивных мутаций (доминирование на уровне организма, а не клетки). Это возможно в двух нижеследующих случаях.
10
1)Для проявления фенотипа необходимо, помимо одной унаследованной всеми клетками организма мутации, возникновение второй мутации на уровне соматической клетки. Например, при семейной ретинобластоме гетерозиготное носительство рецессивной мутации в гене RB1 не сопровождается какой-либо патологией. Однако возникновение на протяжении жизни еще одной мутации RB1 хотя бы в единственной клетке сетчатки (что само по себе является относительно нередким событием) приводит к полной инактивации гена и развитию ретинобластомы из мутантного клеточного клона.
2)В случае геномного импринтинга ген транскрибируется только с хромосомы, унаследованной от одного из родителей (отца либо матери). В такой ситуации «доминантная» передача болезни в родословной будет зависит от пола родителя, передающего болезнь.
Аутосомно-рецессивный тип наследования
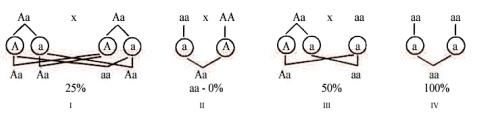
Аутосомно-рецессивный тип наследования наблюдается при заболеваниях, для манифестации которых необходимо присутствие мутантного гена в гомозиготном состоянии, т.е. на обеих гомологичных хромосомах. Гетерозиготные носители мутации остаются клинически здоровыми благодаря тому, что белковый продукт второй (нормальной) копии данного гена синтезируется в количестве, достаточном для поддержания соответствующей функции на физиологическом уровне. В типичных случаях аутосомно-рецессивного наследования генотип родителей больных имеет вид Аа × Аа (где а — рецессивный мутантный ген, А — доминантный нормальный ген) (рис. 1-4). Если больной вступает в брак с другим больным с тем же заболеванием (тип брака аа × аа), либо с гетерозиготным носителем мутации в том же гене (аа × аА), то такой характер передачи аутосомно-рецессивного заболевания называется
псевододоминантным.
Рис. 1-4. Распределение аллелей гена заболевания и вероятность проявления рецессивного признака у потомства различных пар.