

4.1.1. Осаждение
Соединение, в виде которого определяемый компонент осаждается из раствора, называется формой осаждения (или осаждаемой формой). Например, при определении бария в виде сульфата осаждаемая форма – BaSO4, а при определении железа – Fe(OH)3. К форме осаждения предъявляются следующие требования: 1) осадок должен быть мало растворим, т.е. осаждение должно быть достаточно полным; 2) полученный осадок должен быть чистым и легко фильтрующимся.
Полнота осаждения
При образовании в растворе осадка трудно растворимого соединения между насыщенным раствором и осадком устанавливается динамическое равновесие
BaSO4 ↔ Ba2+ + SO42–.
Запишем выражение для константы этого равновесия
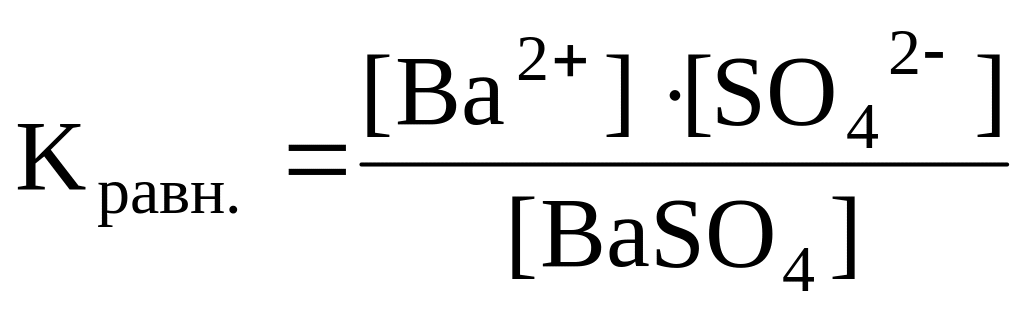 .
.
и, учитывая, что концентрация твердой фазы ([BaSO4]) постоянна, внесем ее в константу равновесия:
Кравн : [BaSO4)] = [Ba2+]·[SO42–] = const = ПР BaSO4.
Эту постоянную величину называют произведением растворимости и обозначают ПР.
В общем случае, если имеем трудно растворимую соль MxRу получим:
MxRу
↔ хMn+
+ уRm-
и
![]() .
.![]()
Произведение концентраций ионов в насыщенном растворе малорастворимой соли при постоянном давлении и температуре является величиной постоянной. При увеличении концентрации катионов осадка уменьшается концентрация анионов и наоборот. Действительно, при увеличении концентрации ионов Мn+ или Rm- увеличивается вероятность встречи между ними. При этом скорость осаждения увеличивается по сравнению со скоростью реакции растворения осадка, и поэтому концентрация других разновидностей ионов в растворе уменьшается.
Произведение растворимости позволяет предвидеть образование (или растворение) осадков при выполнении химических реакций.
Например, при
смешивании растворов соли кальция и
какого-нибудь сульфата (CaCl2
и Na2SO4)
возникают условия для соединения ионов
Са2+
с ионами SO42–
и образования осадка сульфата кальция.
Однако осадок может выпасть только в
том случае, если после смешения раствор
окажется пересыщенным по отношению к
сульфату кальция, т.е. если произведение
концентраций ионов Са2+
и SO42-
превысит
![]() :
ПКИ > ПР – это условие образования
осадка.
:
ПКИ > ПР – это условие образования
осадка.
Определим, выпадет ли осадок при смешивании равных объемов 0,01 М раствора CaCl2 и 0,1 М раствора Na2SO4. В первый момент после смешения концентрации ионов уменьшаются в два раза, так как объем в два раза увеличился, то есть [Ca2+] = 0,005 М, а [SO42–] = 0,05 M. Тогда произведение концентраций ионов
ПКИ = [Ca2+] · [SO42–] = 0,005·0,05 = 2,5·10–4.
Табличное значение = 6,1·10–5 (при 25 оС), поэтому получаем ПКИ > ПР, следовательно, осадок образуется.
Осаждение будет происходить до тех пор, пока произведение концентраций ионов, постепенно уменьшаясь, не станет равным произведению растворимости осадка. Затем снова устанавливается равновесие между осадком и раствором и осаждение прекращается.
Рассмотрим случай, когда в насыщенном растворе малорастворимой соли MxRу находятся также ионы постороннего сильного электролита. Тогда вокруг находящихся в растворе ионов осадка образуется ионная атмосфера – облака из ионов постороннего электролита противоположного знака, которая замедляет передвижение ионов, и, таким образом, уменьшает вероятность встречи их между собой, а также с поверхностью осадка. В результате скорость реакции осаждения уменьшается, тогда как ионы кристаллической решетки в это время продолжают взаимодействовать с растворителем и переходят в раствор с прежней скоростью. Поэтому при введении в насыщенный раствор малорастворимой соли постороннего электролита состояние равновесия нарушается, часть твердой фазы будет переходить в раствор и растворимость осадка увеличится. Процесс растворения твердой фазы проходит до тех пор, пока активность ионов в растворе, т.е. их способность к взаимным столкновениям не станет такой же, как и до введения в раствор постороннего электролита. После этого вновь установится динамическое равновесие между осадком и ионами.
Таким образом, из приведенного примера можно сделать вывод, что постоянной величиной является не произведение концентраций ионов, а произведение их активностей
![]() ,
,
которое в отличие от ПР не зависит от концентрации посторонних ионов в растворе.
Учитывая, что активность a = f · c, где с – концентрация иона, f – коэффициент активности, получим связь между ПР и ПА:
![]() ,
,
где fср– среднеионный коэффициент активности ионов.
Отсюда
![]() .
.
Поскольку в очень разбавленных растворах f ≈ 1, то в приближенных расчетах в большинстве случаев принимают ПА ≈ ПР.
В присутствии посторонних электролитов коэффициенты активности всегда меньше единицы, поэтому ПР, а также и растворимость малорастворимых соединений увеличивается с повышением концентрации сильных электролитов.
Под растворимостью (Р) обычно понимают концентрацию ионов металла в насыщенном растворе малорастворимой соли
Р = [Mn+].
Поскольку MxRy ↔х Mn+ + у Rm-, то
[Rm-]
=![]() [Mn+]
и
[Mn+]
и
![]() .
.
Отсюда
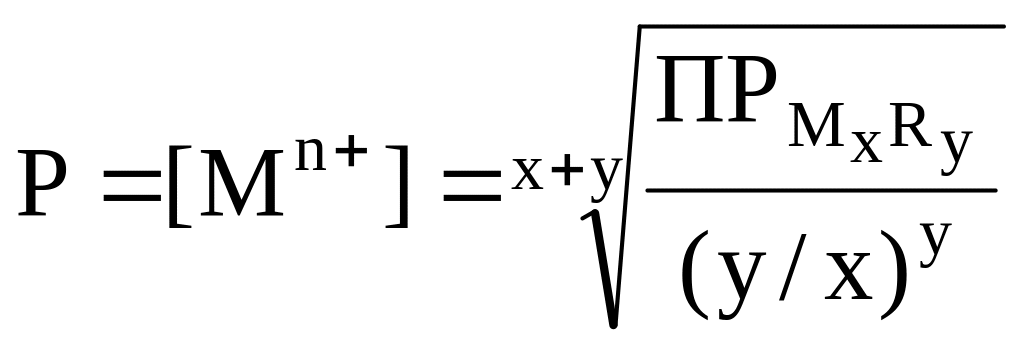 .
.
Например, растворимость хлорида серебра
![]() =
10–5 M.
=
10–5 M.
Введем в раствор такое количество ионов Cl-, чтобы их концентрация стала 0,1 М, тогда для вычисления растворимости используем правило постоянства произведения концентраций ионов ПРAgCl = [Ag+]·[Cl-] и получим
![]() М.
М.
Видим, что увеличение концентрации ионов хлора в 10 000 раз вызвало уменьшение концентрации ионов серебра, т.е. уменьшение растворимости в 10 000 раз.
Явлением уменьшения растворимости осадка при добавлении реактива, содержащего одноименные с ионами осадка ионы, широко пользуются в химическом анализе. Добавляя к исследуемому раствору избыток осадителя, достигают практически полного осаждения определяемых ионов. При этом растворимость осадка уменьшается, количество определяемых ионов становится незначительным и практически не влияет на результаты анализа.
Казалось бы, чем больше концентрация осадителя, тем полнее осаждение. Однако иногда большой избыток одноименных ионов приводит к нежелательным результатам из-за каких-либо посторонних процессов. Например, чтобы более полно отделить Ва2+ от Са2+ с помощью (NH4)2SO4, желательно прибавить большой избыток осадителя. В то же время при большом избытке осадителя кроме BaSO4 в осадок частично переходит и сульфат кальция.
Известны также случаи, когда с увеличением концентрации осадителя растворимость осадка увеличивается. Так, при осаждении PbSO4 раствором H2SO4 осадок заметно растворяется в избытке осадителя из-за образования растворимой кислой соли Pb(HSO4)2. Тиоцианат серебра заметно растворяется в избытке SCN–, что связано с образованием растворимого комплексного иона [Ag(SCN)2]–.
На практике обычно используют полуторный избыток осадителя. Его можно рассчитать по уравнению реакции осаждения. Пусть раствор содержит 0,235 г FeCl3, определим количество раствора аммиака, необходимое для осаждения гидроксида железа (III)
FeCl3 + 3NH3·H2O → Fe(OH)3↓ + 3 NH4Cl.
Молярная масса FeCl3 составляет 162,21 г/моль, а NH3·H2O – 35,04 г/моль. Составим пропорцию:
![]()
![]()
и, учитывая, что необходимо применить полуторное количество осадителя, получим х1 = 1,5·0,1521=0,2280 г.
Осаждение считают полным, если в растворе остается масса вещества, меньшая чувствительности аналитических весов, т.е. меньше чем 1,0·10-4 г. Это значение не превышает погрешность взвешивания и, таким образом, не может изменить показаний аналитических весов.
В большинстве случаев ПР растет с ростом температуры, поэтому для обеспечения полноты осаждения благоприятным условием является низкая температура.
Итак, для достижения полноты осаждения необходим избыток осадителя и нежелательно присутствие посторонних электролитов.
В практических условиях полнота осаждения контролируется добавлением небольшого количества осадителя в прозрачный раствор над отстоявшимся осадком до фильтрования.
Чистота осадка
Посторонние вещества могут попасть в осадок в результате совместного осаждения, соосаждения и последующего осаждения. При гравиметрических определениях совместное и последующее осаждение сводят к минимуму путем предварительного разделения и правильного выбора условий осаждения. Основной причиной загрязнения осадка является соосаждение.
Соосаждение – это переход в осадок присутствующих в растворе посторонних веществ, которые в данных условиях должны оставаться в растворе; другими словами, при соосаждении происходит захват примесей из ненасыщенного раствора. Например, осадок BaSO4, полученный при добавлении BaCl2 к раствору Na2SO4, содержит примеси ионов, входящих в состав и осадителя и осаждаемого вещества.
Соосаждение происходит в процессе образования осадка, а не после его образования. Так, осаждая BaSO4 из растворов, содержащих MnO4-, получают осадок розового или фиолетового цвета из-за соосаждения. Однако, если взять готовый препарат BaSO4 и взбалтывать его с раствором KMnO4, соосаждения не происходит, осадок остается неокрашенным.
По механизму захвата посторонних веществ различают два основных вида соосаждения: адсорбцию и окклюзию.
Адсорбция наиболее ярко выражена в случае аморфных (мелкодисперсных) осадков, которые отличаются сильно развитой общей поверхностью.
При окклюзии посторонние ионы находятся внутри кристаллов осадка и более или менее равномерно распределяются по всему объему твердой фазы. Окклюзия характерна в основном для кристаллических осадков. В реальных условиях анализа один из типов соосаждения не присутствует в чистом виде, а только преобладает.
Методы устранения (или хотя бы уменьшения) соосаждения зависят от его типа. Так, окклюдированные примеси нельзя отмыть, но их можно в значительной мере уменьшить в процессе осаждения. Для этого осаждение рекомендуется вести в условиях, при которых растворимость трудно растворимой соли достигает максимума. Осадитель необходимо прибавлять медленно, интенсивно перемешивая раствор. В этих условиях происходит медленный рост небольшого количества крупных кристаллов. Кристаллические осадки осаждают из горячих кислых растворов (условия их максимального растворения).
Еще один прием борьбы с окклюдированными примесями – переосаждение. Осадок растворяют в подходящем растворителе (растворе кислоты и т.п.) и снова осаждают. Так как основной причиной окклюзии является захват посторонних веществ в процессе роста, то ясно, что количество примесей в повторно осажденном осадке будет меньше.
Для уменьшения адсорбции необходимо увеличение среднего размера частиц, повышение температуры и промывание осадка промывными жидкостями. При этом можно использовать обменную адсорбцию, заменив адсорбированную нелетучую примесь на летучую. Например, при промывании осадка AgCl, загрязненного ионами Na+, азотной кислотой поверхность оказывается загрязненной летучей HCl. В случае аморфных осадков уменьшить содержание примесей можно так. Осаждение проводят из горячих концентрированных растворов, а затем быстро разбавляют горячей водой. При этом образовавшийся осадок имеет не очень большую поверхность, а разбавление горячей водой приводит к отмывке захваченных поверхностью осадка примесей.
Осадок очищается также за счет настаивания: кристаллический осадок вместе с маточным раствором оставляют на какое-то время, в течение которого происходят физико-химические процессы, называемые старением, созреванием осадка. В процессе старения происходит рекристаллизация первичных частиц, растворение мелких и рост крупных кристаллов, превращение осадка в устойчивую модификацию и т.д.