

Нефропатии
.pdfМИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ РЕСПУБЛИКИ БЕЛАРУСЬ БЕЛОРУССКАЯ МЕДИЦИНСКАЯ АКАДЕМИЯ ПОСЛЕДИПЛОМНОГО ОБРАЗОВАНИЯ
КАФЕДРА ПЕДИАТРИИ
Л.М.БЕЛЯЕВА, Е.А.КОЛУПАЕВА, Е.К.ХРУСТАЛЕВА, С.М.КОРОЛЬ
ДИЗМЕТАБОЛИЧЕСКИЕ НЕФРОПАТИИ
Учебно-методическое пособие
Минск БелМАПО
2010
ÓÄÊ 616.61-053.2(075.8) ÁÁÊ 56.9
Ì 54
Утверждено на заседании Учебно-методического совета Государственного учреждения образования «Белорусская медицинская академия
последипломного образования» в качестве учебно-методического пособия (протокол ¹ 7 от «20» октября 2005 г.)
Авторы:
заведующая кафедрой педиатрии БелМАПО, профессор, д.м.н. Л.М.Беляева, доцент кафедры педиатрии БелМАПО, к.м.н. Е.А.Колупаева, доцент кафедры педиатрии БелМАПО, к.м.н. Е.К.Хрусталева, доцент кафедры педиатрии БелМАПО, к.м.н. С.М.Король
Рецензенты:
заведующий кафедрой пропедевтики детских болезней БГМУ, доцент, к.м.н. В.И.Твардовский; доцент кафедры общей врачебной практики БелМАПО, к.м.н. Л.Г.Соловьева.
Беляева Л.М.
М 54 Л.М.Беляева, Е.А.Колупаева, Е.К.Хрусталева, С.М.Король. Дизметаболи- ческие нефропатии: учебно-методическое пособие. Изд. 2-е, перераб. и доп – Минск.: БелМАПО, 2010. - 24 с.
В данном пособии изложены современные представления об этиологии, патогенезе, клинических проявлениях дизметаболических нефропатий у детей и подростков. Рассматриваются вопросы диагностики метаболиче- ских нарушений. Приведены принципы диетических и лечебных мероприятий при дизметаболических нефропатиях.
Пособие предназначено для педиатров, детских нефрологов, студентов медицинских ВУЗов.
ÓÄÊ 616.61-053.2(075.8) ÁÁÊ 56.9
© Кафедра педиатрии, 2010 © БелМАПО, 2010
СОДЕРЖАНИЕ |
|
ВВЕДЕНИЕ ................................................................................. |
3 |
ПАТОГЕНЕЗ КРИСТАЛЛУРИИ.......................................................... |
6 |
НАРУШЕНИЯ ОБМЕНА ОКСАЛАТОВ................................................. |
8 |
НАРУШЕНИЯ ОБМЕНА МОЧЕВОЙ КИСЛОТЫ................................... |
14 |
НАРУШЕНИЯ ОБМЕНА ЦИСТИНА................................................... |
18 |
ПРИЛОЖЕНИЯ............................................................................ |
21 |
ЛИТЕРАТУРА.............................................................................. |
23 |
Введение
Âпоследние годы во всем мире отмечается увеличение частоты заболеваний органов мочевой системы у детей.
Âпрактической работе врача-педиатра нередко встречаются заболевания органов мочевой системы, связанные с обменной патологией, требующие специальных методов диагностики, лечения и профилактики. В структуре нефропатий у детей преобладают заболевания врожденного и наследственного генеза, а также болезни, связанные с наследственной предрасположенностью, имеющие скрытое начало и торпидное течение, среди которых наиболее значительную группу составляют больные с обменными нефропатиями различного генеза.
Под обменными нефропатиями понимается гетерогенная группа заболеваний врожденного, наследственного, мультифакториального и приобретенного генеза, характеризующаяся поражением почек различными продуктами обмена как за счет их накопления в почечной ткани, так и обусловленная непосредственным их токсическим воздействием на различные структурнофункциональные элементы нефрона. В подавляющем большинстве случаев обменных нефропатий выявляется дизметаболическая нефропатия, в основе которой лежит поражение цитомембран и интерстициальной ткани почек. Выраженные метаболические нарушения нередко приводят к развитию мо- чекаменной болезни уже в детском возрасте.
Дизметаболические нефропатии (ДН) – группа заболеваний с различ- ной этиологией и патогенезом, которые характеризуются интерстициальным процессом с поражением канальцев почек вследствие нарушения обмена веществ.
Âшироком смысле слова к ДН относятся любые заболевания, связанные
ñразличными нарушениями обмена, которые приводят к изменениям функционального состояния почек или структурным сдвигам на уровне различных элементов нефрона. Поэтому ДН могут носить транзиторный характер, например при отравлениях, нарушениях гемодинамики, заболеваниях желудоч- но-кишечного тракта и др.
Дизметаболическая нефропатия, как правило, генетически детерминирована. При данной патологии четко прослеживаются признаки, характерные для полигенно наследуемых заболеваний: относительно высокая частота болезни в общей популяции (от 1 до 32–40 на 1000 населения) и семейная пред-
4
расположенность. Установлено, что в последующих поколениях отмечается более раннее начало заболевания и утяжеление его клинических симптомов при однотипности проявлений болезни у ребенка и ближайших родственников (Ю.Е.Вельтищев, Э.А.Юрьева, 1989; В.В.Длин и соавт., 2005). Кроме ДН с наследственной предрасположенностью, метаболические нарушения (вторичные) часто возникают на фоне других почечных заболеваний.
Âзарубежной литературе термина ДН не существует, рассматриваемая патология диагностируется как оксалурия или гипероксалурия.
У детей при ДН в случае отсутствия коррекции метаболических нарушений возможно прогрессирование мембранно-деструктивных изменений и формирование тубуло-интерстициального нефрита (ТИН) и/или мочекаменной болезни (МКБ), а также развитие микробно-воспалительного процесса в по- чечной ткани (пиелонефрита) со снижением почечной функции и в дальнейшем формированием хронической почечной недостаточности (ХПН). Все это делает необходимым ранее выявление ДН и проведение своевременных и корректных лечебных мероприятий.
Âзависимости от причины выделяют первичные è вторичные ÄÍ. Первичные ДН являются наследственно обусловленными заболеваниями, характеризуются прогрессирующим течением, ранним развитием уролитиаза
èхронической почечной недостаточностью. К ним относятся первичная наследственная гипероксалурия (оксалоз), синдром Леша-Нихана, цистиноз и цистинурия.
Вторичные ÄÍ представляют собой вторичные тубулярные синдромы, иначе называемые дизметаболическими расстройствами с кристаллуриями, которые могут быть полигенно-наследуемыми или мультифакториальными. В зависимости от типа нарушений обмена при дизметаболических диатезах выделяют оксалатный (щавелевокислый), уратный (мочекислый), цистиновый диатезы.
Вторичные ДН могут быть связаны с повышенным поступлением определенных веществ в организм, нарушением их метаболизма на фоне различ- ных заболеваний (гломерулонефрита, системной красной волчанки, пиелонефрита, заболеваний желудочно-кишечного тракта и др.), лекарственной терапии, нестабильностью цитомембран канальцев и др. Течение их более благоприятное, чем первичных ДН.
Любые ДН, независимо от причины, характеризуются мочевым синдромом в виде кристаллурии. Персистирующая кристаллурия может приводить к отложению кристаллов в ткани почки, к их адгезии, «слипанию» друг с другом, что служит основой формирования камня и развитию мочекаменной болезни.
Мочекаменная болезнь (МКБ) тесно связана с дизметаболической нефропатией и является патогенетическим ее продолжением. Т.е., МКБ является вариантом реализации дизметаболической нефропатии.
Наиболее часто в основе кристаллурии лежат нарушения обмена щавелевой кислоты (оксалатов), мочевой кислоты (уратов), кальция, фосфатов, цистина, триптофана и др. Подавляющее большинство кристаллурий и выявляемых камней связано с кальцием (от 70 до 90%), около 85-90% из них
– с оксалатами (в виде оксалата кальция), остальные с фосфатами (фосфаты кальция – 3-10%) или являются смешанными – оксалатно(фосфатно)- уратными. На долю уратной кристаллурии и литиаза приходится около 5%,
5
цистиновой – до 3%. В 5-15% выявляются трипельфосфаты – фосфатные кристаллы, содержащие ион аммония, магний и кальций.
Повышенное выделение почками продуктов нарушенного обмена – оксалатов, уратов и других в растворенном виде или, тем более, в виде кристаллов, конгломератов кристаллов, повреждает, прежде всего, эпителий почечных канальцев. Нередко возникают повреждения внутриклеточных структур эпителиальных клеток, что ведет к нарушению функции канальцев – реабсорбции, секреции. При выраженной кристаллурии наблюдается механическое повреждение стенок канальцев с попаданием кристаллов в интерстициальную ткань, окружающую канальцы. В ответ на повреждение интерстиция инородными телами (кристаллами) реагируют макрофаги, а также другие клетки интерстиция, выделяя ряд интерлейкинов с провоспалительным эффектом, что может привести к развитию ТИН. Выраженная кристаллурия часто приводит к образованию конкрементов (оксалатных, уратных, цистиновых, смешанных), что нередко осложняется присоединением микробно-воспалитель- ного процесса – пиелонефрита.
Специфических клинических симптомов для ДН нет. Необходимо обращать внимание на наличие рецидивирующих болей и животе, дизурических расстройств, аллергического дерматита. Нередко отмечается снижение суточ- ного объема мочи и насыщенный ее характер. Лабораторными признаками обменных нарушений являются упорная микролейкоцитурия, гематурия (от микродо макро), возможна микропротеинурия, постоянная кристаллурия, увеличение экскреции фосфолипидов и N-ацилэтаноламина с мочой, повышение активности фосфолипазы С, наличие в моче перекисей. Возможно появление канальцевых нарушений в виде снижения аммонио- и ацидогенеза, никтурии, изменения концентрационной функции почек. Выявляемая абактериальная микролейкоцитурия, по мнению большинства исследователей, носит защитный характер и направлена на элиминацию солей и восстановление повреждений в мочевых путях.
ПАТОГЕНЕЗ КРИСТАЛЛУРИИ
Патогенез кристаллурии сложен и зависит от ее варианта. В основе любого вида кристаллурии лежит процесс преципитации солей из раствора, образования кристаллов и подчиняется единым физико-химическим закономерностям.
Предполагается, что образование кристаллов происходит в собирательной системе нефрона. Процесс кристаллообразования определяется тремя факторами:
·перенасыщением канальцевой жидкости сверх ее пределов стабильности; ·снижением активности ингибиторов перенасыщения; ·активаторами преципитации.
Перенасыщение мочи. Для образования кристалла необходимо наличие ионной пары – аниона и катиона (например, иона кальция и иона оксалата). Для каждой ионной пары существует свой коэффициент растворимости. Если произведение химической активности компонентов данной ионной пары ниже коэффициента растворимости, происходит их полное растворение (состояние мочи называется недонасыщенным). Для перенасыщенного состояния мочи существует коэффициент формации: если произведение хи-
6
мической активности компонентов ионной пары выше этого коэффициента, раствор становится нестабильным и отмечается спонтанная преципитация осадка, т.е. выпадение кристаллов. Между значениями коэффициентов растворимости и формации выявляется нестойкая стабильность раствора (метастабильность), при которой компоненты ионной пары находятся в растворенном состоянии, но даже при незначительном нарушении физико-химических свойств мочи может произойти их преципитация. Кроме того, метастабильный раствор способен поддерживать рост уже образовавшихся кристаллов.
Таким образом, перенасыщение мочи различными видами ионов в конеч- ном итоге приводит к их преципитации в виде кристаллов, к последующему их росту. рН мочи является важным фактором растворимости ионов. Например, при кислых значениях рН мочевая кислота практически не диссоциирует и легко преципитирует, а фосфат кальция, наоборот, малорастворим при щелочных значениях рН мочи.
Нарушение уродинамики также способствует выпадению кристаллов. Свидетельством этого является повторное образование камней в условиях ча- стичной обструкции на фоне аномалий развития мочевой системы.
Ингибиторы перенасыщения мочи. Одного перенасыщения мочи может быть не достаточно для образования кристаллов. Нередко при перенасыщении мочи не выявляется кристаллурия, а у других пациентов при нормальной концентрации компонентов ионной пары обнаруживается кристаллурия. По-видимому, это обусловлено действием ингибиторов перенасыщения
– веществ, которые повышают способность мочи удерживать кристаллоиды в растворенном состоянии. К ингибиторам перенасыщения мочи относят цитрат, пирофосфат, магний, дифосфонаты, гликозаминогликаны, мочевину, мелкие пептиды и рН мочи. Ингибиторы перенасыщения, с одной стороны, способны задерживать спонтанную преципитацию (например, пирофосфат задерживает преципитацию фосфата кальция), с другой, присоединяясь к поверхности уже образовавшегося кристалла, снижать его агрегацию и рост, делая возможным его вымывание мочой. Важными ингибиторами образования фосфатно-кальциевых кристаллов и камней являются пирофосфаты, цитрат, магний и низкомолекулярные вещества, а оксалатно-кальциевых – пирофосфаты и высокомолекулярные вещества.
Активаторы кристаллизации и роста кристаллов. Некоторые вещества способны стимулировать преципитацию других типов ионов и образование кристаллов. Например, добавление урата натрия в метастабильный раствор оксалата кальция вызывает выпадение преципитата и рост его кристаллов. Важными активаторами кристаллообразования являются инфекция мочевой системы и рН мочи. Среди возбудителей инфекции особое значение имеют микроорганизмы, продуцирующие уреазу и способные расщеплять мочевину, в результате чего увеличивается концентрация иона аммония, моча ощелачи- вается и образуются трипельфосфаты. Трипельфосфаты образуются только вследствие действия уреазоактивной флоры и являются свидетельством инфекции мочевой системы. Стойкое изменение рН мочи вследствие тех или иных заболеваний также может провоцировать кристаллообразование и рост камней. Например, при некоторых заболеваниях ЖКТ рН мочи постоянно сдвинута в кислую сторону, что приводит к образованию мочекислых камней èç-çà снижения растворимости мочевой кислоты в кислой среде.
7
НАРУШЕНИЯ ОБМЕНА ОКСАЛАТОВ
Âнастоящее время многие исследователи отмечают некоторое возрастание доли нефропатий, связанных с нарушением обмена щавелевой кислоты. Щавелевая кислота – это конечный продукт обмена ряда аминокислот (глицина, серина, гидроксиоксипролина), играющих важную роль в обмене соединительной ткани. Соли щавелевой кислоты содержатся в достаточном количестве в продуктах питания. Поступая с пищей, оксалаты пассивно абсорбируются в кишечнике. Ограничение их абсорбции обусловлено образованием нерастворимых комплексов с кальцием. Почками выводится около 90% от общего количества щавелевой кислоты. Только около 10% щавелевой кислоты экзогенного происхождения, в то время как 35–40% экскретируемых оксалатов образуется из аскорбиновой кислоты и 40% – из глиоксиловой кислоты. Оксалаты выводятся путем клубочковой фильтрации и канальцевой секреции через 24–36 часа после поступления с пищей. В норме уровень экскретируемого с мочой оксалата колеблется от 10 до 40 мг в сутки и зависит от возраста, времени суток и времени года. Максимальная концентрация определяется ночью и ранним утром, когда минутный диурез минимальный.
Âтечение года максимум экскреции приходится на осень.
Оксалатно-кальциевая кристаллурия
Âоснове заболевания может быть как нарушение обмена кальция, так и нарушение обмена оксалатов. У большинства пациентов с оксалатно-кальци- евой кристаллурией нет выраженного нарушения метаболизма оксалатов или повышения их экскреции с мочой, но выявляется гиперкальциурия. Однако кристаллы оксалата кальция могут образовываться и при нормальном уровне кальция в моче вследствие повышения содержания оксалатов. Так как все оксалатные кристаллы содержат кальций, термины «оксалатная нефропатия» и «оксалатно-кальциевая нефропатия» употребляются как синонимы.
Гипероксалурические состояния возникают при увеличении всасывания оксалатов в кишечнике либо при повышении их эндогенного образования (Табл. 1).
Табл. 1. Известные причины гипероксалурии (по Д.Фрейтаг, К.Хруска, 1987)
Увеличенная абсорбция оксалатов |
Повышенное эндогенное |
|
образование оксалатов |
Повышенное поступление с пищей |
Аскорбиновая кислота (витамин С) |
Воспалительные заболевания кишечника |
Пиридоксин (дефицит витамина В6) |
(болезнь Крона, язвенный колит) |
Этиленгликоль |
Кишечные анастомозы |
Первичная гипероксалурия |
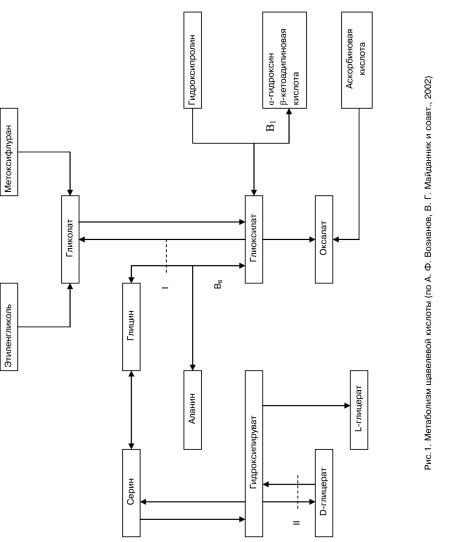
Основное количество оксалатов образуется из глиоксилата (Рис. 1). Наибольшее количество глиоксилата образуется из глицина под воздействием глициноксидазы. Обратное превращение глиоксилата в глицин и аланин катализируется аланин- и глицин-глиоксилаттрансаминазами, коферментом которых является пиридоксин. При его недостатке процесс трансаминирования замедлен, накапливаются большие количества глиоксилата с дальнейшим его переходом в оксалат, развивается гипероксалурия.
Предшественником образования оксалатов является и аскорбиновая кис-
8
лота, но количество оксалатов, образующихся из аскорбиновой кислоты, незначительно и имеет значение только при существующем нарушении обмена оксалатов. В генезе гипероксалурий играет важную роль нарушение почечных цитомембран. Причины мембранопатий разнообразны. Лабильность фосфолипидного слоя цитомембран может приводить к кальцифилаксии – нарушению гомеостаза внутриклеточного кальция, приводящего к патологической кальцификации. Нестабильность цитомембран может быть результатом повышенной активности процессов перекисного окисления липидов как вследствие окислительного стресса, так и снижения факторов антиоксидантной защиты. Такие изменения вызывают ускоренный метаболизм мембранных фосфолипидов в результате активации фосфолипаз и высвобождения компонентов липидной оболочки – фосфатидилэтаноламина, фосфатидилсерина, которые через этаноламин и серин метаболизируются в оксалат.
Кроме того, образование кристаллов оксалата кальция возможно вследствие эпитаксии, т.е. из-за индукции другими солями, в частности, при экскреции урата натрия.
По происхождению выделяют первичную гипероксалурию (оксалоз) è вторичную (оксалатную нефропатию).
Первичная гипероксалурия (оксалоз)
Первичное нарушение обмена щавелевой кислоты носит наследственный характер и обусловлено ферментативными нарушениями. При отсутствии или глубокой недостаточности активности ферментов развивается тяжелая патология – оксалоз. Это редкое наследственное заболевание, связанное с отсутствием фермента обмена глиоксиловой кислоты. Тип наследования
– аутосомно-рециссивный, описаны случаи доминантного наследования. Выделяют два типа первичной гипероксалурии.
I тип оксалоза обусловлен низкой активностью фермента аланин-глиокси- латаминотрансферазы в клетках печени, который обеспечивает трансаминирование глиоксилата. В результате возникающего блока глиоксилат накапливается, и его метаболизм идет по пути превращения в гликолат и оксалат. Таким образом увеличивается экскреция с мочой гликолата, глиоксилата и оксалата в виде оксалата кальция.
II тип оксалоза развивается из-за недостаточности фермента D-глицерат- дегидрогеназы, катализирующего переход гидроксипирувата в D-глицерат. Избыточное накопление гидроксипирувата приводит к избытку глиоксилата, оксалата кальция и L-глицерата, которые выделяются с мочой. Экскреция гликолата снижается практически до нуля.
Такое системное нарушение обмена приводит к отложению оксалата кальция в различных органах и тканях: почках, головном мозге, хрящах, костях, связках, стенках сосудов, лимфоузлах, эндокринных железах, селезенке и др. В почках оксалаты кальция откладываются прежде всего в проксимальных канальцах, интиме сосудов, интерстиции. В результате развивается деградация клеток канальцев, сужается просвет сосудов. По мере прогрессирования заболевания развивается интерстициальный нефрит, гидронефроз, нефролитиаз, что в конечном итоге приводит к хронической почечной недостаточности (ХПН).
Клиника. Первые симптомы заболевания начинаются в раннем возрасте, у 2/3 больных в 3-5 лет. Выраженность клинических проявлений зависит от
9
10