

Лекция_10
.docЛекция 10.
-
Ориентация и реакционная способность в реакциях присоединения
-
Реакционная способность
Электрофильное присоединение
Как и в случае электрофильного ароматического замещения, электронодонорные заместители при двойной связи повышают ее реакционную способность в реакциях электрофильного присоединения, а электроноакцепторные – понижают ее.


Влияние алкильных групп на скорость электрофильного присоединения
Алкильные группы, как правило, способствуют увеличению скорости электрофильного присоединения.
Если в реакциях электрофильного присоединения образуется циклический интермедиат 2, то введение алкильных заместителей в молекулу этилена приводит к аддитивному ускорению скорости реакции до тех пор, пока все 4 атома водорода не будут замещены на алкильные группы. Наблюдаемая аддитивность является одним из доказательств образования циклического интермедиата типа 2:

В реакциях электрофильного присоединения галогеноводорода HX ускоряющий эффект не аддитивен. Замена двух водородов при одном из атомов углерода будет значительно ускорять скорость реакции, но дополнительные алкильные заместители у другого атома углерода практически не влияют на скорость реакции. Это есть следствие того, что: 1) если E=H, то циклический карбокатион не может образовываться (так как у водорода нет электронной пары); 2) промежуточно образующиеся открытые карбокатионы типа 1 имеют следующую относительную стабильность: первичные < вторичные < третичные:


Нуклеофильное присоединение
Нуклеофильное присоединение происходит тем быстрее, чем более двойная связь обеднена электронной плотностью. Эта реакция лучше всего идет с субстратами, содержащим 3 или 4 акцепторных группы. Влияние заместителей настолько велико, что можно утверждать:
простые олефины вообще не реагируют по нуклеофильному механизму, а полигалогено- или полицианоолефины обычно не вступают в реакции, идущие по механизму электрофильного присоединения:

Такие реагенты, как аммиак NH3 и амины NR3, реагируют только как нуклеофилы и присоединяются только к субстратам, чувствительным к нуклеофильной атаке.
Есть реагенты, которые атакуют только как электрофилы, поэтому такие субстраты, как тетрафторэтилен, не будут вступать в реакции с ними.
Известны случаи, когда один и тот же реагент с простыми олефинами реагирует только по электрофильному механизму, а с полигалогено- или полицианоолефинами – по нуклеофильному:


Субстраты Михаэля почти всегда реагируют по нуклеофильному механизму (1,4-присоединение):

Чем больше акцепторные свойства заместителя Z, тем легче нуклеофильное присоединение.
Активирующая способность заместителей Z в субстратах Михаэля снижается в ряду:
![]()
Почему соединения с тройной связью реагируют как электрофилы, а не как нуклеофилы?
Кажется очевидным, что акцепторные заместители способствуют нуклеофильному присоединению и замедляют электрофильное в результате понижения электронной плотности двойной связи. Однако между атомами, связанными тройной связью СС, «концентрация» электронов выше, чем между атомами С=С, и тем не менее тройные связи менее склонны к реакциям электрофильного присоединения и легче вступают в реакции нуклеофильного присоединения, чем двойные связи (-ВЗМО и *-НСМО ацетиленов лежат ниже соответствующих орбиталей алкенов). Это не универсально, но справедливо в большинстве случаев. Все реагенты, способные образовывать мостиковые интермедиаты типа 2, с двойными связями реагируют быстрее, чем с тройными. В то же время присоединение электрофильного протона Н (кислотно-катализируемая гидратация, присоединение галогеноводородов) идет примерно с одинаковыми скоростями в случае алкенов и соответствующих алкинов [Марч, т. 3, с. 150; см., однако, новый Сайкс, с. 84: винилкатионы менее стабильны, чем алкилкатионы (см. ниже), и если следовать этой логике, то присоединение Н к алкенам должно идти быстрее, чем к алкинам].

Другим фактором, определяющим различие в реакционной способности алкенов и алкинов в реакциях электрофильного присоединения, является относительная стабильность положительно заряженных интермедиатов.
Если электрофильное присоединение идет через открытый карбокатион (например, присоединение гидрогалогенидов), то винильный карбокатион 9 энергетически менее выгоден по сравнению с алкильным карбокатионом 1:

В образующемся при протонировании ацетилена винильном катионе 9 -связь не участвует в стабилизации карбокатиона, так как ее р-электроны ортогональны р-орбитали, несущей положительный заряд. С учетом того, что в винильном катионе 9 несущий положительный заряд углерод находится в состоянии sp-гибридизации и обладает более высокой электроотрицательностью по сравнению с sp2-гибридизованным положительно заряженным углеродом в алкильном катионе 1, винильный катион 9 должен быть менее стабилен, чем алкильный 1.
В тех случаях, когда электрофильное присоединение идет через промежуточный мостиковый интермедиат типа 2 (Br, I, SR и т. д.), интермедиаты 8 оказываются более напряженными (энергетически менее выгодными) по сравнению с соответствующими интермедиатами 2 (углы между связями при sp2-гибридизованном углероде 120):


Еще одно объяснение более медленного присоединения таких электрофилов к алкинам по сравнению с алкенами исходит из того, что система 8 антиароматична.

Различной реакционной способности алкинов и алкенов в реакциях электрофильного присоединения могут даны и другие, в принципе сходные объяснения. Одно из них состоит в том, что электроотрицательность sp-гибридизованных углеродов тройной связи больше, чем электроотрицательность sp2-гибридизованных углеродов двойной связи (вспомните: соединения с тройной связью обладают большей кислотностью, ацетиленид-анионы более стабильны, чем этиленид-анионы). Поэтому атакующему электрофилу труднее оторвать пару электронов от такой связи. Другое объяснение заключается в том, что электроны тройной связи более прочно удерживаются из-за меньшего расстояния между атомами углерода.
Как и следовало ожидать, тройные связи с активирующей группой Z вступают в реакции нуклеофильного присоединения особенно хорошо.
Свободнорадикальное присоединение
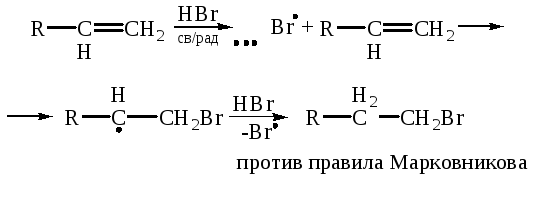
Свободнорадикальное присоединение может происходить с субстратами любых типов. Главное условие такого присоединения – присутствие в реакционной смеси свободнорадикальных частиц. Такие реагенты, как, например HBr, RSH атакуют по ионным механизмам в отсутствие инициатора, но в его присутствии механизм присоединения меняется на свободнорадикальный:


-
Ориентация
Электрофильное присоединение
Для электрофильного присоединения к простым и галогенированным олефинам и к ацетиленам обычно соблюдается правило Марковникова:
положительно заряженная часть реагента, т. е. электрофильная частица, присоединяется к наименее замещенному (наиболее гидрогенизированному) атому двойной или тройной связи
Правило Марковникова является феноменологическим правилом (основанным на множестве наблюдений). Обычно его объясняют рядом стабильности карбокатионов:
третичный КК вторичный КК первичный КК

Но откуда электрофил Е «знает», присоединение к какому атому даст наиболее стабильный катион?
Если возможно образование двух альтернативных интермедиатов (в данном случае – карбокатионов), то реакция протекает по двум параллельным путям. Кинетическая схема процесса выглядит следующим образом:

Если скоростьопределяющей является 1-ая стадия процесса, то для такой кинетической схемы (1-ая стадия – сложная реакция, состоящая их двух параллельно протекающих бимолекулярных элементарных стадий) отношение выходов продуктов 1 и 2 (или отношении концентраций этих продуктов в реакционной смеси) равно отношению констант скоростей параллельных элементарных стадий. Таким образом, приведенное выше объяснение по умолчанию предполагает, что более стабильный карбокатион должен образовываться быстрее менее стабильного, т. е. что между термодинамикой и кинетикой (между равновесием и динамикой) процесса имеется определенное соответствие, а именно: чем меньше энергия карбокатиона, тем меньше энергия активации его образования (т. е. меньше энергия ведущего к нему переходного состояния).
Действительно, для многих подобных ситуаций такая зависимость действительно существует и, более того, для однотипных элементарных процессов (для реакционных серий) является линейной.
Принцип Белла – Эванса – Поляни (БЭП)
Он гласит:
Для однотипных элементарных процессов имеется тесное соответствие между кинетическими и термодинамическими параметрами процесса, а именно существует линейная зависимость между тепловым эффектом H и энергией активации E процесса:
E=A+BH – линейная функция
Этот принцип, как и уравнение Гаммета, связывает термодинамику и кинетику. Также как и уравнение Гаммета, он является экстратермодинамическим соотношением, т. е. не вытекает из законов термодинамики. Подобно уравнению Гаммета, этот феноменологический принцип имеет разумное объяснение при условии определенных допущений, в данном случае – касающихся формы профиля реакции (формы поверхности потенциальной энергии реакции).
Чем более экзотермичен процесс (H<0), тем меньше энергия активации. Для эндотермичных процессов(H>0) – наоборот: чем более эндотермичен процесс, тем больше энергия активации.
На рисунке 1 представлен упрощенный «геометрический» вывод принципа БЭП. Уровень энергии 1 соответствует гетеролитическому разрыву двойной связи или отщеплению электрофила от карбокатиона. – теплота образования первичного карбокатиона, – теплота образования вторичного карбокатиона.
Из рисунка хорошо видно, что при условии примерно одинаковой крутизны (непересечения) обеих ветвей энергетической диаграммы реакция, которая ведет к термодинамически менее стабильному продукту, имеет более высокую энергию активации. Реакция, которая ведет к термодинамически более стабильному продукту, имеет меньшую энергию активации.

Рисунок 1. «Геометрический» вывод принципа Белла-Эванса-Поляни
Из рисунка можно сделать еще один вывод: по мере увеличения экзотермичности процесса (уменьшения эндотермичности) переходное состояние сдвигается по координате реакции в сторону исходного состояния (в сторону реагентов); наоборот по мере роста эндотермичности (уменьшения экзотермичности) процесса – в сторону конечного состояния (в сторону продуктов реакции). Это утверждение является одной из формулировок постулата Хэммонда.
Постулат Хэммонда
Оригинальная формулировка гласит:
«Если два состояния, например переходное состояние и нестабильный интермедиат, образуются последовательно в ходе реакции и имеют приблизительно одинаковую энергию, то их взаимопревращения будут включать только небольшие изменения молекулярной структуры». Это возможно лишь при соблюдении еще одного «геометрического» условия: приблизительно одинаковой крутизны восходящей и нисходящей ветвей энергетической диаграммы (энергетического профиля) реакции.
Из данной формулировки постулата Хэммонда следует, что структурные изменения в субстрате и/или реагенте будут влиять на энергию интермедиата и энергию переходного состояния (ПС) сходным образом, т. е. структурные факторы, стабилизирующие интермедиат, должны понижать и энергию ПС. Это очень важный постулат, на котором базируется множество выводов о влиянии структуры на реакционную способность.

Рисунок 2. Постулат Хэммонда для экзотермическойой реакции
-H – велика, переходное состояние близко к исходному состоянию (рис. 2). Чем больше экзотермичность процесса (при параллельном изображении потенциальных кривых), тем ближе переходное состояние к исходному состоянию и тем меньше энергия активации E
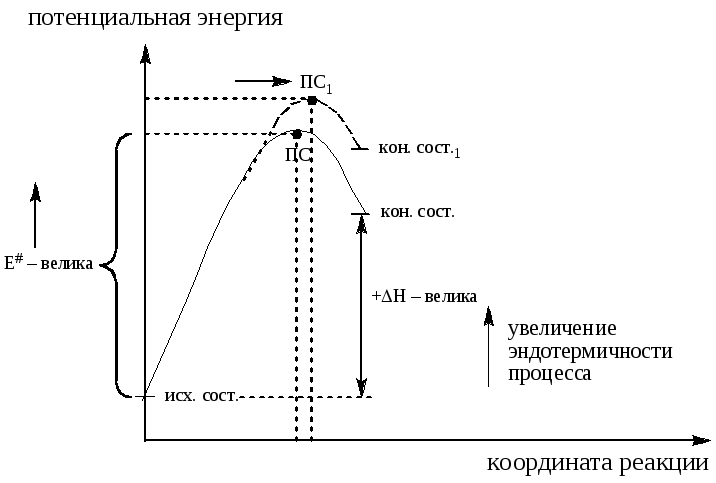
Рисунок 3. Постулат Хэммонда для эндотермическойой реакции
+H – велика, переходное состояние близко к конечному состоянию или продуктоподобно (рис. 3). Чем больше эндотермичность процесса (при параллельном изображении потенциальных кривых), тем ближе переходное состояние к конечному состоянию и тем больше энергия активации E

Рисунок 4. Постулат Хэммонда для реакций с небольшим тепловым эффектом
При небольшом тепловом эффекте и значительной энергии активации ПС сильно отличается по структуре как от исходного, так и от конечного состояния (рис. 4).
«Геометрические» условия соблюдения принципа БЭП и постулата Хэммонда отражают свойства поверхностей потенциальной энергии для однотипных элементарных процессов (сходных по строению частиц).
Таким образом, на основании принципа Белла-Эванса-Поляни или на основании постулата Хэммонда можно утверждать, что карбокатиону с более низкой энергией предшествует переходное состояние с более низкой энергией.
Правило Марковникова для несимметричных мостиковых катионов
Правило Марковникова соблюдается и для циклических интермедиатов типа несимметричного бромониевого иона 3:

Правило Марковникова для галогенозамещенных олефинов
Правило Марковникова соблюдается также для галогенозамещенных олефинов.

Нуклеофильное присоединение
Направление атаки нуклеофила изучено мало, кроме присоединения по Михаэлю, когда отрицательно заряженная часть реагента региоселективно атакует атом углерода, более удаленный от акцепторного заместителя Z:

Свободнорадикальное присоединение
Основное влияние на региоселективность присоединения к кратным связям по свободнорадикальному механизму оказывают стерические факторы. Это объясняется очень высокой реакционной способностью радикалов, которые «не различают» положения с различной электронной плотностью в субстрате, и является одной из иллюстраций общей закономерности падения региоселективности при увеличении реакционной способности.

Все субстраты указанного типа, независимо от природы атакующего радикала и заместителя Х, атакуются по стерически более доступному атому, т. е. тому, у которого нет заместителя.
Стабильность свободнорадикальных интермедиатов изменяется в таком же порядке, как и стабильность соответствующих карбокатионов:
третичные > вторичные > первичные

Однако этот фактор, действуя в том же направлении, оказывает, по-видимому, меньшее влияние, чем стерический.
Олефины с неконцевой двойной связью, не содержащие групп, способных стабилизировать радикал, дают обычно эквимолярную смесь изомерных продуктов.
Сопряженные диены
Атака E, Y и R всегда происходит по концу сопряженной системы, так как в каждом случае это приводит к интермедиату, стабилизированному резонансом. По этой же причине сопряжение увеличивает реакционную способность двойных связей в реакциях с электрофилами по сравнению с несопряженными (изолированными) двойными связями. В случае несимметричных диенов состав продуктов реакции определяется относительной стабильностью промежуточно образующихся ионов (Марч: «… наиболее устойчивым является ион, который действительно образуется»).