

4. Ориентирующее действие заместителей в бензольном ядре
Все заместители по характеру своего ориентирующего действия при реакциях электрофильного замещения в бензольном ядре делят на две группы.
1. Заместители первого рода, или орто-пара-ориентанты смещают электронную плотность в сторону кольца, т. е. обладают электронодонорными свойствами.
2. Заместители второго рода, или мета-ориентанты, смещают электронную плотность от бензольного кольца, т. е. обладают электроноакцепторными свойствами.
При наличии двух заместителей разного типа место вступления следующего электрофильного заместителя определяют заместитель первого рода, так как он активирует бензольное ядро к электрофильной атаке (Бейльштейн).
5. Применение бензола и его гомологов. Ароматические углеводороды являются важным сырьем для производства различных синтетических материалов, красителей, физиологически активных веществ.
Толуолв большом количестве используется как сырье для синтеза взрывчатых веществ; является полупродуктом для анилино-красочной и фармацевтической промышленности, а также используется в качестве растворителя. Тринитротолуол (тротил) был наиболее распространенным бризантным взрывчатым веществом во время Великой Отечественной войны.
Изопропилбензол (кумол)применяется для синтеза фенола и ацетилена, а гидропероксид кумола используется как инициатор полимеризации.
Винилбензол (стирол)применяется для получения полимерного материала - полистирола.
Производные ароматических углеводородов
А. Ароматические гидроксисоединения. К числу ароматических гидроксисоединений относят фенолы и ароматические спирты. В фенолах гидроксильная группа связана непосредственно с ядром, а в ароматических спиртах находится в боковой цепи. Наибольший интерес представляют фенолы. Фенолы были открыты в 1834 г. Рунге. Их общая формула Аr-ОН. В зависимости от числа гидроксильных групп различают одно-, двух- и многоатомные фенолы. Двухатомные фенолы известны в виде трех изомеров: орто-, мета- и пара-.
Способы получения. 1. Фенолы извлекают в виде фенолятов из среднего масла каменноугольной смолы. 2. Важным промышленным способом получения фенола является окисление кумола (изопропилбензола) кислородом воздуха.
Физические свойства. Фенолы – легкоплавкие кристаллические вещества. Чистые фенолы – бесцветны, на воздухе розовеют. При обычной температуре плохо растворяются в воде, при нагревании – хорошо. Обладают резким запахом и бактерицидными свойствами.
Химические свойства. Химические свойства фенолов несколько отличны от спиртов.
1. Взаимодействие со щелочами. 2. Образование простых эфиров. 3. Образование сложных эфиров. 4. Реакции за счет бензольного ядра. 5. Реакции конденсации фенолов с альдегидами. 6. Реакция гидрирования фенолов. 7. Реакция окисления.
Применение. Фенолы используются как дезинфицирующие средства; они идут на изготовление красителей, лекарств (салициловая кислота), взрывчатых веществ (пикриновая кислота), пластических масс, синтетических волокон (капрон, нейлон), гербицидов и флотореагентов.
Б.
Ароматические альдегиды и
кетоны содержат в своем составе
карбонильную группу![]() ,
связанную с атомом углерода бензольного
ядра или боковой цепи. Ароматические
альдегиды – это производные ароматических
углеводородов, в боковой цепи которых
имеется альдегидная группа
,
связанную с атомом углерода бензольного
ядра или боковой цепи. Ароматические
альдегиды – это производные ароматических
углеводородов, в боковой цепи которых
имеется альдегидная группа![]() .
.
Способы получения. 1. Окисление ароматических углеводородов кислородом воздуха на ванадиевом катализаторе. 2. Окисление ароматических спиртов. 3. Гидролиз ароматических дигалогенопроизводных. 4. Восстановление хлорангидридов ароматических кислот на палладиевых или никелевых катализаторах.
Физические свойства. Ароматические альдегиды – преимущественно жидкости с приятным запахом, трудно растворимые в воде.
Химические свойства. Ароматические альдегиды вступают во все химические реакции, характерные для альдегидов алифатического ряда. В отличие от альдегидов жирного ряда ароматические альдегиды вступают в окислительно-восстановительное взаимодействие с образованием соответствующего спирта и соли кислоты (реакция Канниццаро, 1853).
Применение.
Бензойный альдегид (бензальдегид) –
жидкость с т. пл. -260С, т. кип. 179,20С;![]() ÷1,046.
На воздухе быстро окисляется в бензойную
кислоту. Содержится во многих эфирных
маслах, в косточках вишни,
персика, абрикоса. Имеет запах горького
миндаля. Применяется в производстве
душистых веществ и красителей.
÷1,046.
На воздухе быстро окисляется в бензойную
кислоту. Содержится во многих эфирных
маслах, в косточках вишни,
персика, абрикоса. Имеет запах горького
миндаля. Применяется в производстве
душистых веществ и красителей.
В.
Ароматические кетоны –
производные ароматических углеводородов,
содержащие карбонильную группу![]() ,
соединенную с двумя радикалами, из
которых один обязательно ароматический.
При этом различают две группы кетонов:
а) жирно-ароматические (смешанные), когда
один из радикалов алкил, а второй арил;
б) чисто ароматические кетоны,
в которых оба радикала ароматические.
,
соединенную с двумя радикалами, из
которых один обязательно ароматический.
При этом различают две группы кетонов:
а) жирно-ароматические (смешанные), когда
один из радикалов алкил, а второй арил;
б) чисто ароматические кетоны,
в которых оба радикала ароматические.
Способы получения. 1. Жирноароматические кетоны получают рекцией Фриделя -Крафтса – Густавсона действием хлорангидрида уксусной кислоты на бензол в присутствии хлорида алюминия. 2. Ароматические кетоны получают на бензола действием хлористого бензоила в присутствии хлорида алюминия. 3. Непредельные ароматические кетоны получают конденсацией ароматических альдегидов с жирными кетонами.
Физические свойства.Ароматические кетоны – жидкости или твердые вещества с приятным цветочным запахом. Нерастворимы в воде.
Химические свойства.Ароматические кетоны дают все реакции, характерные для кетонов жирного ряда: окисления, восстановления (не реагирует с NaHSO3), присоединения спиртов и синильной кислоты, галогенирования, замещения кислорода на галоген, конденсации, образования фенилгидразинов и оксимов и др.
Применение. Кетоны используют в парфюмерной и кондитерской промышленности как душистые вещества, а также для защиты тканей и пластмасс от разрушающего действия света.
Г. Ароматические кислоты. Ароматические карбоновые кислоты – это производные ароматических углеводородов, имеющие карбоксильную группу –С(О)-ОН, связанную с углеродным атомом бензольного ядра.
Строение.Изомерия.Номенклатура. Простейшая одноосновная ароматическая кислота С6Н5С(О)-ОН называетсябензойнойилибензолкарбоновой кислотой. Для карбоновых кислот наиболее употребительны тривиальные названия. Радикал бензойной кислоты С6Н5С(О)-О называетсябензоилом. Простейшие ароматические двухосновные кислоты, содержащие в соединении с бензольным ядром два карбоксила С6Н4(СООН)2, называютфталевыми кислотами. Они существуют в виде трех изомеров:орто-,мета- ипара-. Наибольшее практическое значение имеюто- ип-фталевая (терефталевая) кислоты.
Способы получения. 1. Окисление алкильных групп гомологов бензола перманганатом или дихроматом калия в присутствии катализатора оксида ванадия (V). 2. Окислением нафталина дымящей серной кислотой. 3. Нитрильный синтез. 4. Гидролиз тригалогенопроизводных с галогенами у одного атома углерода. 5. Реакция Гриньяра.
Физические свойства. Ароматические кислоты – твердые кристаллические вещества, трудно растворимые в воде. Они кипят и плавятся при более высоких температурах, чем кислоты жирного ряда с тем же числом атомов углерода.
Химические свойства. Некоторые примеры химических реакций: 1. Образование соли. 2. Образование галогенангидридов. 3. Получение ангидридов ароматических кислот. 4. Реакция этерификации. 5. Образование амида и нитрила кислоты. 6. При действии натрия на хлористый бензоил. 7. При взаимодействии терефталевой кислоты с этиленгликолем.
Важнейшие представители.Бензойная кислотавстречается в свободном состоянии в некоторых смолах. Ее получают окислением толуола азотной кислотой. Применяется в качестве антисептического и консервирующего средства, в производстве красителей, лекарственных и душистых веществ.
Фталевая кислотаиспользуется в производстве красителей и пластификаторов (дибутилфталат).Терефталевая кислотав большом количестве идет для изготовления полиэтилентерефталата – полимерного материала, из которого получают волокно лавсан (заменитель шерсти).
Д. Ароматическими аминами называют производные ароматических углеводородов, в бензольном ядре которых вместо атома водорода содержится группа –NH2. Амины с аминогруппой в боковой цепи обладают свойствами алифатических аминов.
Номенклатура и изомерия. Для ароматических аминов наиболее употребительны тривиальные названия. Простейший ароматический амин, являющийся производным бензола, называют фениламином, аминобензолом или анилином. Последнее название происходит от – испанского названия индиго, красителя растительного происхождения, разложением которого анилин был получен в 1826 г. Ближайшими гомологами анилина являются аминопроизводные толуола, называемыетолуидинами. Они существуют в виде трех изомеров.
Классификация. Ароматические амины, как и амины жирного ряда, могут быть первичными, когда азот аминогруппы соединен только с одним ароматическим радикалом (анилин, толуидины), так и вторичными и третичными, если с азотом соединены соответственно два или три радикала. При этом такие амины могут быть двух типов: жирноароматическиеи чисто ароматические.
Способы получения. 1. Способ Н.Н. Зинина (1842) – восстановление ароматических нитросоединений. 2. Реакция взаимодействия галогенопроизводных ароматического ряда с аммиаком. 3. Чисто ароматические амины получают нагреванием аминов с их солями. 4. Третичные ароматические амины получают алкилированием или арилированием первичных или вторичных аминов.
Физические свойства. Низшие амины и в том числе анилин – жидкости со своеобразным запахом. Высшие амины – твердые вещества. Амины сильно токсичны. В воде растворяются плохо. Увеличение числа аминогрупп приводит к возрастанию растворимости.
Химические свойства. Важнейшие реакции аминов перечислены ниже.
1. Реакция присоединения алкилгалогенидов. 2. При нагревании ароматических первичных аминов с ароматическими альдегидами. 3. Реакции ароматических аминов с азотистой кислотой. 4. Ацилирование аминов. 5. Сульфирование и нитрование.
Важнейшие
представители.Анилин(аминобензол,
фениламин) С6Н5NН2– важнейший из ароматических аминов.
Это бесцветная, трудно растворимая в
воде маслянистая жидкость, темнеющая
на свету и воздухе, имеющая т. пл. -5,890С
и т. кип. 184,40С,![]() .
Анилин растворим в спирте, эфире, ацетоне,
бензоле, хлороформе и др. Получают анилин
каталитическим восстановлением
нитробензола. Он применяется в парфюмерии
для изготовления душистых веществ, для
производства лекарственных препаратов:
стрептоцида, норсульфазола, сульфадемизина,
фталазола и др., сернистых красителей,
антиоксидантов для каучука и резины,
стабилизаторов химической стойкости
полиамидных и полиуретановых смол,
порохов, изготовления взрывчатых веществ
и самовоспламеняющихся ракетных топлив.
.
Анилин растворим в спирте, эфире, ацетоне,
бензоле, хлороформе и др. Получают анилин
каталитическим восстановлением
нитробензола. Он применяется в парфюмерии
для изготовления душистых веществ, для
производства лекарственных препаратов:
стрептоцида, норсульфазола, сульфадемизина,
фталазола и др., сернистых красителей,
антиоксидантов для каучука и резины,
стабилизаторов химической стойкости
полиамидных и полиуретановых смол,
порохов, изготовления взрывчатых веществ
и самовоспламеняющихся ракетных топлив.
ДиметиланилинС6Н5N(CH3)2– желтая жидкость с дегтярным запахом;
т. пл. 2,50С, т. кип. 194,150С,![]() ;
плохо растворяется в спирте, ацетоне,
бензоле и хлороформе. Получают
диметиланилин обработкой анилина
метиловым спиртом под давлением.
Применяется в производстве красителей,
взрывчатых веществ (тетрила) и в качестве
проявителя для цветной фотографии.
;
плохо растворяется в спирте, ацетоне,
бензоле и хлороформе. Получают
диметиланилин обработкой анилина
метиловым спиртом под давлением.
Применяется в производстве красителей,
взрывчатых веществ (тетрила) и в качестве
проявителя для цветной фотографии.
Е. Диазо- и азосоединения. Среди производныхпервичных ароматических аминов наиболее важными являются диазосоединения и азосоединения. Те и другие содержат в молекуле группу из двух атомов азота –N=N-, называемуюазогруппой. Азогруппа является хромофором.
В диазосоединениях азогруппа соединена, с одной стороны, с ароматическим радикалом, а с другой – с какой-нибудь группой, например с гидроксилом, галогеном и др. В этом случае соединение носит приставку диазо-: C6H5-N=N-OH- фенилдиазогидрат. В диазосоединениях азогруппа группа связана с двумя ароматическими углеводородными радикалами.
Способы получения. Диазосоединения получают реакцией диазотирования, т.е. действием на первичные ароматические амины азотистой кислоты
Химические свойства. Реакции могут идти с выделением или без выделения азота.
IРеакции, идущие с выделением азота
1. Реакция разложения солей диазония с образованием фенолов. 2. Реакция А.Н. Несмеянова. 3. Реакция Гаттермана – Зандмейера. 4. Реакция Меервейна.
IIРеакции, идущие без выделения азота
1. Реакция образования фенилгидразина (восстановление солей диазония). 2. Реакция азосочетания.
Ж. Многоядерными ароматическими углеводородами называют соединения, в молекулах которых содержится два или более бензольных ядра. При этом различают две группы соединений:
а) соединения с изолированными бензольными ядрами, в которых бензольные ядра не имеют общих углеродных атомов;
б) соединения с конденсированными бензольными ядрами, когда два или несколько бензольных ядер имеют общие углеродные атомы.
1. Соединения с изолированными бензольными ядрами. Простейший многоядерный ароматический углеводород с двумя изолированными бензольными ядрами – дифенил (бифенил). Он имеет состав С12Н10 и строение С6Н5-С6Н5. Существуют многоядерные соединения, бензольные ядра в которых связаны друг с другом через углеродные атомы, не входящие в цикл. Большую группу производных образует углеводород с тремя бензольными ядрами, называемойтрифенилметаном.
Физические
и химические свойства трифенилметана.
Трифенилметан (С6Н5)3СН
– кристаллическое вещество с т. пл. 920С, т. кип. 3590С,![]() растворяется
в спирте, эфире, ацетоне, бензоле,
хлороформе, сероуглероде, не растворяется
в воде. Применяется в качестве стабилизатора
химической стойкости полимеров, топлива
и для производства различных красителей.
растворяется
в спирте, эфире, ацетоне, бензоле,
хлороформе, сероуглероде, не растворяется
в воде. Применяется в качестве стабилизатора
химической стойкости полимеров, топлива
и для производства различных красителей.
Трифенилметановые красители. На основе производных трифенилметана синтезированы красители трифенилметанового ряда. Важнейшие из них малахитовый зеленый, кристаллический фиолетовый. К группе трифенилметановых красителей относится известный индикатор фенолфталеин.
2. Соединения с конденсированными бензольными ядрами. Простейшими ароматическими соединениями с конденсированными бензольными ядрами являются нафталин (нафтален), антрацен и фенантрен.
Нафталин был открыт в каменноугольной смоле в 1819 г. Состав его установил А.А. Воскресенский (1858), а строение – Эрленмейер (1866). Строение молекулы нафталина выражается формулой, в которой соединены два конденсированных бензольных ядра, имеющих два общих соседних углеродных атома. Кроме известных обозначений о-, м-, п—существуеют также особые названия для положения заместителей: в 1,8 – пери- (от греч. peri - близкий), в 2,6 – амфи- (от греч.amphi – с двух сторон).
По химическим свойствам нафталин, как и его гомологи, проявляет ароматические свойства. Поэтому для него характерны реакции замещения, однако, хотя и с трудом, он вступает в реакции присоединения; нитрируется; окисляется (это имеет промышленное значение).
Антрацен и фенантрен– многоядерные ароматические углеводороды состава С14Н10с тремяконденсированными бензольными ядрами плоского строения. В молекуле антрацена бензольные ядра конденсированы линейно, а в молекуле фенантрена – ангулярно (под углом).
Фенантрен
- кристаллическое вещество с голубой
флуоресценцией; т. пл. 101 0С, т. кип.
340,10С,![]() растворяется
в спирте, эфире, ацетоне, бензоле,
хлороформе. Применяется в качестве
стабилизатора нитратов целлюлозы и
нитроглицерина, компонент дымовых
составов и физиологически активных
веществ.
растворяется
в спирте, эфире, ацетоне, бензоле,
хлороформе. Применяется в качестве
стабилизатора нитратов целлюлозы и
нитроглицерина, компонент дымовых
составов и физиологически активных
веществ.
Наряду с антраценом и фенантреном в каменной смоле содержатся пиренС16Н10,хризенС18Н12,бензпиренС20Н12(содержится в выхлопных газах автотранспорта и в табачном дыме и является причиной рака легких у курильщиков) и др.
Основная литература [1] (199-207; 247-300)
Контрольные вопросы:
1. Какие соединения называются элементорганическими? Дайте определение, приведите примеры металлорганических соединений и назовите их.
2. Назовите закономерности образования металлорганических соединений в зависимости от места элемента в периодической системе элементов Д. И. Менделеева.
3. Охарактеризуйте общие способы получения металлорганических соединений.
4. Опишите основные характерные физические и химические свойства магнийорганических соединений. Назовите синтезы Гриньяра и напишите уравнения соответствующих химических реакций.
5. Приведите примеры промышленного использования алюминийорганических соединений в самолето- и ракетостроении.
6. Расскажите о физических и химических свойствах кремнийорганических соединений: силанов, силоксанов, силазанов, полисилоксанов?
7. Что такое карбоциклические углеводороды? Дайте определение предельных циклоалканов. Какова их классификация?
8. Расскажите о составе и строении циклоалканов; назовите простейшие из них.
9. От таких факторов зависят прочность и устойчивость циклов?
10. Назовите основные физические и химические свойства циклоалканов.
11. Дайте определение и классификацию циклических терпенов (циклоолефинов).
12. Назовите способы получения алифатических, моноциклических и бициклических терпенов. Укажите их характерные свойства.
13. Что такое ароматические углеводороды? Дайте краткую химическую характеристику бензола и его гомологов.
14. Изложите основные способы получения бензола и его гомологов из каменног угля,
нефти и синтетическими методами.
15. Назовите основные химические свойства бензола и его гомологов.
16. Каково ориентирующее действие заместителей в бензольном ядре и влияние дополнительных ядер в многоядерных соединениях на их ориентирующее действие?
17.Опишите особенности получения ихарактерные химические свойства ароматических оксисоединений (фенолов), альдегидов и кетонов, кислот, аминов, азо- и диазосоединений и многоядерных соединений.
18. Перечислите области использования ароматических углеводородов в качестве реагентов при флотации руд.
19. Покажите роль отечественных и советских ученых в развитии химии ароматических углеводородов.
Тема лекции №6:.Основные понятия химии высокомолекулярных соединений
В природе распространены макромолекулы, состоящие из десятков и сотен тысяч и даже миллионов атомов. Такие соединения имеют большую относительную молекулярную массу. Поэтому их называют высокомолекулярными или иначе полимерами. Полимерами называются соединения, молекулы которых состоят из большого числа атомных группировок, соединенных химическими связями в длинные цепи. Среди полимеров линейные макромолекулы представляют собой атомные цепи с многократно повторяющимися звеньями, которые образовались в процессе полимеризации из молекул низкомолекулярных соединении - мономера.
Простейший органический полимер – полиэтилен - продукт полимеризации этилен. Этилен – ненасыщенный углеводород, легко вступающий в реакции присоединения. Две молекулы этилена, соединяясь, образуют молекулу бутена:
СН2=СН2 + СН2 = СН2 -СН3 –СН2 –СН-СН3-.
Исходное вещество – этилен - называется мономером, образующийся бутилен-димером. При соединении трех молекул этилена образуется тример, четырех – тетрамер и т.д. Если соединяются n молекул мономера, образуется полимер (от слова «поли»- много):
nСН2= СН2 [-СН2 –СН2-]n.
Многократно повторяющиеся группировки, которые являются остатками мономеров, называются звеньями, или химическими единицами, или мономерными звеньями; большая молекула, составленная из звеньев, называется макромолекулой или полимерной цепью.
Число звеньев в цепи называется степенью полимеризации и обозначается буквами n или Р. Произведение степени полимеризации n на молекулярный вес звена Мзв равно молекулярному весу полимера:
Мпол=nМзв.
Величина n может варьировать в широких пределах: от n, равным нескольким единицам, до n, равным 5000-10000 и даже больше. Полимеры с высокой степенью полимеризации называются высокополимерами, полимеры с низкой степенью полимеризации - олигомерами.
Условно: М 5103 - полимер; М 5103 и М=5000500 – олигомер; М 500 - НМС, где М - молекулярная масса ВМС.
Высокополимеры имеют очень большие значения молекулярных весов, порядков 104-106. Поэтому высокополимерные вещества являются и высокомолекулярными. Однако не каждое высокомолекулярное вещество имеет полимерное строение, например, белки не полимеры, т.к. в них не наблюдается черодования одинаковых группировок атомов.
Полимерные соединения, цепи которых содержат несколько типов мономерных звеньев, называются сополимерами или смешанными полимерами.
Особенности полимеров:
- полимеры обладают высокоэластичным состоянием, которое объясняется не только структурой полимерных молекул, но и свойством внутреннего вращения известным для простых молекул, это состояние основывается на применении конформационной статистики макромолекул;
-аморфные полимеры по структуре сложнее, чем низкомолекулярное вещество, но в их ближнем порядке примыкают к строению жидкостей;
-релаксационные и тепловые свойства расплавов полимеров и жидкостей во многом аналогичны (процесс стеклования, реология);
-кристаллические полимеры на ряду с кристаллической фазой имеют в объеме и аморфную фазу с межфазными слоями;
-по электрическим свойствам полимеры – диэлектрики и для них характерно электретное состояние;
-по магнитным свойствам полимеры – диамагнетики;
-по оптическим свойствам они характеризуются ярко выраженным двойным лучепреломлением при молекулярной ориентации.
Главная особенность строения полимерного соединения – это наличие цепных молекул, в которых последовательно связано большое число атомов. Для такого соединения характерны два типа связей – химические и межмолекулярные, резко различающиеся по энергии и длине. В самой цепи атомы соединяются между собой прочными химическими связями длиной порядка 1-1,5 Å. Между цепями действуют значительно более слабые межмолекулярные силы на расстояниях пределах 3-4 Å.
Полимеры, построенные из одинаковых мономеров, называются гомополимерами, а из разных мономеров - гетерополимерами или смешанными (из различных атомов).
Остатки мономеров могут соединяться в макромолекуле друг с другом с образованием полимеров (и сополимеров) линейного, разветвленного или сетчатого (пространственного) строения.
Линейными полимерами называются полимеры, макромолекулы которых представляют собой длинные цепи с очень высокой степенью асимметрии, остаток мономера – А:
… А – А –А – А –А – А - …
К типичным линейным полимерам относятся; полиизобутилен, полиизопрен, поливинилхлорид, полиэтилен (иногда называют полиметиленом, так как повторяющимся звеном является метиленовая группа – СН2-):
[-СН2-С(СН3)2-]n,
[-СН2-СН3-СН-СН2-]n,
[-CH2C![]() -CH-]n,
[-CH2-CH2-]n.
-CH-]n,
[-CH2-CH2-]n.
Разветленный полимер представляет собой длинную цепь (называемую обычно главной, или основной) с боковыми ответвлениями (боковыми цепями), причем число этих ответвлений и их длина могут варьировать в очень широких пределах:
.
:
… -А – А – А – А – А – А – А – основная цепь
А А
А А - боковая цепь
. А
:
.
:
Разветленные сополимеры – привитые сополимеры; по строению главной цепи все полимеры делятся на гомоцепные (пример, карбоцепные) и гетероцепные (из различных атомов).
Полимеры: природные и синтетические, подавляющее число их полимеров содержит в своем составе углерод и водород.
Полимеры: кремнийорганические (построены из атомов кремния и кислорода, а боковые группы содержат атомы углерода, водорода или других химических элементов; смазки, каучуки, покрытия), органические [биополимеры, волокнообразующие, твердые (смолы, пластмассы), высокоэластичные (каучуки, резины)], неорганические (сера, неорганические стеклы, асбест).
Классификация полимеров
Способность элемента образовывать полимерные соединения зависит от его положения в периодической системе Д.И.Менделеева.
Элементы первой группы, а также одновалентные элементы других групп (Н, Не) вообще не способны образовывать полимеры, так как для образования цепи элемент должен иметь по крайной мере две валентности. Все остальные элементы могут давать гомоцепные или гетероцепные полимерные соединения, устойчивость которых зависит от прочности связи между атомами.
Наиболее прочная связь между С – С; наименее прочная N – О.
Выше указывалось, что полимеры делятся на органические, элементоорганические и неорганические. Однако точное разграничение этих трех классов очень затруднительно, так как между ними имеется много различных промежуточных соединений.
К
органическим полимерам относятся
соединения, содержащие С, Н, О, N,
S,
Ha![]() и те в главную цепь которых входят О, N
и S,
также соли органических кислот.
и те в главную цепь которых входят О, N
и S,
также соли органических кислот.
Неорганические полимеры – это полимеры, не содержащие атома углерода; металлы, соли в разбавленных растворах – не полимеры;
Элементы I группы не образуют полимеров
Элементы II группы (Bе, Cd и т. д.).
Большое значение имеют элементы III и IV группы.
Основная литература [2] (5-17; 40-46); [3] (7; 8)
Контрольные вопросы:
1. Дайте определение понятий ВМС, полимеры, сополимеры, олигомеры, мономеры.
2. Расскажите о соотношений между этими понятиями.
3. Назовите принципиальные особенности строений их основные отличия от низкого низкомолекулярных веществ.
4. Дайте классификацию ВМС.
Тема лекции №7: Структура полимеров и особенности строения ВМС.
Структура полимеров. Исследование структуры полимеров является одним из актуальных направлений химии высокомолекулярных соединений. Фундаментальные работы, проводимые в этой области, имеют своей целью выяснение взаимосвязи между молекулярной и надмолекулярной структурами полимеров и их физико-механическими свойствами. Еще в ранних работах Флори, Вундерлиха, Каргина предпологалось, что цепное строение полимеров и их химическая природа предопределяеют как геометрию самой молекулы, так и дальний порядок в конденсированной фазе кристаллических высокомолекулярных соединений.
В 60-х годах ХХ-го века была экспекриментальна доказана возможность образования множества разнообразных надмолекулярных структур не только в кристаллизующихся полимерах, но и в аморфных состояниях. Было установлено, что все структурные возможности реальных полимерных систем закодированы в микроструктуре отдельной макромолекулы, но реализуются они на уровне надмолекулярной организации.
Успехи, достигнутые мировой наукой, нашли многообразные приложения в химической технологии при создании новейших полмерных материалов. Достаточно упомянуть, что сверхпрочные волокна появились в результате исследования жидкокристаллических структур в растворах полимеров. Созданию сверхмолекулярых пленок и волокон из пропилена предшествовало исследования надмолекулярных структур, образующихся в условиях формирования их расплава и раствора.
Таким образом, эксплуатационные и технологические свойства полимеров и материалов на их основе могут изменяться в широком интарвале не только за счет химического строения, но и за счет их надмолекулярной структурой модификации полимеров является одной из проблем структурной фихико-химии высокомолекулярных соединений, так как до настоящего времени отсутствуют исчерпывающие сведения об условиях возникновения определенных молекулярных и надмолекулярных образований в полимерах различного химического строения и их влиянии на физические и механические свойства полимерных материалов.
Начало развития фундаментального направления химии высокомолекулярных соединений – структуры полимеров можно отнести к пятидесятым годам, когда был открыт синтез стереорегулярных полимеров Циглером (ФРГ) и Натта (Италия).
Следует отметить, что только по исследованиям структуры высокомолекулярных соединений в разные годы удостоены Нобелевских премий Полинг, Крик, Уотсон, Циглер, Натта, Флори, Керл, Смолли. Последние трое – за открытие фуллеровской структуры олигомеров углерода С60 и С70.
В Московском государственном университете им. М. В. Ломоносова в шестидесятые годы впервые начали читать спецкурс «Структура полимеров» академик В.А. Каргин. В Казахском национальном техническом университете этот курс введен профессором Асаубековым М. А., выпускником МГУ им. М. В. Ломоносова в 1991 году.
Структура – термин, принятый из биологии. Там определены первичные, вторичные и третичные структуры. Первичная структура – это последовательность присоединения атомов и звеньев – конфигурация макромолекул. Вторичная – конформация макроцепей: спиральная, зигзагообразная, клубковая, глобулярная, складчатая и полипептидная. Последняя – это знаменитая двойная спираль Крика-Уотсона. Третичная структура полимеров представляет собой различные ассоциаты или ансамбля из многих макромолекул, которых называют надмолекулярными структурами (HМC). В настоящее время известны следующие НМС полимеров: фибриллы, глобулы (из нескольких свернутых на себя макромолекул), ламели, сферолиты, дендриты, эдриты и полосатые структуры («мозги»).
До открытия синтеза стереорегулярных полимеров Циглером (ФРГ) и Натта (Италия) в 50-х полимеры считались бесструктурными. Господствовала теория «бахромчатой мицеллы», согласно которой полимер представляет собой систему из длинных и запутанных макромолекул, проходящих через микрокристалличекские участки. Из-за большого набора времн релаксации макромолекул для их распутывания и упорядоченного расположения в кристаллическую решетку теоретически требовалось бы многие годы, а стереорегулярные полиолефины легко и быстро кристаллизовались. За это открытие Циглер и Натта получили Нобелевскую премию. До этого американский химик Лайнус Полинг был удостоен звания лауреата Нобелевской премии за открытие вторичной структуры белков. Затем он получил Нобелевскую премию мира.
Обычная вторичная структуцра ДНК открыта в 1953 г. Джеймсом Уотсоном из США и Френсисом Криком из Англии. Это знаменитая «двойная спираль Уотсона-Крика». За открытие двойной спирали Уотсон и Крик удостоены Нобелевской премии. Это открытие приобрело ивердую и вполне определенную репутацию одного из выдающихся достижений в истории науки. Восхитительная красота двухспиральной модели ДНК – в той непринужденности, с которой объясняется одно из подлинных чудес – способность живого и самовоспроизводимого: стоит двум комплектарным нитям разойтись, как тут же будут готовы две «типографские» матрицы для создания двух идентичных копий. Это и есть молекулярная основа биологической наследовательности.
Вторичные стуктуры возникают в биополимерных цепочках не при любых условиях, и их физика очень интересна. Увеличивая температуру или добавляя к воде некотрые низкомолкулярные вещества, можно вызвать, например, раскручивание спиралей – при этом происходит так называемый переход спираль-клубок. Название этого перехода связано с тем, что спиральная полимерная цепь довольно жесткая, а деспирализованная – клубок часто еще называют плавлением спирали. Это фазовый переход.
Аналогия с обычным плавлением твердого вещества усуглубляется тем, что переход спираль-клубок при изменении температуры происходит очень резко – в узком температурном интервале - и сопровождается заметным поглощением теплоты, которая уходит на разрыв стабилизирующих спираль водородных связей; при плавлении разрушаются не отдельные нитки спирали (что было бы аналогично выполнению отдельных ячеек кристаллической решетки), а сразу весьма длинные куски – это кооперативность.
Пример третичный структуры – глобулярный белок триазофосфатизомеразы. Его третичная структура имеет вид плотно сжатого комка и называется глобулой.
Третичная структура ДНК изучена мало, потому что макромолекулы ДНК очень длинные (числом мономерных звеньев достигает до десяти миллиардов) и формируют пространственные структуры не сами по себе, а в комплексе с белками (размером которых по сравнению с ДНК очень малы).
Кроме глобулярных, бывает ещё фибриллярные белки, где молекулярные цепи по несколько штук связываются в «косички» - фибриллы (волокна): например, такое кологен сухомилий.
При ухудшении качества растворителя в парном взаимодействии звеньев начинает превалировать притяжение, т. е. сталкивающиеся попарно звенья как бы на некоторое время слипаются. Ясно, что когда притяжение звеньев достаточно сильны, в макромолекуле джолжны произойти переход типа газ-жидкость: участки полимерной цепи «сконденсирются на себя» и вместо весьма разреженного полимерного клубка возникает плотная сконденсированная «капля» - полимерная глобула. Этот переход и называется переходом клубок-глобула. Глобулярное состояние очень часто встречается не только у белков, но и во многих других системах – от ДНК до макроскопических полимерных сеток.
В пятидесятых годах почти одновременнно появилось теория Каргина-Китайгородского-Слонимского об упорядоченности (структурооброзовании) полимеров и экспериментальные данные (электронномикроскопические микрофотографии) А. Келлера, Е. Фишера, Н. Бакеева и др., подтверждающующих эту теорию.
Таким образом исследования лауреатов Нобелевской премии: Полинга, Крика, Уотсона, Циглера, Натта, Флори, Каргинга, Тобольского, Волькенштейна, Джейла, Манделькорна, Бундерлиха, Френкеля и др. определили начало и значимость структурных исследований полимеров.
Микро- и наноструктуры полимеров. В настоящее время структуру полимеров рассматривают на молекулярном и надмолекулярном уровнях.
Молекулярное строение – это конфигурация и конформация микромолекул.
Надмолекулярная структура (НМС) – это фиксированные многими методами исследования в объеме полимера области разной степени упорядоченности, размер которых существенно превышает размеры отдельных макромолекул конкретного полимера.
Особенности того или иного метода позволяют выявить области гетерогеннности широкого масштаба – от десятков – сотен ангстрем до микронов и миллиметров. Этими областями могут быть кристаллиты, ламели, фибриллы, сферолиты, дендриты, эдрины и т.д., иными словами следует говорить о многоступенчатой структурной организации полимеров.
Структура полимеров – это взаимное расположение в пространстве, внутреннее строение и характер взамодействия между единичными элементами, макроскопическое полимерное тело.
Структуру любого тела рассматривают как набор постепенно усложняющихся подсистем, обладающих ограниченной автономностью. На одном из уровней набора осуществует некая «выделанная» подсистема, которая предопределяет основные свойства рассматривающего тела. В полимерах такая главная подсистема – макромолекула. Макромолекула сама обладает достаточно сложной структурой.
Хотя многие свойства полимеров в конечнрм счете определяется их молекулярным строением, но эти свойства выявляются, передаются через все последующие уровни НМС. В.А. Каргин отмечал, что практически все возможности в реальности свойства полимерных систем закодированы в структуре отдельных макромолекул, но реализуются они на уровне надмолекулярной организации, и следовательно, в области степени зависят от способа конденсации отдельных макромолекул в твердую систему или концентрированный раствор.
Уровни НМС классифицируют по трем признаком: геометрическим, кинетическим и термодинамическим.
Говоря о первых, необходимо прежде всего указать на шкалу масштабов надмолекулярной оргаизации, задаваемой методом измерений, и степень сложности (последовательности) уровней.
Различают рентгено-электронномикроскопический и оптический уорвни НМС: первый охватывает диапазон размеров от десятков до сотен-тысяч ангстрем (кристаллиты, лемали); ко второму можно отнести образование размером в несколько микронов – миллиметров (сферолит).
Для установления последовательности уровней экспериментально нужно выяснить, является ли рассматриваемый элемент НМС первичным структурным образованием, или, в свою очередь, собран из более простых элементов. Следовательно, можно характеризовать термином «сборка» основной универсальный механизм образования более высоких уровней НМС. Например, огромное многообразие надмолекурярных форм в кристаллизующихся полимерах может быть собрано из элементарных ламелей.
По кинетическим и термодинамическим признаком НМС делят на стабильные и нестабильные. Кинетический характер НМС связан с подвижностью соответствующих структурных элементов.
Мера кинетической стабильности – время жизни элемента НМС. Оно зависит от внешних факторов, например, температуры. Введение времени жизни элемента позволяет учесть как длительность проведения эксперимента, используемого для регистрации НМС в полимерах, так и устойчивость структурных элементов к длительности внешних воздействий, например, механических. Если время воздействия силы больше времени жизни структурного элемента, то система ведёт себя как непругая, т. е. перестройка НМС происходит с поглощением энергиии. В противоположном случае структурные элементы упруги и перемещаются (разрушаются) как целое.
В зависимости от того, находятся ли структурные элементы при температуре выше илм ниже температуры фазового перехода, различают термодинамическую стабильность и нестабильность.
НМС по геометрическим признакам можно также разделить организованные и неорганизованные, с точки зрения наличия или отсутствия дальнего порядка в расположении элементов. В свою очередь, организованные структуры могут быть дискретными, т.е. сохраняющими форму, размеры, границы с окружающей средой вр время наблюдения. Дискретным структурам можно противопоставить флуктуационные (статистические) образования в полимерах. Все дискретные структуры, термодинамически стабильны, а флуктуационные – нестабильны и обладают конечным временем жизни, которое, однако, может быть больше времени наблюдения.
Организованные структуры обычно дискретны и термодинамически стабильны. Последние ниже температуры фазового перехода обладают и кинетической стабильностью. При температуре выше фазового перехода термодинамическая стабильность утрачивается, хотя при этом кинетическая стабильность может ещё долго сохраняться.
Кристаллизующиеся полимеры в конденсированном состоянии обычно образуют структуры организованные, дискретные, термодинамически стабильные. Все кристаллизующиеся гипкоцепные полимеры в блоке способны лишь к образованию флуктуационных структур большой или меньшей стабильности. Кристаллы, жидкие кристаллы и суперкристаллы – организованные, дискретные, термодинамически и кинетически стабильные структуры. Глобулы – дискретные, термодинамически стабильные, но неорганизованные образования.
Поскольку значительная роль в описании НМС отводится отдельной макромолекуле, выбранной в качкстве основной подсистемы, поскольку изменение такой важной характеристики макромолекулы, как ее конформационное состояние, позволяет всего судить о наличии НМС.
Рассмотрим конформации молекулы полиэтилена с молекулярной массой 280000. Такая молекула состоит из 20000 групп. Поскольку длина связи С-С равна 1,54 Å, то длина цепи равна 30800 Å. При условии постоянства валентных углов между двумя соседними связями полностью вытянутая цепь должна иметь плоскую зигзагообразную транс-конформацию по всей длине. Эта конформация является наиболее наиболее выгодной в кристалле. Расстояние между концами такой цепи составляет 25300 Å. Расчет показывает, что число поворотных изомеров для такой молекулы имеет порядка 3220000, т.е. около 1010000. это число велико, что в молекуле не реализуются все возможные конформации. Если принять для конформации модель с постоянными валентными углами и заторможенным вращением, то расстояние между концами цепи полиэтилена молекулярной массы 280000 равно 493 Å.
В среде растворителя происходит дополнительное изменение средних размеров макромолекулы. Это связано взаимодействием растворителя и полимера. Если более выгодным является контакт молекул растворителя с макромолекулами (хороший растворитель), то происходит еще большее расширение клубка полимерной цепи вследствие «расталкивания» сегментов цепи молекулами растворителя. если молекулы растворителя отталкиваются от макромолекул (плохой растворитель), то статический клубок сжимается. Экспериментально обнаруженное расстояние между концами молекулы ПЭ в растворе при Ө-температуре оказалось равным 529 Å. Это значение находится в хорошем соответствии с теоретически рассчитанным значением 493 Å, приведенным выше.
Конформационный переход клубок-глобула. Типичная конформация свободно-сочлененной цепи из большого числа звеньев – это клубок. Длина макромолекулы ДНК, входящей в состав хромосом человека, - около метра; если бы не гибкость цепей ДНК, если бы они были жестки, как спицы, то как бы мы упаковывали и хранили их в клеточном ядре размером 10-6 м?
Конформацию клубка макромолекулы принимают в хорошем растворителе, где отталкивание звеньев доминирует. Качество растворителя может ухудшаться, например, при добавлении в раствор плохого растворителя, осадителя или изменении температуры.
Вместе с тем оказалось, что сам по себе переход клубок-глобула весьма своеобразен и необычен, и это стимулировано незатухающий интерес к изучению его проявлений в самых разных полимерных системах. Сегодня мы знаем, что глобулярные состояния очень часто встречаются не только у белков, но и вомногих других системах – от ДНК до макроскопических полимерных сеток, надмолекулярных структур полимеров, и что, соответственно, многие распрастраненные явления в физико-химии полимеров – от образования компактной формы ДНК до коллапса полимерных сеток – могут быть описаны как переходы типа клубок-глобула. Такое широкое понимание роли перехода клубок-глобула стало возможным после появления в 1968 г. работы Лифшица И.М. К настоящему времени имеется достаточно много экспериментальных работ, посвященных наблюдению описанного перехода клубок-глобула, происходящего при понижении температуры несколько ниже – точек или изменении среды. Для наблюдения перехода клубок-глобулав отдельной макромолекуле был использован ряд экспериментальных методик: обычно рассеяние света, измерение вязкости осмотического давления, исследование упругого нейтронного рассеяния, двойного лучепреломления в потоке, ядерный магнитный резонанс высокого разрежения, молекулярная спектроскопия и др. Теоретическое и экспериментальное изучение этого перехода, безусловно очень важно с фундаментальной точки зрения. В биополимерах обнаружены три типа переходов клубок-глобула: коллапс полимерных сеток, образование контактной формы ДНК и денатурация белков.
Конформационный переход спираль- клубок. В сополимерах метакриловой кислоты с 2-гидроксиэтилметакрилатом и метилметакрилатом, имеющих преимущественно синдиотактическую структуру, обнаружен конформационный переход от комнатной к вытянутой спирали.
При фиксированном рН вытянутая цепь полиакриловой кислоты лучше флокирует, чем клубок.
О стержнеобразной конформации, установленной ранее лишь для полимептидов со спиральной структурой, в водорастворимом синтетическом полиакрилоамиде с заряженными группами и о переходах спираль-клубок сообщено в работах. Всякая макромолекула представляет собой кооперативную систему, ибо из-за ограниченности свободы вращения вокруг ковалентных связей положение ближайших и близких соседей (звеньев) взаимозависимы. Это касается линейных взаимодействий. Но линейная память оказывается в определенной мере фундаментальным кооперативным свойством, ибо накладывает определенные ограничения и на объемные взаимодействия. Особенно отчетливо это проявляется в случае дифильных, или амфифильных сополимеров, имеющих мономеры двух сортов, А и Б, обладающие различной растворимостью. Допустим, что мономер А растворим хорошо, а мономер В нерастворим вовсе. Даже если сополимер АВ представляет собой блок-сополимер, нерастворимый блок не может выпасть в сплошной осадок, ибо линейная память включает в себя ближний конфигурационный порядок и при образовании осадка исключаются все конформации с существенными нарушениями валентных углов или связей. Тем более линейная память исключает контакты между слишком близко (вдоль цепи) расположенными звеньями типа В. Этот запрет распространяется и на отдельные нерастворимые звенья и на их олигоады. В результате агрегации, и парных или множественных пространственных взаимодействий нерастворимых звеньев могут возникнуть вторичные макромолекулярные структуры вулканизационного или конденсационного типа: в обоих случаях клубки имеют поджатые конформации.
Птицин и Эзнер, предлагая теорию перехода клубок-глобула, учли не только парные взаимодействия, но и тройные взаимодействия, и предсказали существование перехода клубок-глобула несколько ниже Ө-температуры, причем этот переход носил характер фазового перехода первого рода.
Развивая теорию глобула- клубок Лифшиц, Гросберг и Хохлов решили задачу о компатизации макромолекулы. Компактизацию клубков наблюдали на дифильных сополимерах в различных средах. Термодинамически образование глобул в аморфных полимерах энергетически не выгодно, но кинетически оно весьма вероятно. Чем и объясняется относительно частое наблюдение надмолекулярных структур глобулярного характера. Свойства разбавленных водных растворов полиамфолитов изучены в зависимости от состава сополимеров, температуры, рН среды, добавки низкомолекулярных электролитов и комплексообразования с другими полимерами. Показано, что сополимеры, содержащие приблизительно эквимолярное количество кислотных и основных мономеров показывают минимум вязкости в воде, что объясняется образованием внутримолекулярных ионных контактов между противоположными зарядами. В зависимости от рн среды полиамфолиты ведут себя либо как поликислоты, либо как полиоснования. Вблизи изоэлектрической точки (ИЭТ) вязкость проходит через минимум, где силы притяжения противоположено заряженных групп настолько велики, что обусловливают образования более компактных частиц, чем конформации невозмущенных гауссовых клубков. В результате этого вязкость полиамфолитов вблизи ИЭТ имеет гораздо меньше, чем вязкость ионных полимеров в Ө-точке. По-видимому, в ИЭТ микромолекулы полиамфолита начинают коллапсировать. Часть мономерных звеньев в середине клубка, выжимая растворитель, конденсируется в компактную глобулу, состоящую в основном только из звеньев полимера. Это обстоятельство позволяет рассматривать полиамфолитов в ИЭТ в качестве удобного объекта для изучения фазового состояния полимерных молекул.
Надмолекулярная структура аморфных полимеров. Впервые идея о структурной неоднородности аморфных полимеров была выдвинута в работах Каргина, Слонимского, Китайгородского и Бакеева еще в конце 50-х годов. На основании, главным образом данных электронно-микроскопических наблюдений была создана пачечная теория строения аморфных полимеров. В дальнейшем эта идея получила развитие в работах Аржакова, Иеха, Джойля, Робертсона и др.
Необходимо подчеркнуть, что единственном экспериментальным фактом, противоречащим идее о структурной упроядоченности аморфных полимеров, является идентичность среднеквадратичных расстояний между концами полимерной цепи аморфного полистирола в блоке (по данным нейтронного рассеяния) и в растворе. Однако это противоречие может быть устранено, если принять компромиссные модели упорядоченного состояния аморфных полимеров Аржакова, Бакеева и Кабанова или Линденмейера, в которых соединены элементы, присущие статистическому клубку и упорядоченной (гетерогенной) структуре.
Совокупность известных экспериментальных данных, полученных с привлечением различных физических методов, позволяет рассматривать аморфные полимеры как микрогетерогенные образования, в которых существуют упорядоченные области с размерами порядка десятков нанометров. Эти микронеоднородности носят флуктуационный характер, поэтому они не имеют ярко выраженных фазовых границ, что сильно затрудняют их обнаружение при использовании многих физико-химических методов исследования. Видимо, этим главным образом и объясняется отсутствие в настоящее время единой модели строения аморфных полимеров.
Совершенно очевидно, что кристаллические полимеры являются микронеоднородными системами.
Неоднородность строения полимеров не может не отразиться на механических свойствах, подобно тому, как неоднородности низкомолекулярных кристаллов (дислокации) сильно влияют на их механические свойства. Это влияние появляется в том, вследствие существования дислокаций реальные кристаллы способны пластически деформироваться и разрушаться под действием напряжений, значение которых на несколько порядков меньше, чем предел прочности идеальных кристаллов. Более того, оказывается, что в случае полимеров структурная неоднородность является одним из необходимых условий, обеспечивающих их способность и пластической деформации.
Для кристаллизующихся полимеров под простыми элементами часто понимают кристаллиты, в аморфных полимерах – домены или посторонние включения или даже физические зацепления макромолекул.
Непрерывно растущие потребности народного хозяйства требуют все большего количества полимерных материалов для решения самых разнообразных практических задач. Эти потребности могут быть удовлетворены в основном двумя путями: синтезом новых полимеров с новыми свойствами и модификацией, свойств уже известных полимеров; производство которых освоено промышленностью. Вряд ли следует ждать, что в скором времени будут синтезированы какие-то новые полимеры, выпуск которых сможет быть организован в таких масштабах, как производство полиэтилена, поливинилхлорида, полистирола, полиамидов, полиэфиров. Поэтому можно ожидать, что структурная модификация свойств полимеров, в ближайшее время получит большое распространение.
Особенности строения ВМС. Неоднократность полимера по химическому составу в том, что в одной и той же цепи содержится звенья различного состава. Например, у промышленных образцов вторичных ацетатов целлюлозы (вторичными называются ацетаты целлюлозы, полученные частичным омылением триацетата целлюлозы и содержащие меньшее, чем в триацетате, количество ацетильных групп) одни звенья могут быть проэтерифицированы полностью, в то время как в других звеньях имеются свободные гидроксильные группы. Химическая неоднородность наблюдается у всех промышленных образцов эфиров целлюлозы, поливинилового спирта и некоторых других полимеров. Химический состав таких полимеров принято характеризовать средним процентным содержанием имеющихся в них функциональных групп (например, ацетильных) или содержанием азота и т.д.
В
триацетате целлюлозы содержание
ацетильных групп, выражаемое отношением
![]() , равно 61,5% . В
промышленности применяют ацетаты
целлюлозы, содержащие 54-58% групп ОСОСН3.
При
таком среднем составе число
проацетилированных гидроксильных групп
в разных звеньях может значительно
различаться.
, равно 61,5% . В
промышленности применяют ацетаты
целлюлозы, содержащие 54-58% групп ОСОСН3.
При
таком среднем составе число
проацетилированных гидроксильных групп
в разных звеньях может значительно
различаться.
Теоретическое количество азота в тринитрате целлюлозы составляет 14%. На практике применяются нитраты целлюлозы, содержащие в среднем от 10 до 13,5% азота (отдельные звенья содержат разное число нитратных групп).
Наиболее характерной чертой полимерных соединений является высокое значение молекулярного веса, т.е. очень большой размер молекул. Однако практически не существует таких полимеров, у которых все молекулы имели бы строго одинаковые размеры, или, другими словами, одинаковую степень полимеризации. Наряду с очень большими молекулами в полимере могут быть и небольшие (молекулярный вес порядка 1000) молекулы промежуточных размеров. Следовательно, любой полимер в той или иной степени неоднороден по величине молекулярного веса или, как говорят, полимолекулярен (иногда пользуются термином «полидисперсность»). Поэтому в химии полимеров пользуются понятием среднего молекулярного веса. Ряд полимерных соединений одинакового химического строения, отличающихся только по молекулярным весам, называется полимергомологическим рядом. Полимеры моно- и полимолекулярные (моно- и полидисперсные). Макромолекула способна изменять форму в результате теплового движения или действия внешних сил; - молекула полимера.
Изомерия – явление, заключающееся в существовании химических соединений с одинаковым качественным и количественным составом, но с различными свойствами.
Геометрическая изомерия, 1. Диастереометрия, возникающая вокруг двойной связи и малых циклов; 2. Изомерия комплексных соединений, вызванная неодинаковым расположением лигандов во внутренней сфере комплекса.
Оптическая изомерия, пространственная изомерия, проявляющаяся в способности энантиомеров ( - молекулы которых относятся друг к другу, как несимметричный предмет к своему зеркальному изображению) вращать плоскость поляризации света в противоположные стороны.
Полимеры могут быть регулярными и нерегулярными, если в цепи полимера наблюдается монотонное чередование звеньев, т.е. соблюдается совершенный, дальний порядок звеньев по цепи, то полимер построен регулярно. Нарушение этого порядка ведет к нерегулярности строения цепи полимера.
Причиной нерегулярности являются, во-первых, следствие различного способа последовательного присоединения друг к другу одних и тех же мономерных звеньев. Так, при реакции полимеризации звенья могут соединяться по схеме I или по схеме II:
…-СН2-СН-СН2-СН-СН2-СН-… (I)
Х Х Х
…-СН2-СН-СН-СН2-СН2-СН-CH-CH2-… (II)
Х Х Х Х
Присоединение по первой схеме называется «голова к хвосту», по второй - «голова к голове».
При полимеризации мономеры большей частью соединяются по типу «голова к хвосту», однако отдельные мономеры в некоторых участках цепи могут соединяться по второму типу, что нарушает регулярность цепи, например у поливинилхлорида
…- СН2 – СН – СН2 –СН – СН – СН2 – СН2 – СН-…
C![]() C
C![]() C
C![]() C
C![]()
полистирола и др.
Во-вторых, нерегулярность цепи может обусловливаться разной степенью разветвленности, так как места присоединении боковой цепи, число ответвлений и их длина могут быть различными, пример, разветвленные полимеры.
В–третьих, нерегулярность цепи может быть следствием беспорядочного чередования мономеров различного химического строения, пример, сополимеры.
Основная литература [2] (5-17; 40-46), [5] (3-18)
Контрольные вопросы:
1. Расскажите о структуре полимеров. Понятие о молекулярном строении.
2. Что представляет собой понятие «надмолекулярная структура - НМС»? Расскажите о трех признаках уровни НМС.
3. Расскажите о конформационных переходах: клубок-глобула и спираль-клубок.
4. Назовите типы неоднородностей в полимерах.
5. Расскажите о неоднородности полимеров по химическому составу.
6. Изложите неоднократность макромолекул по строению, понятие о конфигурации цепей.
7. Укажите возможные присоединения мономерных звеньев в цепи.
8. Что такое геометрическая и оптическая изомерия макромолекул?
9. Дайте понятие нерегулярные, регулярные и стереорегулярные полимеры.
10. Расскажите о неоднородности ВМС по молекулярным массам.
11. Что такое моно- и полидисперсные полимеры?
Тема лекции №8:Исследование структуры полимеров физическими методами.
Необходимым условием повышения эффективности качества научных исследований является широкое внедрение современных физических методов исследования. Одним из таких новых методов является спектроскопия ядерного магнитного резонанса (ЯМР).
Усовершенствование ЯМР-спектров высокого разрешения, работающих при высоких частотах (до 300 МГц) и температурах, использование техники импульсного возбуждения и преобразования Фурье ГТ-ЯМР, применение ЭВМ для плохоразрешенных спектров, вращение образца под «магическим» углом и т.д. значительно разрешил возможности метода ЯМР. Осветим основные направления интенсивно развивающихся методов ЯМР. Это ЯМР высокого разрешения различных изотопов: 1Н,13С,19Ғ, 31Р и др.; ЯМР широких линий; ЯМР-релаксометрия и др.
Ядерный магнитный резонанс. Количество работ с применением этого метода в исследовании полимеров в последние годы увеличивается. Это связано с тем, что ЯМР 13С обладает большими потенциальными возможностями для исследования полимерных систем, позволяющими добиться значительно лучшего разрешения, поскольку диапазон химических сдвигов резонансного углерода в органических соединениях достигает 600 м.д. по сравнению с 20 м.д. для протонов. Используя современные методы регистрации, в ЯМР13С можно получить более узкие линии, чем в спектрах ЯМР1Н, потому что ядра углерода, расположенные в цепи молекулы, существенно экранированы от вляния межмолекулярных, а также и некоторых внутримолекулярных взаимодействий С-Н, что уменьшает роль диполь-дипольного взаимодействия и сужает линии. Спектры ЯМР13С обычно не содержать спиновых мультиплетов, что упрощает спектр и уменьшает вероятность перекрывания сигналов. ЯМР13С регистрирует индивидуальные сигналы от каждого из атомов углерода соединения, молекулярный вес которого составляет 300-500. Для таких сложных молекул протонный магнитный резонас (ПМР) использовался для идентификации по принципу «отпечатков пальцев».
Еще одно преимущество ЯМР 13С состоит в том, что он позволяет непосредственно наблюдать молекулярный скелет, углеродсодержащие функциональные группы, не имеющие протонов (например, карбонильную, нитрильную) и интересующие исследователя углеродные реакционные центры.
ЯМР 13С для исследования полимеров применяется в следующих направлениях:
Спектральная характеристика полимеров – это определение химических сдвигов,
анализ различных спин-спиновых взаимодействий с участием ядер углерода, определение интенсивностей сигналов.
Исследование распределения звеньев в полимерной цепи. С помощью ЯМР 13С
можно получить информацию о микроструктуре, недоступную ни одному методу. Если ПМР позволяет проводить надежные измерения содержания триад (60 МГц) и тетрад (220 МГц) звеньев, а также оценить содержание пентал (300 МГц), то ЯМР 13С увеличивает длину надежное определяемых фрагментов до пентад, причем, данные, полученные из углеродных и протонных спектров, совпадают в пределах 1-2%.
3. Анализ полимеров в блоке и гелей. Важная особенность углеродной спектроскопии заключается в том, что многие полимеры и сополимеры в блоке дают спектры высокого разрушения. Анализируя эффект Оверхаузера, времена релаксации и ширину линий в спектрах высокого разрешения полимеров в блоке, а также применяя технику вращения образца под так называемым «магическим» углом, можно глубоко исследовать структурную модификацию полимеров, пластификацию и решить ряд других проблем, связанных с изучением тонкой структуры полимера в блоке. При гелеобразовании растворов полимеров подвижность молекулярных цепей ограничена и спектры ЯМР1Н этих образцов не обнаруживают структуры, свойственной спектрам высокого разрешения. Спектры же ЯМР13С достаточно хорошо разрешены и поэтому позволяют изучить микроструктуры гелей полимеров.
4. Релаксация. Исследование релаксации ядер 13С в полимерных системах, по-видимому, будет со временем столь же полезно, как и исследование протонной релаксации по временам релаксации ядер13С. Особый интерес в этом плане представляет изучение влияния сшивания полимерных цепей на затрудненность молекулярного движения.
5. Одна из наиболее важных областей применения спектроскопии ЯМР 13С – исследование природных соединений и биополимеров (углеродов, терпенов, стероидов, аминокислот, пептидов, белков, нуклеотидов и др.). Исследование структур белков методом ПМР в существенной мере осложнено большой шириной линии, характерной для спектров ПМР этих соединений, а также малыми различиями химических сдвигов1Н различных аминокислотных остатков. Аллерханд и др. сообщил результаты исследования спектра ЯМР13С рибонуклеазы А. Полная интерпретация спектра не была сделана, однако пришлось отнести несколько полос и индивидуальным группам аминокислотных остатков. Измерены времена спин-решеточной релаксации Т1для растворов нативной и денатурированной рибонуклеазы. Это дало возможность оценить характер сегментального движения и конформационное состояние в обеих формах. Для нативного фермента время спин-решеточной релаксации было существенно меньше, а время молекулярной корреляции на два порядка больше, чем для денатурированного. Эти результаты указывают на потери конформационной подвижности в денатурированной форме. Ограничение внутреннего движения в нативном ферменте приводит к существенному диполь-дипольному уширению линии спектров ЯМР13С –Т.
ЯМР широких линий. Ширина резонансных линий для твердых тел велика, так как из-за ограниченности свободы движения молекул магнитные поля дипольного взаимодействия практически не усредняются. Обычно спектры ЯМР в блоке представляют собой одну линию колоколообразной формы, что объясняется диполь-дипольными взаимодействиями большого числа ядер. Если эта линия приобретает более сложную форму с более или менее четко выраженной структурой, то это может быть вызвано разными причинами: наличием химического сдвига, наличием двух фаз в системе.
Особенности формы линии четко проявляются при записи второй функции поглощения. Если линия колоколообразная, то по второй производной можно судить о том, близка ли форма линии к лоренцевой или гауссовой. Оснощение высоты боковых максимумов к высоте центрального минимума для линии лоренцевой равно 0,25, а для гауссовой – 0,45.
По
величине и форме сигнала можно определить
степень кристалличности полимера.
Величина второго момента (![]() )
широких линий позволяет изучить строение
цепи полимера, присоединение звенев в
цепи «голова к хвосту» или «голова к
голове» и др.
)
широких линий позволяет изучить строение
цепи полимера, присоединение звенев в
цепи «голова к хвосту» или «голова к
голове» и др.
Линия спектра ЯМР полиэтилена с разветвленными цепями уже, а следовательно и подвижность протонов больше, чем в линейном полиэтилене. Применение ЯМР широких линий, в исследовании полимеров подробно описано И.Я. Слонимом и А.Н. Любимовым. Установлено, что этот метод позволяет исследовать также химические процессы (полимеризация, сшивание, действие облучения, деструкция), молекулярные движения в полимерах и т.д.
Изучение
зависимости формы линии ЯМР от температуры
позволяет судить о характере межмолекулярных
сил, которые определяют молекулярную
подвижность в полимере. Роль межмолекулярных
взаимодействий проявляется, например,
в изменении ширины линии от температуры
с увеличением числа СН2–групп
между амидными группами в ряду полиамидов.
В полиэтилене сужение линий ЯМР
наблюдается при – 730, а в полиамидах
при температуре от 52 до 1020, так
как замена СН2–группы на амидную
способствует появлению водородных
связей между цепями. Чем меньше длина
парафиновых сегментов между амидными
группами, тем чаще в цепи водородные
связи, тем меньше подвижность и выше
температура сужения линии ЯМР. Второй
момент является наиболее важной
характеристикой линии ЯМР в полимере,
так как его величину можно рассчитать
для жесткой структуры и для структуры
с определенными подвижными молекулярными
группами и сравнить экспериментальные
результаты с теоретическими. Изменение
второго момента с температурой позволяет
судить о движениях главной цепи и боковых
групп полимера. Теоретическое значение![]() ,
рассчитанное для жесткой структуры
поликарбоната известного строения,
равно 20-25 эрг2. Экспериментальная
же величина
,
рассчитанное для жесткой структуры
поликарбоната известного строения,
равно 20-25 эрг2. Экспериментальная
же величина![]() при – 1960равна 16-17 эрг2.
Расхождение объясняется, повидимому.
Тем, что и при этой температуре еще
сохраняется некоторая подвижность
метильных групп. Падение
при – 1960равна 16-17 эрг2.
Расхождение объясняется, повидимому.
Тем, что и при этой температуре еще
сохраняется некоторая подвижность
метильных групп. Падение![]() с температурой в области – 1500до
800 обусловлено вращением СН3–групп.
При температуре – 150-1700
с температурой в области – 1500до
800 обусловлено вращением СН3–групп.
При температуре – 150-1700![]() уменьшается потому, что начинается
движение сегментов цепных молекул:
полимер переходит из стеклообразного
состояния высокоэластичное. Таким
образом, методом ЯМР можно определить
такую важную характеристику как
температура стеклования полимера.
уменьшается потому, что начинается
движение сегментов цепных молекул:
полимер переходит из стеклообразного
состояния высокоэластичное. Таким
образом, методом ЯМР можно определить
такую важную характеристику как
температура стеклования полимера.
Ядерный магнитный резонанс - 1Н высокого разрешения (ЯМР 1Н-ВР). Микроструктура полимеров. Конфигурацию макромолекул можно определить методом ЯМР-ВР. При этом особое внимание обращается на наличие химически различающихся протонов. Экспериментальное подтверждение одной или нескольких структур определяется совпадением предсказываемых и экспериментально наблюдаемых химических сдвигов. Спектры часто осложняются спин-спиновыми взаимодействиями с электронами.
Как уже сказано выше, на ядро влияет несколько атомов в цепи макромолекул и соответственно по химическим сдвигам можно определить различные комбинации друг относительно друга пяти звеньев (пентады). По интенсивности сигналов вычисляют содержание различных триад, тетрад, пентад и макроцепи и таким образом можно наиболее полно расшифровать микроструктуру макромолекулы, особенно при использовании совершенствованных ЯМР-методов (высокая частота – до 300 МГц, высокая температура, варирование растворителей, применение парамагнитных сдвигающих реактивов для разделения полос спектров, использование ЭВМ для трудно разрешимых спектров и т.д.)
В области исследоания микроструктуры полимеров методом ЯМР следует отметить работы Н. А. Платэ и Л. Б. Строганова, которые разработали и успешно применяют метод машинного анализа плохо разрешенных спектров ЯМР.
Перспективно применение парамагнитных сдвигающих реактивов (ПСР) – комплексов лантанидов с дикетонами, которые являются льюсовыми кислотами. В растворе они образуют аддукты с субстратами (органические молекулы, полимеры), содержащими атомы с неподеленной парой электронов (О,N,S,P) и обладающими основными свойствами. Под влиянием парамагнитного иона линии в спектре ЯМР заметно сдвигаются. Вследствие быстрого обмена между свободными и связанными в комплекс макромолекулами наблюдаемый спектр является средним между спектром связанного и свободного субстрата.
В случае полимеров ПСР используются для изучения стереохимического строения и последовательности распределения звеньев. При добавлении ПСР в растворе полимера наблюдается расщепление сигналов, например протонов СН3-групп. Это позволяет количественно оценить соответствующую структурув цепи, например, количество изотактических, синдиотактических и гетеротактических триад в цепи полимера. Так, спектр полиэтиленгликоля в воде представляет собой единичный пик, и его приняли за сигнал протонов СН2-групп. При добавлении ПСР к водному раствору полиэтиленгликоля разделяются сигналы концевых, предконцевых и центральных СН2-групп.
ПСР резко увеличивает возможности ЯМР-спектроскопии. Во многих случаях информация, получаемая из спектров ЯМР, снятых на частоте 220 МГц, меньше, чем могут дать спектры с добавлением СПР на частоте 60 МГц. В настоящее время проводятся поиски пиковых ПСР, в частности, обладающих избирательностьюпо отношению к определенным функциональным группам и пригодных для применения в полярных ПСР-полимер-растворитель и позволили бы ещё больше расширить область применения этого метода.
Избирательная сольватация полимеров. Люти удалось из данных ЯМР определить число молекул растворителей в сольватном слое полимера и охарактеризовать преимущественную сольватацию одним из двух растворителей. Полученное значение константы сольватационного равновесия дает удовлетворительное описание избирательной сольватации для широкого интервала составов. Измеряемым параметрам при использовании ЯМР было время спин-решеточной релаксации Т1. Известно, что при нарушении больцманского распределения ядерных спинов время Т1определяет промежуток времени, который требуется для того, чтобы они вернулись к первоначальному больцмановскому распределению.
Сведения относительно стркутуры тройного раствора или локального расположения молекул растворителя не могут быть получены термодинамическими методами. Метод ЯМР дает возможность оценить различие между молекулами растворителя вблизи полимерной цепи и вдали от нее из-за большой разницы в их скоростях релаксации. Таким образом, ЯМР – это прямой метод, позволяющий описать состояние макромолекулы и ее ближайшего окружения в растворе.
Исследование конформации синтетических полимеров. Известно, что конформация многих биополимеров в растворах определяется природой растворителя, в которой они растворены. Относительно мало работ по ПМР-ВР синтетических полимеров.
Начнем с «простого» полимера – полиэтилена. Здесь нет определенных функциональных групп, которые способны к специфическим взаимодействиям. Поэтому структура полиэтилена в растворе казалось бы должна представлять собой беспорядочный клубок. Но детальное изучение полиэтилена в растворе метода ПМР-ВР показало, что все внутренние метиленовые протоны алканов с числом углеродаn≤16 дают единичный пик, алканы сn17 – два пика. Это означает, что высокомолекулярные алканы образуют дополнительную структуру, которая отсутствует в низших углеводородах. Образование этой новой структуры в высокомолекулярных парафинах зависит от растворителя. В ароматических растворителях (хлорнафталин) резонансная полоса внутренних метиленовых протонов расщепляется приn17. В четыреххлористом углероде или дейтерированном н-алкане расщепления резонансного пика нет. По мнению Кан Чен Лю, новое образование представляет собой упорядоченную складчатую структуру макромолекул, которая обычно встречается в кристаллических полимерах.
Методом ПМР-ВР изучена внутримолекулярная структура полиэтиленоксида (ПЭО). Изучали спектры ПМР-ВР в зависимости от длины макромолекул в разных растворителях и при разных концентрациях. В 50%-ных растворах ПЭО макромолекулы находятся в ассоциированном состоянии и поэтому получаются плохо разрешенные спектры. В основном снимали ПМР-ВР спектры 5 %-ных растворов ПЭО. Для примера, тетрамера и пентамера появляется единичный пик. В случае гексамера образуется дуплет, причем интенсивность нового пика растет с увеличением длины цепи. Таким образом, новая структура обнаруживается в олигомерах, содержащих семь мономерных единиц.
Таким образом, мы рассмотрели влияние различных факторов (молекулярный вес полимера, температура, строение цепи) на конформацию макромолекулы в растворе. На изменение конформации полимера существенно влияет растворитель.
Основная литература [5] (52-60)
Контрольные вопросы:
1. Теоретические основы метода ядерного магнитного резонанса и его применеие для исследования структуры полимеров.
2. Что представляет собойЯМР широких линий?
3. Что Вы знаете о микроструктуре полимеров?
4. Расскажите о избирательной сольватации полимеров.
5. Расскажите о исследовании конформации синтетических полимеров.
6. Что такое ПМР-ВР?
Тема лекции №9: Получение ВМС методами радикальной и ионной полимеризации
Общие принципы и подходы к получению ВМС. Реакции образования молекул полимеров называются полимеризацией и поликонденсацией. Большинство полимеров, которые синтезированы в лаборатории или нашли практическое применение, имеет молекулярную массу 5000-200 000.
В процессе развития полимерной науки использовались две классификации полимеров. Одна из них делит (Карозерс, 1929 г.) все полимеры конденсационные и полимеризационные (аддиционные), другая – на ступенчатые и цепные полимеры. Термины «конденсационные» и «ступенчатые» иногда считают синонимами, так же как термины «полимеризационные» и «цепные». Хотя эти термины действительно во многих случаях равнозначны, их не следует путать друг с другом, так как в основе их лежит два различных принципа классификации. Конденсационно-аддиционная классификация имеет в виду главным образом состав или структуру полимеров, тогда как ступенчато-цепная классификация основана на механизме реакций полимеризации. Конденсационными были названы полимеры, которые образуются из полифункциональных мономеров различными реакциями конденсации, известными в органической химии и протекающими с выделением НМ продуктов, например воды. Типичным примером такого конденсационного полимера являются полиамиды, получаемые из диаминов и дикарбоновых кислот с выделением воды по схеме
nН2 N –R–NH2 + nНООС –R’-CООН
Н-(-NН–R–NНСО–R’–СО-)n – ОН + (2n - 1)Н2О,
где R и R’ – алифатические или ароматические группы. Группировка в скобках в формуле полиамида многократно повторяется в полимерной цепи и называется поэтому повторяющейся единицей или элементарным звеном. Состав элементарного звена отличается от состава суммы двух мономеров на молекулу воды. Полиамид на основе гексаметилендиамина R = (СН2)6 и адипиновой кислоты на R’ = (СН2)4 в настоящее время широко используется в производстве волокна и пластмасс и хорошо известен под названием «найлон-6,6» или «полигексаметиленадипамид». В качестве других примеров конденсационных полимеров можно привести сложные полиэфиры, получаемые из дикарбоновых кислот и диолов с выделением воды:
nНО– R–ОH + nНООС –R’- CООН НО-(–R–ОСО–R’–СОО -)n–Н+(2n -1)Н2О,
и поликарбонаты, образующиеся при взаимодействии ароматических бифенолов и фосгена с выделением хлористого водорода
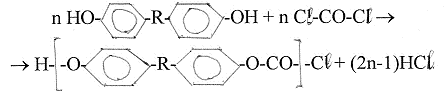
Ряд природных полимеров, и в их числе целлюлоза, шерсть, крахмал и натуральный шелк, относится к разряду конденсационных полимеров на основании того, что гипотетическая схема их синтеза предполагает выделение воды: так, можно полагать, что целлюлоза получается при дегидратации глюкозы.
По классификации Карозерса полимеризационными полимерами называют полимеры, которые образуются из мономеров без выделения НМ побочных продуктов. В отлтичие от конденсационных полимеров элементарный состав такого полимера и его мономера одинаков. Главные представители аддиционных полимеров – полимеры винильных мономеров. При взаимодействии таких мономеров друг с другом образуются полимеры, а двойная связь их переходит в насыщенную по схеме
nCH2=CHY -(-CH2-CHY-)n-,
где Y – водород, алкил, арил, нитрил, сложный эфир, карбоксильная группа, кетон, простая эфирная группа, галоген и др. Другим типом мономеров, образующих аддиционные полимеры, являются альдегиды.
На основании механизма процесса, полимеризация делится на ступенчатую и цепную. Таким образом, ступенчатые полимеры – это полимеры, получающиеся ступенчатой полимеризацией (поликонденсацией), а цепные полимеры – это полимеры, образующиеся цепной полимеризацией. По своим характеристикам эти два процесса сильно различаются. При этом различие заключается главным образом в разной зависимости параметров реакции от времени. Если говорить более конкретно, то при поликонденсации и полимеризации требуется различный промежуток времени для получения высокомолекулярных полимеров, т. е. для завершения роста цепи микромолекулы.
Поликонденсация представляет собой ступенчатую реакцию между функциональными группами исходных веществ.
При поликонденсации размер молекулы полимера увеличивается с относительно низкой скоростью. Сначала из мономера медленно образуется димер, тример, тетрамер, пентамер и т. д. до тех пор, пока не будет достигнут сравнительно высокий молекулярный вес, т. е. полимер будет содержать много молекул мономера. Совершенно иная картина имеет место при цепной полимеризации, когда высокомолекулярный полимер получается почти сразу после начала реакции.
Поскольку основной особенностью радикальной реакции является ее цепной механизм, следует дать краткую характеристику цепных процессов.
Цепными называются такие процессы, в которых превращение исходных веществ, в продукты реакции осуществляется регулярным чередованием нескольких реакций с участием свободных радикалов или ионов (активных центров), идущих с сохранением свободной валентности (или с сохранением структуры активного центра).
Следует различать цепи двух видов: материальную и кинетическую. Материальная цепь характеризуется числом составных звеньев в макромолекуле, т.е. степенью полимеризации «n».
Кинетическая цепь характеризуется числом элементарных актов присоединения молекул мономера к активному центру в расчете на один образовавшийся в процессе зарождения цепи свободный радикал. Эти величины не всегда совпадают.
По цепному механизму протекают различные химические реакции – горение, окисление, хлорирование и бромирование, реакции разложения. Важнейшей цепной реакцией является полимеризация.
Радикальной полимеризацией называется цепная полимеризация, в которой растущие цепи представляют собой свободные радикалы. В ней могут принимать участие различные мономеры винильного и диенового рядов.
Особенности радикальной полимеризации как цепной реакции:
- свободные радикалы являются чрезвычайно активными частицами, время их жизни 10-6-10-9 с, поэтому макромолекулы образуются практически мгновенно, скорость роста цепи велика;
в каждый данный момент реакционной смеси находятся только мономер и полимер, количество активных цепей мало;
концентрация мономера изменяется постепенно в ходе реакции;
увеличение времени реакции приводит к росту выхода полимера без существенного изменения молекулярной массы.
Основные вопросы, возникающие при изучении процессов радикальной полимеризации:
связь строения и реакционной способности мономеров и
радикалов;
кинетические закономерности процесса и различных факторов на
скорость реакции полимеризации и молекулярную массу полимера.
Следует рассмотреть основные кинетические закономерности элементарных стадий процесса.
Любая цепная реакция включает три основных стадий: зарождение, продолжение и обрыв цепи.
Зарождение цепи (инициирование, образование активного центра) – стадия цепной реакции, на которой образуются свободные радикалы (или другие активные частицы) из валентно-насыщенных молекул исходных веществ (мономеров). Например, если молекулу вещества обозначить АХ, а молекулу мономера – М, то зарождение цепи может быть представлено следующей схемой:
АХ А*+Х,
А*+МАМ*,
где А*- активная частица; АМ*- активный центр.
Активная частица может иметь структуру свободного радикала (R), катиона (К*) или аниона (А-). Соответствующую структуру имеет и активный центр.
Под эффективностью инициирования понимают отношение количества радикалов, вступивших во взаимодействие с мономером, к общему числу образовавшихся радикалов.
С учетом эффективности инициирования уравнение скорости реакции инициирования приобретает вид Vин= k инf[J].
Продолжение (рост) цепи – стадия цепного процесса, идущая с сохранением свободной валентности (структуры активного центра) и сопровождающаяся расходованием исходных веществ (мономеров) и образованием продуктов реакции (полимеров).
При полимеризации рост цепи заключается в последовательном присоединении молекул мономера к активному центру в соответствии со схемой
АМ*+МАММ*,
АММ*+МАМММ*,
и т. д.
Если принять допущение о независимости реакционной способности растущего радикала от его длины, то скорость роста цепи подчиняется выражению
Vp=kp[M*]/[M],
где kp – константа скорости реакции роста цепи; [M*] - концентрация растущих радикалов; [M] - концентрация мономера.
Рост цепи является быстрой реакцией, обычно имеет величину порядка 104 л/мольс.
Энергия активации роста цепи невелика, составляет 20-35 кДж/моль, что в несколько раз меньше энергии активации инициирования перекисными инициаторами.
Обрыв цепи (гибель активного центра) – стадия цепного процесса, на которой исчезают свободная валентность или заряд.
При полимеризации обрыв цепи может происходить различными способами, простейшим из которых в случае радикального процесса является рекомбинация (соединение ) по схеме
А-(М)n-1- М* + *М-(М)n-1-А А-(М)2n-А.
Истинный обрыв цепи приводит к обрыву материальной и кинетической цепи. Эта реакция может произойти на любой стадии полимеризации, что приводит к получению макромолекул различной длины.
Различают два основных способа осуществления обрыва цепи: рекомбинацию и диспропорционирование.
Рекомбинация – это соединение растущих макро радикалов друг с другом с образованием валентной связи.
Если
k![]() –
константа скорости реакции обрыва
рекомбинацией, а [M*]
- концентрация растущих макрорадикалов,
то скорость реакции обрыва подчиняется
уравнению
–
константа скорости реакции обрыва
рекомбинацией, а [M*]
- концентрация растущих макрорадикалов,
то скорость реакции обрыва подчиняется
уравнению
V![]() =
k
=
k![]() [M*]2.
[M*]2.
Диспропорционирование – превращение макрорадикалов в неактивные полимерные молекулы в результате межмолекулярной перегруппировки, передачи атома Н от одного радикала к другому.
Если
k![]() –
константа скорости реакции обрыва
диспропорционированием, а [M*]
- концентрация растущих макрорадикалов,
то скорость реакции обрыва подчиняется
уравнению
–
константа скорости реакции обрыва
диспропорционированием, а [M*]
- концентрация растущих макрорадикалов,
то скорость реакции обрыва подчиняется
уравнению
V![]() =
k
=
k![]() [M*]2.
[M*]2.
При полимеризации большинства мономеров возможен обрыв цепи по обоим механизмам, причем доля реакции обрыва диспропорционированием () тем больше, чем выше реакционная способность растущего макрорадикала.
В общем случае
kоб
=
k![]() +
k
+
k![]() ,
,
тогда
Vоб = kоб [M*]2.
Приведенное выражение характеризует скорость реакции обрыва цепи вследствие бимолекулярного обрыва цепи при радикальной полимеризации.
Обрыв цепи является реакцией, протекающей со скоростью, на несколько порядка более высокой, чем скорость роста цепи. Энергия активации обрыва цепи обычно близка к нулю. Истинный обрыв цепи часто сопровождается обрывом только материальной цепи при сохранении кинетической, т.е. свободнорадикального активного центра. Такие реакции называется реакциями передача цепи.
Инициирование радикальной полимеризации возможно различными способами.
Различают термическое инициирование, фотоинициирование, радиационное инициирование, а также химическое инициирование с помощью специально добавленных веществ – инициаторов.
Термическое инициирование - процесс возбуждения цепного свободнорадикального процесса только под действием тепла, без введения специальных веществ – инициаторов.
Чисто термическое инициирование встречается редко, сопровождается обычно побочными процессами. Механизм реакции исследован недостаточно.
В наибольшей степени исследовано термическое инициирование полимеризации стирола, метилметакрилата.
Термическое инициирование характеризуется высоким значением энергии активации (Еин), низкой скоростью, сильно зависящей от температуры.
Фотоинициирование – процесс образования свободных радикалов из мономерных молекул при действии света.
Молекула мономера, поглотившая квант света, переходит в возбужденное (триплетное) состояние, после чего может распасться на радикалы либо отщепить электрон. Фотоинициирование без добавления специальных веществ (фотоинициаторов) возможно для тех мономеров, которые имеют основную полосу поглощения в ультрафиолетовой области (стирол и его производные, метилметакрилат).
Эффективность фотоинициирования зависит от различных факторов: длины волны ультрафиалетового излучения, строения мономера, наличия растворителей и т.д.
Вседствие возможности дезактивации не все поглощенные кванты вызывает инициирование. Степень использования световой энергии характеризуется коэффициентом эффективности, квантовым выходом фотоинициирования.
Радиационное инициирование – процесс образования свободных радикалов при действии ионизирующего излучения, которое характеризуется более высокими энергиями, чем ультрафиалетовое.
При действии электромагнитного излучения (-кванты, рентгеновские лучи) или радиоактивных частиц (электроны или -частицы, нейтроны, -частицы) происходит возбуждение молекулы мономера М с последующей ионизацией, сопровождающейся выбросом электрона:
М М* М+ + е.
При дальнейшей диссоциации катиона может образоваться радикал:
М* Х+ + Y+.
Выброшенный электрон может присоединяться к катиону с образованием другого радикала:
Y+ + е Y-.
Радиационное инициирование имеет ряд преимуществ по сравнению с другими способами инициирования – высокую чистоту образующихся полимеров, возможность проведения реакции при обычных и низких температурах и т.д. Радиационное инициирование возможно для любых мономеров, способных к полимеризации.
Химическое инициирование – процесс образования свободных радикалов путем термического или фотохимического распада различных соединений, содержащих лабильные связи (инициаторов), или при окислительно-восстановительных реакциях.
Вещества, легко образующие свободные радикалы в результате гомолитического разрыва лабильных связей, называются инициаторами. К их числам относятся соединения различных классов, например перекиси (бензоила, диалкильные, гидроперекиси, неорганические, водорода) характеризуются наличием лабильной связи –О-О-; комплексы металлов переменной валентности (ацетилацетонаты), азо-соединения (динитрил азодиизомасляной кислоты; «диниз», «порофор-N») и др.
Оценим влияние различных факторов на скорость полимеризации и молекулярную массу полимера.
Повышение температуры приводит к увеличению констант скоростей всех элементарных стадий процесса полимеризации. Оно оказывает существенное влияние на стадию инициирования, поскольку ее энергия активации является наибольшей по сравнению с другими стадиями.
Зависимость скорости полимеризации и молекулярной массы полимера от концентрации инициатора определяется «правилом квадратного корня». Чем больше концентрация инициатора, тем выше скорость полимеризации, но ниже молекулярная масса полимера.
С увеличением концентрации мономера повышается скорость полимеризации и увеличивается средняя степень полимеризации.
Обычно с повышением давления повышается скорость и молекулярная масса полимера. Это следует из зависимости констант скоростей элементарных стадий полимеризации от давления. Экспериментальные результаты показывают, что константа скорости полимеризации растет с увеличением давления, скорость инициирования несколько уменьшается, константа обрыва kоб также уменьшается из-за повышения вязкости реакционной среды.
Сополимеризация - процесс совместной полимеризации двух или более мономеров. Получаемые при этом ВМС называются сополимерами. Макромолекула сополимера состоит из двух или более различных составных звеньев, находящихся в том или ином количественном соотношении, определяемом условиями реакции. Можно проводить одновременную сополимеризацию смеси трех и более различных мономеров. Такие процессы полимеризации обычно называют многокомпонентной сополимеризацией. Применительно к системам из трех мономеров употребляется термин терполимеризация.
Сополимеризация позволяет осуществить синтез практически неограниченных числа различных продуктов путем изменения природы и относительных количества звеньев двух мономеров, содержащихся в сополимере.
Большое практическое значение реакций сополимеризации объясняется тем, что сополимеры часто обладают более ценными техническими свойствами, чем полимеры, образованные одним мономером (полистирол, полиэтилен). Кроме того, свойства сополимеров в значительной степени зависят от их состава, что определяет возможность их широкого изменения. Изучение сополимеризации важно также и в кинетическом отношении, так как оно дает значительно более точные сведения о реакционной способности мономеров и радикалов, чем изучение гомополимеризации.
Типы сополимеров. Продукты с типом полимера имеющие структурные звенья двух мономеров распределены вдоль полимерной цепи беспорядочно, называют статистическими сополимерами. Известны еще три другие основные структуры сополимера.
Регулярно чередующийся сополимер содержит звенья обоих мономеров в эквимолярных количествах и с правильным чередованием их вдоль цепи
- М1М2М1М2М1М2М1М2М1М2М1М2М1 -.
Блок-сополимер представляет собой линейный полимер с длинными
последовательностями (блоками) одинаковых звеньев, разделенными блоками других звеньев:
М1М1М1М1М1М1М1М1М1М1М1М1М1М1М1М2М2М2М2М2М2М2 -.
Привитой сополимер, называемый также иногда графт-сополимером, -является разветвленным сополимером, у которого основная цепь состоит из звеньев одного мономера, а к ней присоединены одна или больше боковых цепей, построенных из звеньев другого мономера
М1М1М1М1М1М1М1М1М1М1М1М1М1М1М1М1М1М1М1М1 –
\
-М2М2М2М2М2М2М2 -.
Структурные различия между статистическими, регулярно чередующимися, блочными и привытыми сополимерами можно обозначить соответственно приставками: -со-, -чер-, -б- и –пр-, например поли (метилметакрилат-со-стирол), поли (метилметакрилат-чер-стирол), поли (метилметакрилат-б-стирол), поли (метилметакрилат-пр-стирол).
Наиболее изученной является реакция бинарной радикальной сополимеризации, в которой принимают участие два мономера – М1 и М2, а активные растущие частицы представляют собой свободные радикалы.
Главный вопрос, возникающий при изучении сополимеризации – зависимость состава образующегося сополимера от состава смеси мономеров и условий проведения процесса. Решение его возможно путем анализа уравнений состава сополимера.
В зависимости от природы мономеров, типа инициирующей системы возможно проведение сополимеризации по радикальному и ионному механизмам. Процесс может быть осуществлен в массе мономеров, в эмульсии, суспензии и растворителе.
Известно несколько способов полимеризации:
Полимеризация в газовой фазе.
Блочная полимеризация, или полимеризация в массе.
Полимеризация в растворе.
Эмульсионная полимеризация.
Полимеризация в суспензии.
Полимеризация в твердой фазе.
Полимеризация в газовой фазе. Полимеризацией в газовой фазе называют процессы, в которых мономер находится в газообразном состоянии. Образование полимера начинается на стенах реакционного сосуда и происходит на поверхности или в объеме частиц уже получившегося полимера. Полимеризация в газовой фазе используется, например, для получения натрий-бутадиенового каучука.
Блочная полимеризация. Блочную полимеризацию проводят в массе жидкого мономера при определенном давлении и температуре. Если образующийся полимер растворим в мономере, то постепенно по мере полимеризации вязкость среды увеличивается, и в результате образуется сплошной монолитный блок полимера. Если полимер нерастворим в мономере, то полимер получается в виде порошка или пористого тела.
Блочным способом можно получать полибутадиен, полистирол, полихлоропрен, полиметилметакрилат и другие полимеры. Существенным недостатком этого способа является плохой теплообмен в системе, а так как полимеризация – сильноэкзотермический процесс, то возникают значительные местные перегревы продукта, вследствие чего получается полимер, неоднородный по молекулярному весу. Кроме того, при блочной полимеризации мономер не полностью превращается в полимер, а присутствие мономера вредно сказывается на свойствах полимера.
Полимеризация в растворе. По этому способу в качестве среды применяют растворители (главным образом органические); кроме того, вводят инициатор и регулятор полимеризации. Возможны два варианта этого способа:
1. В растворителе растворяется и мономер, и образующийся полимер. При полимеризации получается раствор полимера – лак, поэтому и способ называется «лаковым». Такой лак можно непосредственно использовать для получения покрытий или выделить полимер из раствора осаждением.
2. Мономер растворяется в жидкости, в которой нерастворим образующийся полимер. По мере образования полимер выпадает в осадок, который затем отделяют от жидкости.
При полимеризации в растворе образуются полимеры меньшего молекулярного веса, однако этот способ обладает рядом преимуществ по сравнению с блочной полимеризацией: интенсивное перемешивание существенно улучшает теплообмен, поэтому получаемый полимер более однороден по молекулярному весу и, кроме того, значительно легче отмывается от мономера.
Эмульсионная полимеризация. При полимеризации в эмульсии жидкий мономер диспергируется в не смешивающейся с ним жидкости, образуя эмульсию. Обычно в качестве дисперсионной среды применяется вода. Эмульсии термодинамически неустойчивы и поэтому в случае концентрированных эмульсий в систему вводят эмульгатор. Эмульгаторы – это поверхностно-активные вещества, адсорбирующиеся на поверхности раздела двух фаз (вода - мономер). Роль эмульгатора сводится к образованию механически прочных адсорбционных слоев, предотвращающих слияние (коалесценцию) капель мономера или полимера. Поэтому в качестве эмульгаторов обычно применяются вещества, содержащие полярную группу и сравнительно большой углеводородный радикал. К таким веществам относятся мыла (соли высших органических кислот), органические сульфокислоты и их соли и т.д.
Эмульсии полимера в воде, полученные в результате эмульсионной полимеризации, называются синтетическими латексами. Латексы применяют непосредственно или выделяют из них полимер обычными способами коагуляции коллоидных систем. На практике коагуляцию латексов производят различными электролитами.
Гетерогенная полимеризация в суспензии широко используется для синтеза различных полимеров. Таким способом получают полтвинилхлорид, полистирол, полиметилметакрилат, поливинилацетат. Этот процесс часто называют биссерной, жемчужной или гранульной полимеризацией. При полимеризации по этому способу мономер диспергируют в воде в виде мелких капелек, диаметр которых 0,01-0,5 см. Устойчивость дисперсии достигается механическим перемешиванием или введением в реакционную систему специальных добавок (стабилизаторов).
Для полимеризации в суспензии применяются инициаторы, растворимые в мономере. Такие инициаторы часто называют малорастворимыми катализаторами. Преимуществом полимеризации в суспензии является хороший отвод тепла. Недостаток этого способа состоит в том, что полимер может быть загрязнен остатками стабилизатора, а также необходима промывка и сушка полученных гранул полимера.
Полимеризация в твердой фазе. Многие мономеры способны полимеризоваться не только в жидкой фазе, но и в кристаллическом состоянии, ниже температур плавления. Такую полимеризацию называют твердофазной. Чаще всего ее инициируют, облучая кристаллы мономера рентгеновскими или -лучами, быстрыми электронами и другими частицами высокой энергии. Кристаллическая решетка мономера может оказывать влияние на скорость роста цепей, на строение образующихся макромолекул и на их взаимную упаковку.
При приближении к температуре плавления (даже если она весьма низка) скорость полимеризации твердого мономера резко возрастает и часто оказывается больше скорости полимеризации этого же мономера в жидкой фазе при существенно более высоких температурах. Полагают, что это обусловлено возникновением в кристаллах вблизи температуры плавления так называемых «лабильных заготовок», т.е. коллективов мономерных молекул, обладающих достаточной подвижностью, но сохранивших упорядоченное взаимное расположение. Сочетание двух последних качеств, способствует быстрому росту цепей в заготовках.
В отдельных случаях кристаллы мономера при полимеризации непосредственно превращаются в пучки высокоориентированных полимерных волокон (например, кристаллический триоксан).
Ионная полимеризация – процесс, в котором элементарный акт роста цепи является гетеролитическим. Разрыв двойной связи может происходить под влиянием излучений и различных веществ – катализаторов ионной полимеризации, образующих ионы
А- + СН2=CH ACH2-CH-;
R R
K+
+ CH2-CH2
KOCH2
– CH2
–CH![]() .
.
CH2-O
Классы веществ, способных к ионной полимеризации, очень разнообразны: винильные и диеновые мономеры, карбонильные группы (C=O), неустойчивые гетероциклические соединения и т.д. В целом ионные процессы являются более универсальными, чем радикальные.
Закономерности ионной полимеризации изучены недостаточно, и до настоящего времени нет единой теории процесса. Это связано главным образом со сложностью и многообразием влияния различных факторов. Так, катализаторы, возбуждающиеся ионную полимеризацию, влияют не только на стадию инициирования, а в значительной степени и на стадию роста (влияние противоиона). Растворитель или любая добавка, присутствующая в системе, существенно воздействуют на активные центры, ведущие процесс, чем иногда приводит к изменению его механизма. Процесс ионной полимеризации в большинстве случаев являются цепным, проходящим в три стадии: инициирование (образование активного центра), рост цепи и ограничение роста цепи. В отличие от радикальных процессов здесь на стадии инициирования возможно образования нескольких центров полимеризации разной активности. Обрыв является более сложным, зависит от типа противоиона, и иногда вообще не имеет места, полимеризация протекает вплоть до израсходования мономера (безобрывные процессы).
Катионной полимеризацией называется процесс получения ВМС, при котором растущая цепь имеет положительный заряд, являясь катионом.
Катализаторы катионной полимеризации: протоны, карбкатионы или другие электрофильные частицы – кислоты Льюиса. Можно выделить несколько групп веществ, являющихся катализаторами:
протонные кислоты: H2SO4, H3PO4, CF3COOH и др.;
апротонные
кислоты (реагенты Фриделя-Крафтса) общей
формулы МХn,
где М – металл, а Х - галоген (ВF3,
A![]() C
C![]() 3,
FeC
3,
FeC![]() 3,
SnC
3,
SnC![]() 3,
TiC
3,
TiC![]() 4,
ZnC
4,
ZnC![]() 2,
и др.) или смешанные галогениды;
2,
и др.) или смешанные галогениды;
галогены:
I2,
IC![]() ,
IBr;
,
IBr;
соли
карбония phC+A-,
где А – C![]() ,
SbC
,
SbC![]() 6
и др.;
6
и др.;
соли
оксония типа RO+A-,
где А - BF3(OH),
SbC![]() 6,
и др.;
6,
и др.;
алкилпроизводные металлов R2Al, R2Zn.
Единого механизма катионной полимеризации предположить нельзя. Это связано с многообразием активных центров, образующихся на стадии инициирования и сосуществующих в реакционной массе, с разной их активностью на стадии роста и своеобразием ограничения цепи.
Некоторые общие замечания позволяют лишь приблизительно подойти к уравнению скорости процесса.
Инициирование включает все реакции, приводящие к образованию активного центра. В зависимости от типа катализатора, полярности мономера и среды процесс может идти по-разному.
Процессы инициирования можно подразделить на типы в зависимости от компонентов, участвующих в образовании активных центров: катализаторы; катализатор-сокатализатор, обычно комплекс галогенида с донорной добавкой; катализатор-мономер; катализатор-сокатализатор-мономер.
С другой стороны, можно классифицировать катионные процессы по структуре активных центров, независимо от условий и механизма их образования. Так, можно выделить несколько типов активных центров: свободные катионы (К+) или действующие в ионной паре; цвиттер-ионы (К+-А-); ион радикалы К+; координационные комплексы, не имеющие ярко выраженного ионного характера.
Следует учитывать, что наряду с реакциями комплексообразования имеют место сольватация образующихся частиц молекулами мономера и растворителя, находящимися в большом избытке по отношению к катализатору.
Таким образом, строение активного центра, следовательно, и его активность определяются не только природой катализатора, а зависит также от свойств реакционной среды и температуры реакции.
Изомеризация приводит к образованию нового центра полимеризации, отличающегося меньшей активностью, что должно отразиться на кинетике роста цепи.
Увеличение полярности среды увеличивает скорость инициирования и снижает скорость обрыва цепи. Большое влияние имеет сольватация. Растворитель, способный к комплексообразованию (сольватация), может изменить активность центра роста.
Стабилизация за счет взаимодействия с молекулами среды компенсирует энергетические затраты при диссоциации и повышает устойчивость активных центров. Эффект стабилизации активных центов способствует увеличению молекулярной массы полимеров.
Процессы полимеризации, в которых растущая цепь представляет собой отрицательно заряженную частицу (анион), называют анионной полимеризацией.
Катализаторами, инициирующими этот процесс, являются частицы, несущие отрицательные заряды определенной активности. Образование анионов происходит при диссоциации катализатора (АК) по уравнению, которое моделирует и состояние активных центров растущей цепи:
АК
![]() А-,
К
А-,
К
![]() А
А![]() +
К+.
+
К+.
Из приведенной схемы видно значительное сходство в процессах катализа катионной и анионной полимеризации. Оно заключается в многообразии активных центров, ведущих процесс, в значительной роли среды и противоиона в кинетике и механизме процесса. Основные катализаторы, возбуждающие анионную полимеризацию, - это амиды металлов, алкоголяты, алкилы и арилы металлов, гидроокиси и цианиды металлов.
По анионному механизму полимеризуются ненасыщенные соединения винильного и диенового ряда, карбонилсодержащие мономеры, гетероциклические соединения – окиси, лактоны, лактамы, силоксаны и др. Наиболее активны в анионной полимеризации непредельные мономеры, двойная связь которых обеднена электронной плотностью, т.е. имеющие электроноакцепторные заместители. Поэтому этилен трудно, а олефины совсем не полимеризуются по типично анионному механизму.
Анионная полимеризация развивается по цепному механизму, в котором можно выделить три этапа; инициирование, рост и обрыв цепи.
На каждой стадии присоединения молекулы мономера образуется металлорганические соединение, содержащее поляризованную металлуглеродную связь. Они различаются лишь молекулярной массой. Относительная устойчивость промежуточных металлорганических соединений в углеродной среде объясняет типичную для анионной полимеризации безобрывность процесса.
Промежуточные соединения можно выделить в том случае, если скорость роста цепи устойчивость активных центров приводит и к другой очень важной характеристике процесса – рост цепей продолжается достаточно долго, что приводит к значительному возрастанию степени полимеризации в ходе процесса.
Достаточно устойчивые растущие полимерные анионы называют «живущими» полимерами. При добавлении к такому полимеру новой порции мономера полимеризация возобновляется. Это открывает очень большие перспективы – получение блок-сополимеров добавлением мономеров, способных полимеризоваться на «живых» концах.
Кинетика анионной полимеризации зависит от типа катализатора и исследована лишь для простейших систем. Так, например, можно составить кинетическое уравнение при полимеризации стирола, инициированной амидом калия.
Основная литература [2] (120-264), [3] (8-49, 59-114)
Контрольные вопросы:
1. Общие принципы и подходы к получению ВМС.
2. Дайте понятие о полимеризации и поликонденсации.
3. Назовите основные типы мономеров, вступающих в реакции полимеризации.
4. Что такое цепная и ступенчатая полимеризация?
5. Расскажите о природе активного центра.
6. Расскажите о закономерности радикальной полимеризации.
7. Изложите стадии процесса.
8. Назовите типы инициирования радикальной полимеризации.
9. Перечислите факторы, влияющие на процесс радикальной полимеризации.
10. Расскажите о закономерностях радикальной сополимеризации, механизме и кинетике процесса.
11. Назовите типы сополимеров.
12. Изложите технические способы проведения полимеризации и сополимеризации.
13. Что такое блочная полимеризация, полимеризация в растворе.
14. Дайте понятие эмульсионная (латексная) и суспензионная (гранульная) полимеризация.
15. Изложите закономерности ионной полимеризации.
16. Дайте понятие катионная полимеризация.
17. Расскажите о природе активного центра, используемых мономеров и катализаторов, механизме и кинетику процессов.
18. Изложите анионную полимеризацию, понятие о «живых цепях».
19. Укажите возможности получения сверхвысокомолекулярных монодисперсных полимеров и блоксополимеров в процессах анионной полимеризации.
Тема лекции №10: Получение ВМС методом поликонденсации
Поликонденсацией называется процесс соединения нескольких молекул одинакового или различного строения, сопровождающийся, как правило, выделением простейших НМВ. Исходные мономеры должны содержать в молекуле не менее двух функциональных групп (группы ОН, СООН, NН2 и др.). Функциональность исходных соединений оказывает большое влияние на строение и свойства получаемых полимеров. Так, монофункциональные соединения образуют лишь НМ продукты. При поликонденсации бифункциональных соединений получаются линейные или циклические ВМ продукты, а из три- и тетрафункциональных соединений – полимеры пространственного строения. Для синтеза полимеров поликонденсацией могут быть использованы самые различные химические реакции. К ним относятся: этерификация, амидирование, образование уретанов, ароматическое замещение и другие. Исследование закономерностей процессов поликонденсации привело к установлению двух типов поликонденсационных реакций – равновесной и неравновесной.
По характеру мономеров поликонденсацию можно разделить на два типа. К первому типу относятся реакции с использованием двух различных полуфункциональных мономеров, каждый из которых содержит только один тип функциональных групп. Ко второму типу относятся реакции с участием одного мономера, содержащего оба типа функциональных групп.
На примере синтеза полиамидов можно проиллюстрировать оба типа реакций. Так, полиамиды можно получить взаимодействием диаминов с дикарбоновыми кислотами
nH2N-R –NH2 + nHOOC –R’-COOH H-(-NH-R-NHCO-R’-CO-)n-OH + (2n-1)H2O
или самоконденсацией аминокислот
nH2N-R –COOH H-(-NH-R-CO-)n-OH + (n-1)H2O.
Схематически оба типа реакций могут быть представлены следующими уравнениями: nA-A + nB-B –(-A-AB-B-)n-,
nA-B –(-A-B-)n-,
где А и В – функциональные группы различного вида. Закономерности указанных двух типов поликонденсации очень схожи между собой. Для синтеза ВМ полимеров (т.е. полимеров достаточно высокого молекулярного веса, чтобы быть практически полезными) необходимы те же самые условия и предосторожности.
Конденсация монофункциональных веществ, приводит к образованию только НМ продуктов, поскольку при таком взаимодействии получаются вещества, лишенные функциональных групп. Например, реакции между этиловым спиртом и уксусной кислотой происходит с образованием этилацетата:
С2Н5ОН + СН2СООН С2Н5-О-С-СН3 + Н2О.
О
Взаимодействие бифункциональных соединений приводят к образованию линейных полимеров. В результате каждого акта взаимодействия образуется продукт, имеющий на концах функциональные группы и способный поэтому к дальнейшему взаимодействию. Бифункциональные вещества могут обладать функциональными группами одинакового или различного строения. Например, взаимодействие этиленгликоля и терефталевой кислоты приводит к образованию линейного полимера – полиэтилентерефталата. Процесс происходит по стадиям:
НО-С2Н4 -ОН + НООС-С6Н4СООН Н2О + НО-С2Н4-О-С-С6Н4 –СООН;
О
образование более высокомолекулярных продуктов:
НО-С2Н4-О-С-С6Н4 –СООН + НО-С2Н4-О-С-С6Н4 –СООН
О О
Н2О + НО-С2Н4-О-С-С6Н4 –С--О-С2Н4-О-С-С6Н4 –СООН и т.д.
О О О
Бифункциональные вещества могут содержать в одной молекуле функциональные группы различного строения. Трифункциональные и тетрафункциональные вещества, а также их смеси с бифункциональными соединениями образуют при поликонденсации, разветвленные или трехмерные продукты. При этом обычно первой стадией реакции является образование НМ олигомерных продуктов линейного и разветвленного строения, способных при углублении процесса переходить в сшитые трехмерные структуры. Например, конденсация глицерина с фталевой кислотой протекает по схеме:
образование димера:
СН2–СН–СН2 + НООС–С6Н4-СООН НО-СН2–СН-СН2-О-С-С6Н4-СООН + Н2О;
ОН ОН ОН ОН О
образование разветвленных продуктов; образование трехмерных структур из разветвленных продуктов, полученных в соответствии с предыдущей стадией.
Конденсацию
проводят при высоких концентрациях
исходных веществ, что благоприятствует
образованию линейного продукта.
Циклизация – мономолекулярная
(внутримолекулярная) реакция, в то время
как линейная поликонденсация – это
бимолекулярная (межмолекулярная)
реакция.
С увеличением концентрации исходных
веществ, скорость последней возрастает
значительно быстрее, чем скорость первой
реакции. Таким образом, фактор концентрации
реагентов накладывается на кинетический
и термодинамический факторы, благоприятно
влияя на протекание линейной
поликонденсации. Именно этим объясняется
отсутствие в реальных поликонденсационных
системах колец с числом членов более
12: циклизация практически не идет, когда
при поликонденсации возможно образование
шести- или семичленных колец. Другим
обстоятельством, которым можно
воспользоваться для смешения равновесия
в сторону линейной поликонденсации,
является невозможность в определенных
условиях превращения линейных структур
в циклические и наоборот. Это позволяет
сдвинуть равновесие между двумя
структурами в сторону образования
линейного полимера. Интересно заметить,
что циклизация практически полностью
исключает линейную реакцию, если возможно
образование пятичленных циклов. Как
правило, такая циклизация идет с большей
скоростью, чем линейная поликонденсация
других исходных веществ, где отсутсвует
циклизация. Однако причины такого
поведения непонятны. (Пятичленные циклы,
которые ранее считались совершенно
инертными, в некоторых случаях
полимеризуются с раскрытием цикла).
Реакция поликонденсации обратима.
Это значит, что одновременно протекают
два процесса: образование продуктов
конденсации и их деструкции. Деструкция
может протекать под действием
функциональных групп исходных мономеров,
а также в результате взаимодействия
продукта конденсации с НМ побочными
продуктами реакции (Н2О,
НС![]() и др.).
и др.).
Вследствие обратимости процесса поликонденсации при некоторых условиях устанавливается равновесие, которому соответствует образование полимерного продукта с определенным молекулярным весом. Как правило, продукты поликонденсации имеют меньший молекулярный вес (20 000 – 50 000) по сравнению с продуктами полимеризации. Для получения полимеров более высокого молекулярного веса необходимо, во-первых удалять из системы НМ побочные продукты и, во-вторых, применять строго эквивалентные соотношения функциональных групп (т.е. исходных веществ). Избыточное количество одного из исходных веществ, всегда приводит к образованию НМ продуктов. Так, при взаимодействии фенола или крезола с альдегидами при десятикратном избытке фенола образуется только диоксифенилметан (ВМ продукты не образуются).
Необратимый процесс – межфазная поликонденсация, которая проходит на границе раздела фаз (жидкой фазой служат вода, а другой – органическое вещество).
Основные принципы уменьшения функциональности по сравнению с теоретической следующие:
влияние условий проведения процесса на реакционную способность функциональных групп;
возможность образования циклов;
возможность химического изменения функциональных групп мономера в процессе различных реакций.
Для определения соотношения между циклизацией и поликонденсацией следует учитывать два фактора: термодинамический (т.е. термодинамическую устойчивость цикла) и кинетический.
Термодинамическая устойчивость циклов зависит от числа атомов в цикле, поэтому для бифункциональных мономеров большое значение имеет количество СН2-групп (или других групп атомов) между функциональными группами. Зависимость между размером цикла и его устойчивостью была установлена на основе анализа теплот сгорания ряда циклических соединений. Термодинамическая устойчивость уменьшается с увеличением напряженности цикла. Наиболее напряженными являются 3- и 4-членные циклы, при переходе к 5-, 6- и 7-членным циклам напряжение резко уменьшается, снова увеличивается для 8-11-членных циклов и уменьшается при переходе к циклам больших размеров вследствие изменения их пространственной структуры. Таким образом, ряд термодинамической устойчивости циклов следующий: 3, 4, 8-11<7, 12 и выше <5<6.
Кинетическая разрешимость циклизации зависит от возможности сближения двух функциональных групп, находящихся на разных концах молекулы. Чем меньше вероятность их встречи, тем ниже кинетическая разрешимость циклизации. Обычно при поликонденсации циклические продукты в той или иной степени сопутствуют полимерным процессам.
Если строение исходных мономеров таково, что существует возможность образования 5-членных циклов, то процесса поликонденсации не происходит, а наблюдается только циклизация. Например, -аминомасляная или -оксимасляная кислоты при конденсации образуют только пирролидон и бутиролактон соответственно:
H2N–(CH2)3-COOH H2O+CH2-CH2.
CH2 C=O
\ /
NH
При возможности образования 6 - 7-членных циклов поликонденсация и циклизация протекают одновременно.
При наличии между функциональными группами 8 и более групп атомов реакция поликонденсации преобладает над циклизацией, хотя последная и не исключается полностью. Известны случаи образования в процессе поликонденсации циклов с числом атомов 20 и более.
Термодинамический и особенно кинетический факторы циклизации определяются концентрацией мономеров. Чем выше концентрация мономеров, тем в большей степени возрастает скорость биомолекулярной реакции. Следовательно, повышение концентрации мономера способствует поликонденсации и предотвращает циклизацию. Повышение температуры процесса приводит к увеличению роли циклизации, поскольку энергия активации циклизации выше энергии активации поликонденсации.
Химическое изменение функциональных групп происходит также в результате циклизации.
Все эти факторы приводят к тому, что практическая величина средней функциональности обычно ниже теоретической. Реакционная способность мономеров в процессе поликонденсации определяется реакционной способностью функциональных групп.
Для достижения высокой молекулярной массы полимера необходимо, чтобы функциональные группы обоих типов содержались в реакционной смеси в равных количествах, т.е. соотношение мономеров должно быть эквивалентным. Действительно, при избытке одного из исходных веществ, процесс поликонденсации протекает до тех пор, пока не будет израсходован мономер, присутствующий в меньшем количестве. В этот момент все макромолекулы будут содержать на обоих концах одинаковые функциональные группы, такие же, как у избыточного компонента. Это приводит к остановке процесса поликонденсации.
Влияние глубины проведения процесса на молекулярную массу полимера характерно для процессов поликонденсации в отличие от реакций цепной полимеризации.
Подобно большинству химических реакций, поликонденсация протекает с большей скоростью при более высокой температуре. Величина энергии активации реакций равновесной поликонденсации составляет 83,8-167,6 кДж/моль.
Повышение температуры ускоряет приближение системы к равновесию, облегчает удаление выделяющегося низкомолекулярного продукта реакции, в результате чего равновесие смещается в сторону образования более высокомолекулярных полимеров. На определенных промежуточных стадиях реакций до достижения системой равновесия молекулярная масса полимера выше при более высокой температуре, но после достижения равновесия молекулярная масса выше при более низкой температуре. Это используется для сокращения времени поликонденсации: сначала реакцию проводят при более высокой температуре , а затем после достижения состояния равновесия – при более низкой.
При полимеризации могут реагировать только мономеры с растущей полимерной цепью, при поликонденсации могут вступать во взаимодействие любые две частицы, находящиеся в системе. При поликонденсации мономер исчезает значительно быстрее, чем при полимеризации, так как первый процесс идет медленно с постепенным переходом от мономера к димеру, тримеру, тетрамеру и т. д. Молекулярный вес возрастает на всем протяжении реакции поликонденсации, причем высокомолекулярный полимер не удается получить, не достигнув высокой степени завершенности реакции. Поликонденсацию необходимо проводить в течение большого промежутка времени для получения полимера с высоким выходом и высоким молекулярным весом.
Поликонденсацию можно проводить различными способами, главными из которых являются поликонденсация в твердой фазе, в расплаве, в растворе.
Поликонденсация в твердой фазе проводится при температуре, меньшей темпеатуры плавления мономеров и полимера на 5-100С. Процесс имеет большое значение для мономеров, разлагающихся при плавлении. Реакция носит автокаталитический характер, ее скорость увеличивается со временем. Скорость реакции зависит от величины кристаллов мономера и возрастает с ростом размера кристаллов. Закономерности поликонденсаци в твердой фазе изучены недостаточно.
Поликонденсация в расплаве является наиболее широко распространенным процессом. Она проводится, если полимер и мономеры устойчивы при температуре плавления (2000С). Для предотвращения окисления мономеров и термической деструкции полимера процесс проводят в токе инертного газа. Завершающую стадию осуществляют под вакуумом для облегчения удаления низкомолекулярных продуктов реакции.
Проведение процесса в расплаве имеет ряд преимуществ перед другими способами поликонденсации. Технологическая схема процесса сравнительно проста, обеспечивается возможость применения мономеров с пониженной реакционной способностью. Недостатки метода - необходимость использования мономеров, устойчивых при высоких температурах, а также трудность создания и поддержания высокой температуры реакции.
Поликонденсация в растворе – способ проведения процесса, при котором мономеры находятся в жидкой фазе в растворенном состоянии. При этом образующийся полимер может быть полностью или частично растворим или нерастворим в реакционной среде.
Особенности поликонденсации в растворе заключаются в возможности осуществления процесса в мягких, легко контролируемых условиях, этот способ может быть использован для поликонденсации мономеров, разлагающихся при плавлении. Недостаток процесса – необходимость регенерации растворителя, использование полимера только в виде раствора или введение дополнительной стадии выделения полимера.
Полимерами наиболее распространенных реакций равновесной поликонденсации являются полиэтерификация (взаимодействие дикарбоновых кислот с гликолями) и полиамидирование (взаимодействие дикарбоновых кислот с диаминами).
Основная литература [2] (288--321), [3] (114-157)
Контрольные вопросы:
1. Изложите общие закономерности поликонденсации.
2. Назовите типы реакции, наиболее типичные реакции линейной и трехмерной поликонденсации.
3. Расскажите основные закономерности процессов обратимой и необратимой поликонденсации.
4. Расскажите о влиянии различных факторов на процесс поликонденсации.
5. Изложите основные отличия реакции полимеризации и поликонденсации.
6. Укажите технические способы проведения поликонденсации: поликонденсация в расплаве, в растворе, в эмульсии, на границе раздела фаз (межфазная) поликонденсация.
Тема лекции №11: Химические превращения полимеров
Химические превращеиия полимеров условно подразделяются на следующие типы:
-реакции в боковых или основных цепях макромолекул, не приводящие к изменению их длины (полимераналогичные превращения);
-реакции деструкции, сопровождающиеся разрывом макромолекул под действием различных факторов (нагревание, облучение и т.п.) или химических агентов;
-реакции, приводящие к соединению (сшивание) макромолекул друг с другом с образованием разветвленной или пространственной сетчатой структуры трехмерного полимера.
Условность этого деления химических превращений полимеров состоит в том, что в реальных случаях каждый из указаннных трех типов реакций в чистом виде встречается сравнительно редко. Чаще полимераналогичные превращения сопровождаются частичной деструкцией цепей или их сшиванием, а многие процессы идут с одновременной деструкцией и сшиванием макромолекул (например, окисление), причем соотношение этих реакций можно регулировать изменением условий.
Неполное превращение в молекулярной реакции может быть следствием стерических затруднений, а в некоторых случаях (когда в реакции участвуют две соседние группы) обусловливается и статическими причинами – образование изолированных функциональных групп.
Основными причинами несоблюдения принципа равной реакционной способности, обозначаемыми суммарно как «полимер–эффект», являются: доступность функциональных групп; влияние соседних групп; конфигурационный и конформационный эффекты; электростатический эффект; надмолекулярный или морфологический эффект; кооперативный эффект; эффект негомогенной активности; влияние длины цепей и концентрации. В чистом виде каждый из перечисленных эффектов в полимераналогичных реакциях появляется редко – обычно реакционная способность функциональных групп определяется совокупностью нескольких из них.
Доступность функциональных групп или «эффект цепи» – обусловлена прежде всего стерическими препятствиями, которые существенно выше при наличии объемистых группировок в полимерной цепи по сравнению с теми же группировками в НМ субстрате.
Влияние соседних групп или «эффект соседа» – наиболее часто наблюдаемый эффект в реакциях полимераналогичного превращения, который может как ускорять, так и замедлять реакции функциональных групп, соединенных в основной цепи макромолекул.
Часто влияние соседних групп проявляется через внутримолекулярные взаимодействия. Так, при омылениии ряда алкилметакрилатов вскоре после начала реакции наблюдается явление самоускорения: вслед за первоначальным образованием карбоксилатных анионов гидролиз сложноэфирных групп идет не в результате их атаки гидроксидным ионом, а под действием соседних карбоксилатных анионов, причем гидролиз осуществляется через промежуточные образования ангидридных циклов.
В случае полимеров интерпретация «эффект соседа» часто осложняется, так как в этом случае приходится учитывать три типа триад, отличающихся окружением реакционноспособного звена:
-ХХХ-, -АХХ- или –ХХА-, -АХА-,
1 2 3
где Х – исходное, А – прореагировавшее мономерное звено. В общем случае коснтанты скорости реакции Х в различном окружении не равны, т.е. k1k2k3. Конечный результат, т.е. предельная глубина превращения Х и время ее достижения зависят от соотношения между k1, k2 и k3.
Вопросы деструкции имеют большое значение для перестройки полимеров и эксплаутации готовых полимерных изделий. Процесс перестройки – это не только придание материалу определенной формы, т.е. определенной модификации. Это создание качества, которое в значительной степени зависит от химических процессов, протекающих под влиянием различных воздействий: механических, нагревания, кислорода воздуха, света и т.д.
Впервые термин «полимераналогичные превращения» как химическую реакцию на полимере, протекающую без изменения степени полимеризации, ввел в химию полимеров в 1934 г. Г. Штаудингер.
В общем полимераналогичное превращение можно предоставить схемой
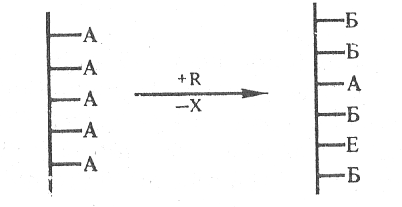
В которой А и Б - исходная и конечная функциональные группы; R – реагент; Х – побочный низкомолекулярный продукт реакции; Е – группа, образовавшаяся вследствие побочного превращения.
В реакции полимераналогичных превращений могут вступать соединения с самыми разнообразными функциональными группами, например:
О О О О О
-С–Н -С-Cl -С-NН2 -С–NН-NH2 -С-NН-OH -SO2Н -SO2Сl -SO2F и др.
На этом методе основаны промышленные способы получения многих полимеров. Различные эфиры целлюлозы (ацетат целлюлозы, нитрат целлюлозы и др.) получаются в промышленности путем ее этерефикации. При этом гидроксильные группы целлюлозы замещаются на – ОСОСН3 и – ОNО2. Помимо известных простых и сложных эфиров целлюлозы, в последнее время получены фениловые эфиры целлюлозы [С6Н7О2(ОН)2ОС6Н5]n, эфиры целлюлозы с аминокислотами [С6Н7О2(ОН)2ОСОС6Н4NН2]n, с фосфорсодержащими кислотами и др.
В результате многих полимераналогичных реакций превращения боковых функциональных групп приводят к изменению строения основной цепи; чаще (но не всегда!) это происходит в результате процессов циклизации.
Циклизация
при химическом превращении возможна и
с участием НМ реагента, например при
взаимодйствии поливинилхлорида с
толуолом в присутствии А![]() С
С![]() 3.
3.
Образование циклов в основной цеи может прозойти и в результате межфрагментной реакции звеньев сополимеров. Так, сополмер стирола и прозводных акриловой кислоты в условиях реакции Фриделя-Крафтса циклизуется с участием ароматического стирольного звена.
Взаимодействия функциональных групп в боковых цепях могут приводить к образованию системы с конденсированными циклами.
Образование циклических структур возможно в диеновых полимерах также и при нагревании в присутствии кислот или хлорида металлов переменной валентности, например, в случае полиизопрена.
Полимераналогичные превращения широко используют для модификации трехмерных полимеров с целью введения в их составные повторяющиеся звенья тех или иных функциональных групп. При этом изменяется лишь молекулярная масса СПЗ, но не изменяется число звеньев во фрагментах между узлами сетки. Химические превращения применяют при получении ионнообменных и хроматографических полимерных материалов, гетерогенных полимерных катализаторов и носителей для иммобилизации биологически активных веществ.
Химические превращения трехмерных полимеров в более широком смысле – это гетерогенные реакции в двухфазных системах: фазы набухшего полимера (полимерная фаза) и жидкой фазы растворимого реагента. Процесс включает стадию диффузии реагента из раствора в полимерную фазу и стадию взаимодействия реагента с активной группой полимера.
Скорость химического превращения трехмерного полимера может лимитироваться:
- внешней диффузией, т.е. диффузией реагента в растворе к поверхности полимерной частицы;
- внутренней диффузией, т.е. перемещением реагента в полимерной фазе к месту взаимодействия с функциональной группой;
- собственно химической реакцией.
На практике превращения чаще проводят при значительном избытке реагента в жидкой фазе, что позволяет исключить вероятность внешнедиффузионного лимитирования и расматривать лишь стадии диффузии реагента внутри полимерной фазы и его химического взаимодействия.
Исследования полимераналогичных превращений показали, что они существенно отличаются от соответствующих превращений НМВ. Так как реакционноспособные группы являются частью макромолекулы, нельзя отделить целевые продукты от исходного вещества, часто не удается достигнуть полного превращения, а побочные реакции протекают на той же макромолекуле.
Основная литература [2] (378-387), [3] (158, 162)
Контрольные вопросы:
1. Назовите типы реакции полимеров.
2. Укажите основные отличия реакции полимеров от реакции низкомолекулярных соединений.
3. Что такое «Эффект цепи» и «Эффект соседа» в химических реакциях полимеров?
4. Изложите возможности химической модификации.
5. Расскажите о химических реакций, не приводящие к изменению степени (коэффициента) полимеризации макромолекул (Полимеранологичные превращения).
Тема лекции №12: Сшивание. Структирование. Деструкция
В результате химических превращений макромолекул между ними могут образоваться поперечные связи, при этом система линейных цепных молекул образует трехмерную полимерную сетку, не способную к растворению или течению. Процессы соединения макромолекул в сетчатую структуру (сшивание) лежат в основе многих промышленных методов производства изделий из полимеров, в частности производства резинотехнических изделий путем вулканизации композиций на основе каучуков.
Соединение линейных макромолекул посредством химических связей может быть осуществлено через многочисленные превращения, которые в общем виде можно разделить на следущие основные типы:
-взаимодействие функциональных групп макромолекул различных полимеров;
-реакции функциональных групп одного и того же полимера;
-реакция макромолекул с полифункциональным НМС (вулканизующим агентом).
Естественно, что осуществление необходимого для сшивания взаимодействия функциональных групп требует соответствующего внешнего воздействия – нагревания, использования катализаторов, УФ- или радиационного облучения.
Реакции разных полимеров друг с другом в принципе, возможны, если они содержат способные к взаимодействию функциональные группы. Например, при взаимодействии сополимеров винилового спирта с полиакриловой кислотой образуются поперечные сложноэфирные связи:
НО
\
СН-ОН
+ С-СН
![]() СН-О-С-СН
СН-О-С-СН
СН2 О СН2 СН2 О СН2
Сшивание за счет реакциий функциональных групп одного и того же полимера. Участвующие в образовании поперечных связей функциональные группы могут быть одинаковыми, например гидроксильными в случае полисилоксанов:
СН3-Si-OH + HO-Si-CH3 CH3 Si-O-Si-CH3
O O O O
Сшивание полимеров реакцией с низкомолекулярными полифункциональными агентами – наиболее распространенный метод превращения линейных полимеров в трехмерные полимеры. Первым из них является открытый более 150 лет тому назад (Гудьир, 1839) процесс вулканизации натурального каучука серой; этот метод в различных модификациях до сих пор широко используют в промышленности для производства эластомеров массового применения (резины).
Поперечные химические связи в полимерах могут образовываться непосредственно между атомами углерода соседних макромолекул без других веществ или при помощи различных соединений, специально вводимых в систему. Такие вещества называются вулканизующими или отвердителями. На практике реакции вулканизации и отверждения проводят обычно при нагревании. В настоящее время известны системы полимеров – вулканизующий агент, отверждающиеся с высокими скоростями и на холод (холодная вулканизация, холодное отверждение). Для придания природным и синтетическим каучукам пространствнной структуры используют взаимодействие каучуков со специальными вулканизующнми атомами (обычно с серой).
При взаимодействии серы с двумя макромолекулами каучуки происходит присоединение серы по месту раскрытия двойных связей с образованием так называемого серного мостика:
СН3 СН3
…-СН2-С=СН-СН2-… …-СН2-С=СН-СН2-…
+ 2 S S S
…-СН2-С=СН-СН2-… …-СН2-С=СН-СН2-…
СН3 СН3
В качестве вулканизующих агентов могут применяться также селен, теллур, кислород.
Вулканизация полибутилена отличается от вулканизации натурального каучука.
При взаимодействии макромолекул бутадиенового каучука с кислородом воздуха в результате реакций сшивания образуются кислородные мостики между винильными группами.
В промышленности пластических масс для получения твердых изделий (т.е. с частой простанственной) отверждают главным образом продукты поликонденсации линейного строения невысокого молекулярного веса. Такие продукты при повышенных температурах представляют собой жидкости, переходящие при охлаждении в хрупкий твердый материал. Полученный материал измельчается в так называемый прессовочный порошок и загружается в форму. При нагревании этого порошка протекают реакции сшивания, в результате которых образуется твердый неплавкий и нерастворимый продукт. Так, феноло-формальдегидные полимеры при нагреванни легко переходят из резольной формы в резитолы – эластичные продукты с редкой пространственной сеткой, которые при дальнейшем нагревании превращаются в резиты – твердые продукты с частой сеткой.
В качестве отвердителей для феноло-формальдегидных смол применяют, например, гексаметилентетрамин (уротропин).
Для отверждения широко используются реакции полимеризации. Например, можно проводить полимеризацию мономера в присутствии НМ ненасыщенных полиэфиров.
Рекциями деструкции называются реакции, протекающие с разрывом химических связей в главной цепи макромолекулы. Деструкция, в результате которой происходит отщепление мономера, называется деполимеризацией. Реакции деструкции иногда используются специально для получения продуктов с более низким молекулярным весом. Кроме того, реакции деструкции имеют большое теоретическое значение для изучения строения ВМС.
В зависимости от природы агента, вызывающего разрыв связей в цепи, различают физическую и химическую деструкцию. Физическая деструкция подразделяется на термическую, механическую, фотохимическую и деструкцию под влиянием ионизирующего излучения (рентгеновские лучи, -, - и -излучение). Химическая деструкция протекает под действием различных химических агентов (вода, кислоты, амины, спирты и т.д.): окислительная – под действием кислорода воздуха и других окислителей; гидролиз – под действием воды и водных растворов кислот, щелочей и солей; алколиз - сольволиз, при котором раствороителем является спирт; ацидолиз – под действием карбоновых кислот, протекающая с образованием более НМ продуктов; аминолиз – под действием аминов.
Основная литература [2] (394-407)
Контрольные вопросы:
1. Изложите химические реакции, приводящие к изменению степени (коэффициента) полимеризации макромолекул (сшивание).
2. Назовите типы макромолекулярных реакций полимеров.
3. Изложите реакции линейного удлинения цепей.
4. Изложите реакции структурирования полимеров.
5. Расскажите о иулканизации каучука и отверждении полифункциональных олигомеров.
6. Изложите реакции деструкции полимеров, виды деструктивных воздействий.
Тема лекции №13: Деструкция. Деградация. Старение и стабилизация
Механическая деструкция – это реакция разрыва цепи, протекающая под влиянием различных механических воздействий, которому подвергается полимер при его переработке (измельчение, вальцевание, смешение, продавливание вязких растворов или расплавов полимеров через капиллярные отверстия и др.) и при эксплуатации изделий. Так, при интенсивном механическом измельчении целлюлозы, крахмала, полистирола, полиизобутилена и др. полимеров наблюдается понижение их молекулярного веса. Механическая деструкция полимеров, как и вообще их разрушение под действием внешнего механического воздействия, обусловлена флюктуациями (флюктуация – статическое отклонение от равномерного распределения) тепловой энергии. Приложенное напряжение создает возможность накопления флюктуаций и обеспечивает направленность процесса разрыва химических связей в основной цепи полимера.
Термическая деструкция полимеров – дестукция полимеров, происходящая при повышении температуры. Термоокислительная деструкция полимеров – деструкция полимеров, происходящая при одновременном воздействии света и кислорода; - одновременное окисление и нагревание полимера.
Термоокислительные и механохимические превращения, неизбежные в условиях переработки, в конечном счете, вызывают резкое ухудшение качества (деградацию) полимерного материала. Деградация – постоянное ухудшение; снижение или утрата положительных качеств.
Переработка полимеров всегда связана с их частичным разрушением, и задача технолога-переработчика состоит в том, чтобы по возможности замедлить химические процессы, приводящие к разрушению полимера. Это можно сделать двумя путями: применением очень чистых мономеров и добавлением специальных веществ, называемых стаблизаторами.
Большинство полимеров перерабатывается с добавкой стаблизаторов, которые в условиях эксплуатации полимерных изделий предотвращают их старение, т.е. изменение свойств во времени. Старение – необратимый процесс естественного изменения во времени первоначальных свойств материалов под воздействием внешних факторов (энергетических – нагревания, окисления, электромагнитных излучений, радиации и механических) или внутренних причин (перегруппировка, циклизация и т.д.).
Стаблизация в широком смысле слова заключается в сохранении исходных свойств полимеров при самых различных воздействиях. Для каждого полимера должны применяться свои специфические стаблизаторы. Но, поскольку все реакции распада являются цепными, для их замедления могут быть использованы три метода: 1) подавление цепных реакций, развивающихся в процессе термо- и термоокислительного распада; 2) создание условий, при которых образующиеся при распаде вещества препятствуют более глубокому разложению полимера; 3) создание условий, при которых распад протекает обратимо. На практике в большинстве случаев используется первый метод.
Основная литература [1] (407-421)
Контрольные вопросы:
1. Дайте понятие механодеструкция, термическая и термоокислительная деструкция.
2. Расскажите о деградации полимеров в условиях эксплуатации и переработки.
3. Расскажите о старении полимеров и методы их защиты.
4. Изложите принципы их стабилизации.
Тема лекции №14: Природные, искусственные и синтетические ВМС
В соответствии с распространенностью в окружающем нас мире полимеры раполагаются в ряд: природные неорганичнские полимеры природные органические полимеры синтетические полимеры.
Природные полимерные материалы: кожа, меха, шерсть, шелк, хлопок и т. п. , используемые для изготовления одежды, различные связующие (цемент, известь, глина), образующие при соответствующей обработке трехмерные полимерные тела, широко используемые как строительные материалы.
Промышленное производство цепных полимеров началось в начале ХХ в., хотя предпосылки для этого создавались ранее. Промышленное производство полимеров развивалось в двух направлениях – путем переработки природных органических полимеров в искусственные полимерные материалы и путем получения синтетических полимеров из органических НМС. В первом случае крупнотоннажное производство базируется на целлюлозе. Первый полимерный материал из физически модицированной целлюлозы – целлулоид – был получен еще в начале ХХ в.
Из целлюлозы получают три основных класса ценных материалов:
сложные эфиры целлюлозы – целлюлоза - простые эфиры целлюлозы
![]()
гидратированная целлюлоза
Целлюлоза
– структурный полисахарид, неразветлённый
полимер D–глюкозы;
главная составная часть клеточных
стенок, составляет 50-99% массы растений,
используется в текстильн![]() ой,
микробиологической промышленности,
для проиводства бумаги, плёнки, лаков,
загустителей и др. Целлюлоза: ацетатная,
белёная (подвергнутая отбелке), вискозная
(волокно, плёнка), техническая
(полуфабрикат). Развитие кино и фотографии
оказалось возможным лишь благодаря
появлению прозрачной пленки из нитрозы.
ой,
микробиологической промышленности,
для проиводства бумаги, плёнки, лаков,
загустителей и др. Целлюлоза: ацетатная,
белёная (подвергнутая отбелке), вискозная
(волокно, плёнка), техническая
(полуфабрикат). Развитие кино и фотографии
оказалось возможным лишь благодаря
появлению прозрачной пленки из нитрозы.
Производство синтетических полимеров началось в 1906 году, когда Л. Бакеланд запатентовал так называмую бакелитовую смолу – продукт конденсации фенола и формальдегида, превращающийся при нагревании в трехмерный полимер. В течение десятилетий он применялся для изготовления корпусов электротехнических приборов, аккумуляторов, телевизоров, розеток и т.п., а в настоящее время чаще используется как связующее и адгезив.
Благодаря усилиям Генри Форда, перед первой мировой войной началось бурное развитие автомобильной промышленности сначала на основе натурального, затем также и синтетического каучука. Производство последного было освоено накануне второй мировой войны. В эти же годы было освоено промышленное производство полистирола и поливинилхлорида, являющихся прекрасными электроизолирующими материалами, а также полиметилметакрилата – без органического стекла под названием «плексиглас» было бы невозможно массовое самолетостроение в годы войны.
После войны возобновилось производство полиамидного волокна и тканей (капрон, нейлон), начатое еще до войны. В течение войны все это производство было переориентировано на выпуск парашютов. В 50-х гг. ХХ в. было разработано полиэфирное волокно и освоено производство тканей на его основе под названием лавсан или полиэтилентерефталат. Полипропилен и нитрон – искусственная шерсть из полиакрилонитрала замыкают список синтетических волокон, которые использует современный человек для одежды и производственной деятельности.
Открытие в середине 50-х годов ХХ столетия и быстрое промышленное освоение катализаторов Циглера – Натта, что привело к появлению полимерных материалов на основе полиолефинов и, прежде всего, полипропилена и полиэтилена низкого давлении (до этого было освоено производство полиэтилена при давлении порядка 1000 атм), а также стереорегулярных полимеров, способных к кристаллизации. Затем были внедрены в массовое производство полиуретаны – наиболее распространенные герметики, адгезивы и пористые мягкие материалы (поролон), а также полисилоксаны – элементорганические полимеры, обладающие более высокими по сравнению с органическими полимерами термостойкостью и эластичностью.
Полиуретаны, синтетические полимеры содержащие в основном цепи макромолекулы уретановые группы.
Имеются так называемые уникальные полимеры, синтезированные в 60-70 гг. ХХ в. К ним относятся ароматические полиамиды, полиимиды, полиэфиры, полиэфиркетоны и др; непременным атрибутом этих полимеров является наличие у них ароматических циклов и (или) ароматических конденсированых структур. Для них характерно сочетание выдающихся значений прочности и термостойкости.
До настоящего времени массовое производство полимерных материалов базируется на цепных полимерах, объемы производства авжнейших из которых в начале 90-х г. ХХ в. составляли (млн. т в год):
полэтилен …………………… …31,9 полиуретаны …………….. 5,2
поливинилхлорид……………… 21,0 фенопласты .……………… 3,5
полистирол………………………12,0 аминопласты……………… 1,0
полипропилен ………………… 12,0 эпоксидные смолы…...…… 0,7
полиамиды …… ……………… 4,5
Природные органические полимеры образуются в растительных и животных организмах. Важнейшими из них являются полисахариды, белки, нуклеиновые кислоты, из которых в значительной степени состоят тела растений и животных (таблица 1) и которые обеспечивают само функциониро-ние жизни на Земле. Считается, что рещающим этапом в возникновении на
Таблица 1
Содержание различных веществ в теле человека
|
Тип вещества |
Массовый, % |
|
Вода Сухой остаток: белки жиры полисахариды НМС нуклеиновые кислоты |
60-80 20-40 15-20 3-20 1-15 2 0,1 |
Земле образование из простых органических молекул более сложных – ВМ.
В животном мире в качестве опорных, структурообразующих полимеров полисахариды «используются» лишь насекомыми и членистоногими. Наиболее часто для этой целей применяется хитин, который служит для построения так называемого внешнего скелета у крабов, раков, креветок. Из хитина деацетилированием получается хитозан, который, в отличие от нерастворимого хитина, растворим в водных растворах муравьиной, уксусной и соляной кислот. В связи с этим, а также благодаря комплексу ценных свойств, сочетающихся с биосовместиместью, хитозан имеет большие перспективы широкого практического применения в ближайшем будущем.
Крахмал относится к числу полисахаридов, выполняющих роль резервного пищевого вещества в растениях. Клубни, плоды, семена содержат до 70% крахмала. Запасаемым полисахаридом животных является гликоген, который содержится преимущественно в печени и мышцах.
Функцию запасаемого питательного продукта выполняет инулин, который содержится в спарже и артишоках, что придает им специфический вкус. Его мономерные звенья пятичленны, поскольку фруктоза относится к кетозам, в целом же этот полимер построен так же, как полимеры глюкозы.
Прочность стволов и стеблей растений, помимо скелета из целлюлозных волокон, определяется соединительной растительной тканью. Значительную ее часть в деревьях составляет лигнин – до 30%. Его строение точно не установлено. Известно, что это относительно низкомолекулярный (М 104) сверхразветвленный полимер, образованный в основном из остатков фенолов, замещенных в орто-положении группами –ОСН3, в пара-положении группами –СН=СН-СН2ОН. В настоящее время накоплено громадное количество лигнинов как отходов целлюлозно-гидролизной промышленности, но проблема их утилизации не решена. К опорным элементам растительной ткани относятся пектиновые вещества и, в частности пектин, находящийся в основном в стенках клеток. Его содержание в кожуре яблок и белой части кожуры цитрусовых доходит до 30%. Пектин относится к гетерополисахаридам, т.е. сополимерам.
В животном мире в качестве опорного, структурообразующего полимера обычно выступают белки. Эти полимеры построены из 20 типов так называемых –аминокислот общей формулы:
H
R-C-COOH
NH2
Из них: аланин R=СН3; аспарагиновая кислота R=НООС-СН2; цистеин R=НS-CH2; глутаминовая кислота R=НООС-СН2-СН2; фенилаланин R=С6Н5-СН2; серин R=НО-СН2 и др.
Нуклеиновые кислоты. В 1868 г. швейцарский ученый Фридрих Мишер выделил из ядер клеток фосфорсодержащее вещество, которое он назвал нуклеином. Позднее это и подобные ему вещества получили название нуклеиновых кислот. Исходными веществами, из которых построены нуклеотиды – звенья макромолекул нуклеиновых кислот, являются: сахар, фосфорная кислота, пуриновые и пиримидиновые соединения.
Природные неорганические полимеры составляют основу земной коры, толщина которой достигает 15 км (литосфера). Литосфера образовалась и образуется при входе на поверхность Земли расплавленной магмы, в которой химические реакции протекают при высокой температуре и давлении. Основной горной породой являются базальты. В таблице 2 приведены данные
Таблица 2
Содержание различных оксидов в базальтовых породах, мас. %
|
Оксид |
Лунный грунт |
Земная кора |
|
SiO2
A FeO TiO2 MgO CaO |
41-46 7-14 18-22 1-12 7-16 8-12 |
44-53 13-19 7-14 0,9-3,3 4-10 8-12 |
по составу базальтовых горных пород, полученных из проб, взятых со дна океанов Земли и лунных морей. Видно, что состав базальтовых пород Земли и ее спутника весьма близок, он состоит в основном из оксида кремния и в значительно меньшей степени из оксидов железа и алюминия. Оксид кремния является типичным полимерным кристаллическим телом, т.е. трехмерным полимерам.
В
природе широко распространены полимеры
алюмосиликатов
– линейные, двухмерно трехмерно сшитые
среди последних наиболее известны
цеолиты
с общей формулой MOA![]() 2O3xSiO2yH2O,
где М – металл I
или II
групп. Цеолиты являются прекрасными
сорбентами, их удельная поверхность
выше, чем угля. Благодаря этому цеолиты
широко используются на практике так
называемых молекулярных
сит.
2O3xSiO2yH2O,
где М – металл I
или II
групп. Цеолиты являются прекрасными
сорбентами, их удельная поверхность
выше, чем угля. Благодаря этому цеолиты
широко используются на практике так
называемых молекулярных
сит.
Основная литература [1] ()
Дополнительная литература [2] (241-279)
Контрольные вопросы:
1. Изложите характеристику основных представителей различных классов ВМС, используемых в производстве полимерных материалов.
2. Расскажите химический состав, способы получения, химические и физикомеханические свойства, области использования, технико-экономические показатели в народном хозяйстве.
Тема лекции №15: Клеи.Композиционные материалы. Красители и краски
Клеи – природные или синтетические вещества, применяемые для соединения различных материалов за счет образования адгезионной связи клеевой плёнки с поверхностями склеиваемых материалов.
Клеи:
глютиновые– животные клеи, получаемые из подкожного слоя шкур, из костей и сухожилий животных; образуют прочные соединения;
животные– клеи, получаемые переработкой материалов животного происхождения - мездры, костей, кожи, молока, крови и др.;
казеиновые – клеи, получаемые на основе казеина (казеин– сложный белок, образующийся при створаживании молока под действием ферментов; применяется в производстве клеев, пластмасс, красок и др.) с добавлением щелочей или натриевых солей;
кремнийорганические – клеи на основе кремнийорганических соединений; используются для соединения металлов, кремнийорганических каучуков и других теплостойких материалов;
природные– клеи, получаемые из природных продуктов органического или минерального происхождения; нестойки к действию влаги, подвержены гниению и относительно быстро теряют прочностные свойства;
растительные– клеи, получаемые на основе камедей (камедиилигумми– высокомолекулярные углеводы, главная составная часть соков и выпотов, выделяемых рядом растений при механических повреждениях и некоторых заболеваниях; содержатся также в некоторых водорослях. Используют в промышленности как клей, стабилизатор эмульсий и суспензий, для производства искусственного волокна и др., а также в медицине в виде слизей, как обволакивающее средство), крахмала, натурального каучука и других продуктов растительного происхождения;
резиновые– клеи на основе натурального или синтетического каучука, вязкие жидкости или пасты;
рыбьи– глютиновые клеи, получаемые переработкой плавательных пузырей, чешуи, костей и других рыбных отходов;
силикатные – клеи на основе водных растворов силикатов натрия или калия с наполнителями и модифицирующими добавками; применяются для склеивания керамики, стёкол, асбеста, металлов и других материалов;
синтетические– клеи, представляющие собой растворы, эмульсии и дисперсии полимеровв органических растворителях или в мономерах, а также не содержащие растворителей смолы, отверждающиеся в присутствии специальных добавок и других композиции на основе продуктов органического синтеза;
эпоксидные– синтетические клеи на основе полиэфиров, содержащих эпоксидные группы; отверждаются при добавлении катализаторов, обладают высокой адгезионной способностью к большинству материалов, обеспечивают высокую прочность и стойкость клеевого соединения.
Клей:
канифольный, получаемый горячей или холодной обработкой канифоли (канифоль– твёрдый продукт переработки смолистых древесины хвойных пород, состоящий из смеси изомерных одноосновных смоляных кислот и сопутствующих им неомыляемых соединений) щёлочью; используются для проклейки бумаги или картона в массе;
пековый, пек, омыленный раствором щёлочи или соды; применяется для проклейки тарного картона и некоторых видов бумаги.
Композиционные материалы – гетерогенные системы, состоящие из разнородных по составу и свойствам компонентов и обладающие полезными потребительскими свойствами.
Композиционные материалы (композиты), материалы из металлической и неметаллической основы (матрицы) с заданным распределением в ней упрочнителей. Прообраз современных композиционных материалов – железобетон. Различают композиционные материалы волокнистые (упрочненные волокнами или нитевидными кристаллами), дисперсно-упрочнённые (упрочнитель в виде дисперсных частиц) и слоистые (полученные прокаткой или прессованием разнородных материалов). К композиционным материалам также относятся сплавы с направленной кристаллизацией эвтектических структур. По прочности, жёсткости и другим свойствам превосходят многие конструкционные материалы.
Красители– индивидуальные органические вещества и их смеси, применяемые для придания цвета различным материалам.
Красители:
азиновые. Группа хинониминовых красителей, производных феназина, имеющих цвета от красного до глубоко-чёрного; применяются для окрашивания нетекстильных материалов, в производстве полиграфических красок, в лазерной технике;
азометиновые нерастворимые в воде; применяются в органическом синтезе, как красители в цветной фотографии, реже для крашения шерсти, шёлка и др.;
акридиновые – производные акридина; применяются для окрашивания натурального шёлка и некоторых текстильных материалов в жёлтый или оранжевый цвет;
активные – красители, содержащие в молекуле атомы или атомные группировки, способные взаимодействовать с функциональными группами макромолекул волокна с образованием ковалентных связей;
ализариновые– производные антрахинона, содержащие оксигруппу; применяются как протравные красители;
аминоксантеновые – производные ксантена (ксантен– гетероциклическое соединение; жёлтые кристаллы, плохо растворимые в воде; структурный фрагмент ксантеновых красителей); применяются для окрашивания нетекстильных материалов, в производстве чернил, карандашей, косметических препаратов и т. п.
антантроновые– производные антантрона; применяются как кубовые красители, образующие ярко-оранжевые окраски на хлопчатобумажных, вискозных и льняных тканях, для окрашивания бумаги, как пигменты для полиграфических красок;
антрахиноназоловые– производные азола, конденсированного с ядром антрахинона, где Х – О,S,NH; используются как кубовые красители бордо, красного и красно-фиолетового цвета для хлопчатобумажных, вискозных и льняных тканей;
антрахиноновые– амино- и оксипроизводные антрахинона; используются для окрашивания синтетических и других волокон, в лакокрасочной и резиновой промышленности, в полиграфии;
антрахинонпиридоновые (смотрите,фталоилакридоновыекрасители);
ариламиновые (смотрите,хинониминовыекрасители);
арилметановые – групповое название диарилметановых, триарилметановых, ксантеновых и акридиновых красителей;
ацедиантроновые – полициклические хиноны; используются как кубовые красители, пигменты для крашения искусственных и синтетических волокон в массе, в производстве полиграфических красок и т.п.;
ацетонорастворимые – нерастворимые в воде красители, применяемые главным образом для крашения ацетатного волокна в массе;
виолантроновые – производные виолантрона; используются как кубовые красители, пигменты (для полиграфических красок, пластмасс), люминофоры;
гемицианиновые – полиметиновые красители с азотсодержащими заместителями на концах цепи, из которых только один входит в состав гетероцикла; используются как катионные красители для крашения полиакрилонитрильных волокон;
диазиновые (смотрите,азиновыекрасители);
диарилметановые – производные главным образом фенилметана, содержащие в параположениях электродонорный и электроакцепторный заместители; используются для крашения полиакрилонитрильных волокон, в множительной технике;
дибензпиренхиноновые
– полициклические хиноны, гдеR–H,C![]() ,Br, жёлтые кристаллы,
нерастворимые в воде: используются как
кубовые красители, образующие окраски
жёлтого цвета;
,Br, жёлтые кристаллы,
нерастворимые в воде: используются как
кубовые красители, образующие окраски
жёлтого цвета;
дисперсные – нерастворимые или слаборастворимые в воде красители, окрашивающие гидрофобные волокна из высокодисперсных водных суспензий;
жирорастворимые– красители, растворимые в неполярных и малополярных средах; применяются для подкрашивания бензинов, восков, жиров и другие;
изовиолантроновые – продукты галогенирования изовиолантрона; применяются как кубовые красители, образующие окраски ярко-фиолетового и красновато-синего цвета, пигменты для пластмасс, резин, ЛКМ;
индигоидные – гетероциклические соединения, где Х-главным образомN,S, А-группа атомов, образующих вместе с атомами углерода пяти- или шестичленный ароматический или гетероциклических остаток; применяются как кубовые красители и пигменты;
индикаторные;
карбоцианиновые (смотрите,триметинцианиновыекрасители);
катионные – красители, содержащие группу аммония; используются для крашения полиакрилонитрильных и других синтетических волокон;
кислотно-протравные – протравные красители, содержащие кислотные группы;
кислотные – соли карбоновых кислот и сульфокислот, растворимые в воде; применяются для окрашивания шерсти, натурального шёлка, полиамидных волокон, кожи и других;
ксантеновые– групповое название окси- и аминоксантеновых красителей;
кубовые– нерастворимые в воде красители, восстанавливающиеся с образованием производных, растворимых в щелочных средах и обладающих сродством к целлюлозным волокнам;
макрогетероциклические– полифункциональные соединения, где А – одинаковые или разные карбо- или гетероциклические ароматические фрагменты; имеют цвета от глубокосинего до жёлтого;
мероцианиновые– полиметиновые красители с азотосодержащим электронодонорным и кислородсодержащим электроноакцепторным заместителями на концах цепи; используются как пигменты и дисперсные красители для синтетических и искусственных волокон, образующие окраски главным образом жёлтого цвета;
металлсодержащие – кислотные красители (преимущественно азокрасители), в молекулы которых атом металла (Cr,Co,Cu) вводят в процессе производства; применяются для крашения шерсти и полиамидных волокон;
монометинцианиновые – цианиновые красители с одной метиновой группой между симметричными азотсодержащими гетероциклическими радикалами; используются главным образом как оптические сенсибилизаторы фотоэмульсий;
оксазиновые – хинониминовые красители, производные феноксазина; применяются главным образом как прямые красители и пигменты с окрасками от красно-фиолетового до синего цвета, а также в лазерной технике;
оксаиновые – полиметиновые красители с кислородосодержащими заместителями на концах цепи; применяются как оптические сенсибилизаторы фотоэмульсий;
оксиксантеновые – производные ксантена; применяются для окрашивания нетекстильных материалов;
основные – соли органических оснований, растворимые в воде; применяются для крашения шерсти, натурального шёлка, полиамидных волокон, бумаги, лент для пишущих машин и т.п., в производстве фаналевых лаков;
периноновые – полифункциональные соединения, продукты взаимодействия ангидридов ароматическихорто- ипери-дикарбоновых кислот с ароматическими моноаминами,орто- ипери-диаминами; используются как пигментами широкого назначения, красители для крашения в массе синтетических волокон;
пиразолантроновые – полициклические соединения, производные пиразолантрона; применяются как кубовые красители, образующие окраски жёлтого, красного, серого цвета;
полиметиновые – органические соединения, содержащие в молекуле цепочку из нечётного числа метиновых групп с электронодонорным и электроноакцепторным заместителями на концах; применяются как красители для ацетатных и полиакрилонитрильных волокон, как оптические сенсибилизаторы фотоэмульсий, в лазерной технике;
полиметинцианиновые
– цианиновые красители с числом
метиновых групп в открытой цепи![]() 5;
применяются как оптические сенсибилизаторы
фотоэмульсий, в лазерной технике;
5;
применяются как оптические сенсибилизаторы
фотоэмульсий, в лазерной технике;
полициклические – кубовые красители, производные карбо- и гетероциклических многоядерных конденсированных соединений, содержащих карбонильные группы, объединённые системой сопряжённых двойных связей; применяются как кубовые красители, пигменты и т.п.;
протравные – красители, окрашивающие текстильные материалы из водных растворов и закрепляющиеся на волокне с помощью металлических протрав-соединений металлов со степенью окисления +ІІІ (хрома, железа и др.);
прямые – красители, способные окрашивать текстильные материалы непосредственно из водных растворов;
сернистые – сложные смеси химических соединений, в молекулах которых содержатся гетероциклические фрагменты, ароматические и хиноидные циклы, связанные между собой дисульфидными, сульфидными и др. группами; не растворяются в воде; применяются для крашения хлопчатобумажных тканей и ниток;
спирторастворимые – красители, растворимые в спирте и аналогичных ему растворителях; применяются для придания окраски нитролакам и спиртовым лакам, пастам для шариковых ручек и т.п.;
субстантивные (смотрите,прямыекраски);
тиазиновые – группа хинониминовых красителей, производных фенотиазина, от фиолетового до зелёного цвета; используются в лазерной технике, гистологии, реже для крашения шерсти, шёлка, хлопчатобумажных тканей;
тиоиндигоидные – группа индигоидных красителей, имеющих в молекуле по крайней мере один серосодержащий гетероциклический остаток; применяются как кубовые красители с широкой гаммой цветов и как пигменты;
триарилметановые – производные главным образом трифенилметана; применяются для окрашивания нетекстильных материалов, в производстве полиграфических красок, чернил, фаналевых лаков, а также в качестве кислотно-основных индикаторов;
триметинцианиновые – цианиновые красители с тремя метиновыми группами в открытой цепи; используются как оптические сенсибилизаторы фотоэмульсий, в лазерной технике;
флуоресцирующие;
фталоилакридоновые – производные фталоилакридона; используются как кубовые красители, образующие окраски с широкой гаммой цветов;
фталоцианиновые – макрогетероциклические красители; внутрикомплексные соединения, гдеM-Cu,Ni,Coи т.п.; имеют цвета от красновато-голубого до зелёного; применяются как пигменты в полиграфии, лакокрасочной промышленности, для крашения резины, пластмасс, в электронной и лазерной технике, а также в качестве прямых красителей;
хинониминовые
– производные вторичных ароматических
аминов, гдеD-N(Alk)2,
ОН или др. электронодонорная группа,
А -![]() (Alk)2О
илиNН; имеют цвета от
красновато-синего до зелёного; используются
главным образом в цветной фотографии.
(Alk)2О
илиNН; имеют цвета от
красновато-синего до зелёного; используются
главным образом в цветной фотографии.
хромирующиеся (смотрите,протравныекрасители);
цианиновые – полиметиновые красители с двумя азотсодержащими гетероциклическими остатками на концах цепи; имеют цвета от жёлтого до синего; используются главным образом как оптические сенсибилизаторы фотоэмульсий.
Краска – кассельская, коричневый природный пигмент, применяемый в полиграфических и художственных красках.
Краски – 1) Общее наименование пигментированных ЛКМ; 2) Наименование некоторых пигментов.
Краски:
акварельные – художственные водорастворимые краски на основе природных плёнкообразующих веществ (например, гуммиарабика; гуммиарабик от латинского gummi–камедь и arabicus– аравийский, - вязкая прозрачная жидкость, выделяемая некоторыми видами акций. Растворяется в воде, образуя клейкий раствор. Ранее применялся как клеящее вещество), образующие прозрачные растворимые плёнки;
битумные – краски на основе битума в качестве плёнкообразующего вещества;
воднодисперсионные – краски на основе водной дисперсии в качестве плёнкообразующего вещества;
водоэмульсионные (смотрите, воднодисперсионныекраски);
краски, готовые к употреблению – масляные краски, выпускаемые с рабочей вязкостью;
гуашевые – художественные водорастворимые краски, образующие матовые непрозрачные плёнки;
густотёртые – масляные высоконаполненные краски, которые перед нанесением разводят олифой до рабочей вязкости;
жидкотёртые (смотрите,краски, готовые к употреблению);
казеиновые – краски на основе казеина в качестве плёнкообразующего вещества;
каучуковые – краски на основе синтетического каучука в качестве плёнкообразующего вещества;
квазиобратимые – термоиндикаторные краски, восстанавливающие первоначальный цвет в покрытии в результате поглощения влаги;
керамические – минеральные вещества, стойкие при высоких температурах; применяются для окрашивания керамических изделий, глазурей, стёкол;
клеевые – краски на основе водного раствора растительного или животного клея;
латексные (смотрите,воднодисперсионныекраски);
люстровые – краски для получения люстра на тонкой керамике;
масляные – краски на основе масла или олифы (плёнкообразующие вещества на основе окисленных или полимеризованных растительных масел или жирных алкидных смол);
микрокапсулированные – краски, содержащие в своём составе микрокапсулированные компоненты (растворители, отвердители и другие);
надглазурные керамические – керамические краски для декорирования тонкой керамики, наносимые на глазурованные обожжённые изделия и закрепляемые обжигом;
необратимые – термоиндикаторные краски, не восстанавливающие первоначальный цвет в покрытии после охлаждения;
обратимые - термоиндикаторные краски, восстанавливающие первоначальный цвет в покрытии после охлаждения;
органодисперсионные – краски на основе дисперсии синтетического полимерного плёнкообразующего вещества в органической жидкости (растворителе, разбавителе);
печатные (смотрите,полиграфическиекраски);
подглазурные керамические – керамические краски для декорирования тонкой керамики, наносимые на неглазурованные обожжённые изделия, которые затем покрывают глазурью и обжигают;
полиграфические – краски для одно- или многоцветной печати на материалах, предназначенных для издательских работ;
порошковые – порошкообразные наполненные полимерные композиции, образующие после напыления на поверхность и отверждения ЛКМ;
силикатные – краски на основе жидкого стекла в качестве плёнкообразующего вещества;
строительные – краски для защитной и/или декоративной отделки зданий и строительных конструкций;
темперные – художественные краски на основе природных плёнкообразующих веществ (яичный желток, казеино-масляная эмульсия и др.);
термоиндикаторные – краски, образующие покрытия, способные изменять цвет или яркость свечения при определённой температуре;
тёртые – общее название густотёртых красок и красок, готовых к употреблению;
тикстропные – краски, обладающие свойствами тиксотропии (тиксотропия – обратимое изменение физико-механических свойств полимерных и дисперсных систем при механическом воздействии в изотермических условиях);
художственные – краски, предназначенные для создания произведений изобразительного искусства и для декоративно-прикладных целей;
эмалевые;
эмульсионные (смотрите,воднодисперсионныекраски).
Основная литература [3] (196, 197, 250; 220-222)
Контрольные вопросы:
1. Дайте понятии клеи, клей, композиционные материалы (композиты), красители и краски.
2. Назовите их виды и разновидности.
3. Расскажите о их применении.