

45.Опухоли мозжечка у детей, общемозговые и очаговые симптомы, диагностика, подходы к хирургическому лечению
Составляют у детей 38% всех новообразований, у взрослых 6%.
При опухолях червя мозжечка симптомы:
- гипертензионная головная боль
- статическая атаксия
- головокружение и нистагм при взгляде в сторону
- нарушение координации в конечностях
При опухолях полушарий мозжечка:
- приступы головной боли ночью или утром
- тошнота и рвота
- координаторные расстройства одностороннего характера, соответственно пораженному полушарию
В случае окклюзии ликворопроводящих путей – гидроцефалия.
Диагностика
- неврологический статус
- осмотр смежных специалистов
- анализ ликвора
- церебральная ангиография
- ЭЭГ
- нейровизуализация (МРТ, МР-трактография, КТ, ПЭТ-КТ)
46.Миопатии Дюшенна, Эрба-Рота, этиопатогенез, клиника, диагностика, лечение.
Прогрессирующая мышечная дистрофия Дюшенна/Беккера – наследственное рецессивное Х-сцепленное (аутосомно-рецессивное) заболевание, с распространенностью 1 случай на 3600-6000 мальчиков, рожденных живыми, возникающее в результате мутации в гене дистрофина, характеризующееся поражением проксимальных групп мышц, кардиологическими, ортопедическими и респираторными осложнениями
Мышечная дистрофия Дюшенна - наследуемая прогрессирующая мышечная дистрофия, характеризующаяся началом в раннем возрасте, симметричной атрофией мышц в сочетании с сердечно-сосудистыми, костно-суставными и психическими нарушениями, злокачественным течением.
Отличия миодистрофии Дюшена и Беккера
Прогрессирующая мышечная дистрофия Дюшенна (миодистрофия Дюшенна) |
Прогрессирующая мышечная дистрофия Беккера (миодистрофия Беккера) |
Тяжелая форма с манифестацией в возрасте 2-5 лет и прогрессирующим злокачественным течением: формированием вялых парезов, параличей и контрактур мышц, обездвиженности. |
доброкачественная форма заболевания с поздним дебютом в 10-20 лет и медленным прогрессированием симптомов мышечной слабости с сохранением способности к самостоятельной ходьбе в течение 15-20 лет от начала заболевания. |
Этиопатогенез
Миодистрофия Дюшена и Беккера наследуется по рецессивному сцепленному с Х-хромосомой типу,поэтому в основном болеют мальчики.
Развитие мышечной дистрофии Дюшенна связано с наличием мутации в 21-ом локусе короткого плеча Х-хромосомы в гене, кодирующем белок дистрофин.
При дистрофии Дюшенна генетические аберрации приводят к сдвигу рамки считывания ДНК и полному прекращению синтеза дистрофина, что обусловливает тяжелое течение заболевания.
Из-за отсутствия дистрофина миофибриллы утрачивают устойчивость к циклическим актам сокращения-расслабления и разрываются.
Саркоплазматические мембраны становятся нестабильными
нарушается работа ионных каналов,в результате повышается концентрация свободного внутриклеточного ионизированного кальция,который оказывает некротизирующие влияние на мышечные волокна,вызывая их лизис..
Клиника
Первые признаки заболевания проявляются в 1-3 года жизнислабостью мыщц тазового пояса.
Уже на 1-м году обращает на себя внимание отставание детей в моторном развитии.
Дети, как правило:
С задержкой начинают садиться, вставать, ходить.
Движения неловкие, при ходьбе дети неустойчивы, часто спотыкаются, падают.
В 2-3 года появляются:
¨мышечная слабость
¨патологическая мышечная утомляемость, проявляющаяся при физической нагрузке - длительной ходьбе, подъеме на лестницу, изменение походки по типу «утиной».
¨взбирание лесенкой» или «взбирание по самому себе» - симптом Говерса
Яркий признак миопатии Дюшена – псевдогипертрофия мышц (“икры гнома”), особенно икроножных: на самом деле происходит не развитие мышц, а их перерождение в жировую и соединительную ткани.
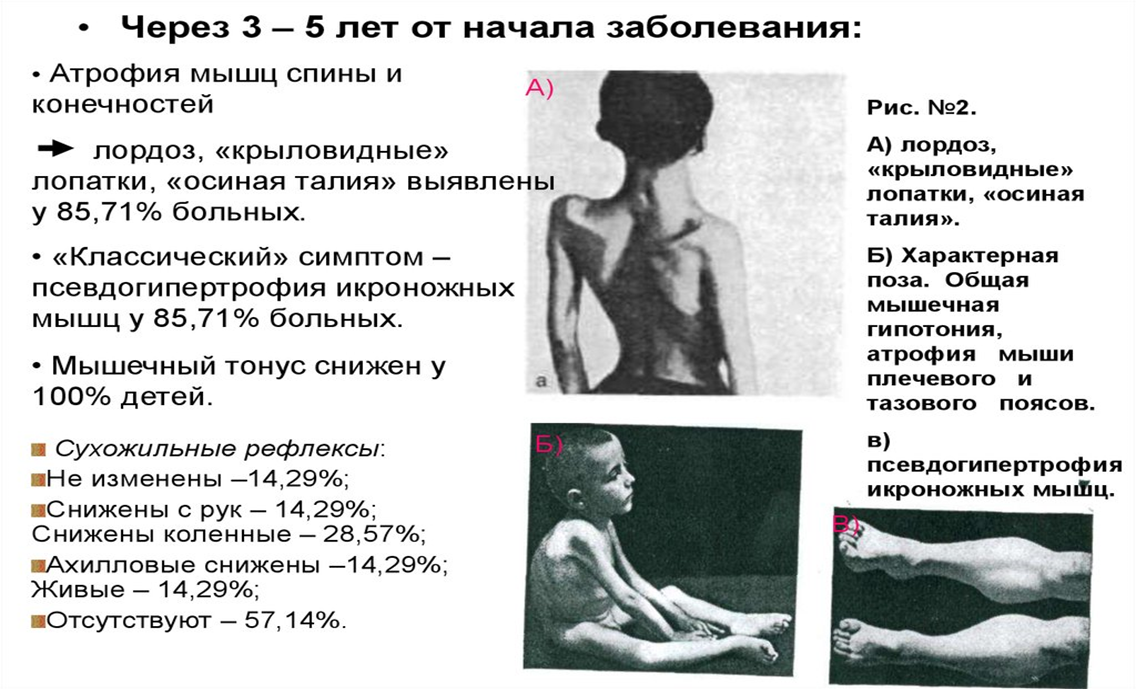
Патология костно-суставной системы и внутренних органов (сердечно-сосудистой и нейроэндокринной систем).
Со временем, по мере прогрессирования мышечной дистрофии развиваются контрактуры крупных суставов, наблюдается эквиноварусная деформация стопы
*¨На рентгенограммах обнаруживают сужение костномозгового канала, истончение коркового слоя длинных диафизов трубчатых костей.
Сердечно-сосудистые расстройства клинически проявляются лабильностью пульса, артериального давления, иногда глухостью тонов и расширением границ сердца.
На ЭКГ регистрируются изменения миокарда (блокада ножек пучка Гиса и др.).
¨Нейроэндокринные нарушения встречаются почти у половины пациентов.
¨Чаще других даются синдром Иценко-Кушинга, адипозогенитальная дистрофия Бабинского-Фрелиха. Интеллект у многих больных в различной степени (от легкой дебильности до имбецильности).
¨Течение быстро прогрессирующее, злокачественное.
К 7-10 годам возникают глубокие двигательные расстройства.
К 14-15 годам наступает обездвиженность.
Диагностика
Диагноз ставится на основании:
данных генеалогического анализа (рецессивный сцепленный с Х-хромосомой тип наследования)
клинических особенностей болезни (раннее начало в 1-3 года, симметричные атрофии проксимальных групп мышц развивающиеся в восходящем направлении, псевдогипертрофии икроножных мышц, грубые соматические и нейроэндокринные расстройства, снижение интеллекта, быстрое злокачественное течение болезни)
Данных лабораторных методов исследования:
Биохимический анализ крови:
· повышение уровня КФК – является облигатным, ранним доклиническим признаком;
· повышение уровня трансаминаз: АЛТ, АСТ;
· повышение уровня ЛДГ- не является облигатным признаком. Инструментальные исследования:
· УЗИ мышц – признаки мышечной дегенерации: замена мышечной ткани жировой или фиброзной тканью;
· ЭМГ – первично-мышечные изменения, миопатический тип ЭМГ: короткие остроконечные многочисленные потенциалы;
· ЭхоКГ – могут быть выявлены признаки гипертрофической или дилятационной кардиомиопатии
Лечение
направлено на поддержание физической активности пациента и улучшения качества его жизни.
ГКС-преднизолон по 0,75 мг/кг/сут. увеличивает мышечную массу у мальчиков, страдающих мышечной дистрофией Дюшенна, замедляя прогрессирование болезни.(по схеме через день)
препараты, улучшающие обмен веществ ( вит группы В,Е, аминокислоты, препараты кальция)
применяется лечение прозерином, галантамином(ингибиторы холинэстеразы)
ЛФК. (особенно замедляющая образование контрактур), так же, как и пассивное растяжение больных мышц, массаж, электрофорез прозерина, лидазы, кальция хлорида, ванны, индуктотермия.
+ наблюдение кардиолога обязательно!!!
При наличии контрактур и фиксации суставов показано ортопедическое вмешательство
Миопатия Эрба-Рота
Прогрессирующая мышечная дистрофия Эрба-Рота — аутосомно-рецессивная наследственная миодистрофия, отличающаяся полиморфизмом клинических проявлений и вариативностью скорости прогрессирования симптомов. Может носить нисходящий характер, т. е. начинаться со слабости в проксимальных отделах рук, но чаще имеет стандартный восходящий тип распространения мышечных изменений.
дебют в детском или юношеском возрасте чаще в 14-16 лет
конечностно-поясная миодистрофия прежде всего атрофии мышц тазового пояса
ранний признак утиная походка и др миопатические феномены
в дальнейшем атрофии мышц плечевого пояса, рук (форма
ЛЕЙДЕНА-МЕБИУСА)
Этиопатогенез
до конца не изучен? Субстратом дистрофии Эрба-Рота являются патологические метаболические и структурные изменения в мышечной ткани (миопатия). Они возникают в результате генетических мутаций, приводящих к недостатку или полному прекращению синтеза белков, являющихся необходимыми структурными компонентами миоцитов.(Согласно существующей на сегодняшней день версии, причины дистрофии Эрба-Рота заключаются в генетическом дефекте, передающемся от одного из здоровых родителей – здорового носителя мутировавшего гена в парных неполовых хромосомах или в Х-хромосоме. Это такие гены, как 13q12, 17q12-q21.33, 4q12 и 5q33)
Клиника
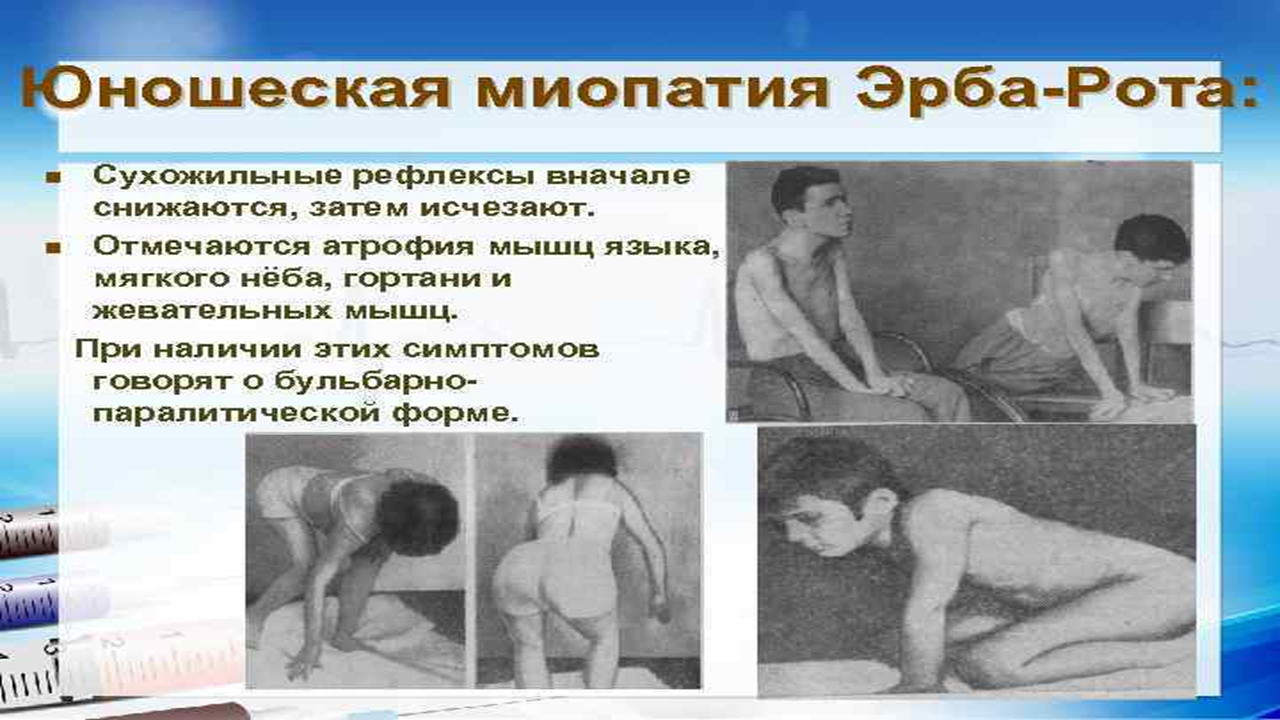
Симптомы заболевания вначале неспецифичны и включают в себя общую слабость, слабость мышц спины. Постепенно заболевание прогрессирует. Пациент перестает удерживать спину в нормальном положении, развивается гиперлордоз – переразгибание поясницы кзади.
Достаточно рано изменяется походка. Она становится похожей на «утиную» — переваливание ног из-за слабости мышц бедра и тазового пояса.
Быстро развивается гипотрофия мышц верхнего плечевого пояса. Также постепенно развивается общая гипотрофия, а затем и атрофия мышц. Иногда имеет псевдогипертрофия голеней – замена мышечной массы жировой и соединительной тканью.
Со временем пациент перестает выполнять многие активные движения. Значимо затрудняется вставание, больным приходится вставать на четвереньки, помогать руками при вставании.
Лицо пациента становится амимично, не полностью смыкаются веки, губы же, напротив, выворачиваются кпереди и нередко утолщаются (губы тапира). Мимику такого пациента ещё иногда называют лицом миопата
Диагностика
Диагностика дистрофии Эрба-Рота основана на физикальном осмотре пациентов, изучении семейного анамнеза и анализе данных исследований:
генетическое тестирование (используется для определения типа мышечной дистрофии);
электронейромиографии (ЭНМГ);
биопсии и биохимического исследования мышечной ткани;
общего анализа кроки;
анализа крови на КФК (креатинфосфокиназу);
анализа мочи.
Лечение
медикаментозные курсы, включающие АТФ, витамины Е и группы В, тиоктовую кислоту и др.
Занятия лечебной физкультурой должны проводиться ежедневно и включать упражнения на все группы мышц.
Регулярно назначаются курсы массажа и физиопроцедуры.
При поражении сердечной мышцы рекомендован инозин, сердечные гликозиды, антиаритмики.
При развитии контрактур может потребоваться ортопедическое лечение.
Выраженное снижение жизненной емкости легких из-за атрофии дыхательных мышц служит показанием к ИВЛ.