

Изучение уф спектров
Электронные уровни наглядно и с высокой точностью описываются в терминах теории молекулярных орбиталей. Исходя из деталей взаимодействия и данных о потенциалах ионизации можно расположить электроны различных молекулярных орбиталей в следующий ряд по их энергии: <<n. - орбитали занимают связывающие электроны всех типов органических молекул, -орбитали заняты электронами двойных и тройных связей, n- орбитали заполняют электроны несвязывающих электронов гетероатомов, например, кислорода или азота. Возбуждение переводит электроны на более высокие разрыхляющие орбитали, энергия которых растет *<*. Таким образом, в электронных спектрах могут проявляться следующие переходы: n* (в карбонильных соединениях); * (в алкенах, алкинах, карбонильных и азосоединениях); * (в карбонильных соединениях); * (в алканах).
Наличие и интенсивность проявления линии в спектре определяется вероятностью или разрешенностью соответствующего перехода. Для описания спектров используют следующие правила:
а) поглощение одного кванта сопровождается возбуждением одного электрона;
б) суммарное спиновое число при электронном переходе должно остаться неизменным.
Есть правила, учитывающие симметрию молекулы и симметрии основного и возбужденного состояний, но они не столь всеобщи. Во всех устойчивых молекулах электроны всегда спарены, возбуждение переносит электрон на более высокий по энергии уровень, но спин его остается противоположным спину электрона оставшегося. Системы, содержащие только спаренные электроны, называют синглетными; системы, в которых присутствуют неспаренные электроны - триплетными. Переходы между синглетными или триплетными уровнями разрешены и проявления в спектрах интенсивны (триплетные уровни заселены слабо и соответствующие линии слабы только поэтому), переходы между синглетным и триплетным уровнями, наоборот, запрещены и линии, им соответствующие, малоинтенсивны.
В молекулах можно выделить структурные фрагменты, обуславливающие избирательное поглощение излучение и называемые хромофорами и фрагменты, вступающие в электронное взаимодействие с хромофорами, изменяющие таким образом интенсивность поглощения и/или положение максимума и называемые ауксохромами. Выделяют следующие типы влияния ауксохрома:
а) батохромный сдвиг - смещение полосы поглощения в сторону более длинных волн (меньших частот) или красный сдвиг;
б) гипсохромный сдвиг - смещение в сторону более коротких волн (больших частот) или синий сдвиг;
в) гиперхромный эффект - увеличение интенсивности поглощения;
г) гипохромный эффект - понижение интенсивности поглощения
УФ спектр органического вещества характеристичен, так как поглощение определяется только собственно хромофором и его ближайшим окружением, т. е. один и тот же хромофор проявляется практически одинаково как в относительно простых, так и в самых сложных молекулах. В зависимости от непосредственного окружения одной и той же хромофорной группировки положение максимума поглощения в УФ спектрах различных соединений может несколько изменяться. Сдвиг максимума в сторону более длинных волн принято называть батохромным сдвигом, а сдвиг в сторону более коротких волн - гипсохромным. Замена растворителя в отдельных случаях может вызвать некоторые изменения как в положении полос (на 2- 10 нм), так и в их интенсивности (на 10-20%).
Как правило, такая замена влияет на спектры полярных веществ и практически не сказывается на УФ спектрах неполярных соединений. Наиболее сильные изменения в спектрах обусловлены химическим взаимодействием вещества с растворителем (в частности, образованием водородной связи), а также изменением степени диссоциации или соотношения таутомерных форм вещества. Во всех таких случаях следует проверить, выполняется ли для данного раствора закон Бугера-Ламберта-Бера.
Таким образом, УФ спектроскопия позволяет определить в исследуемых соединениях группировки-хромофоры и дает прекрасную возможность для количественного анализа веществ, содержащих такие группировки. Как структурно-аналитический метод УФ спектроскопия значительно менее информативна по сравнению с другими методами и носит в основном эмпирический характер, поскольку зависимость между характером поглощения и структурой молекулы не имеет строгого физико-математического обоснования, что, однако, не мешает широкому использованию метода.
Отсутствие в УФ спектре исследуемого вещества максимума поглощения в области 200-800 нм служит надежным доказательством того, что в этом веществе не содержатся сопряженные диеновые или полиеновые системы, ароматические ядра и карбонильные группы. Этот признак часто оказывается полезным при установлении структуры соединения, например, позволяет легко различить изомеры с сопряженными и изолированными двойными связями, как в случае приводимой ниже пары:

УФ спектры основных гетероциклических соединений представлены ниже:
|
макс (в гексане) |
|
фуран |
200 ( 10000) |
252 ( 1) |
тиофен |
|
235 ( 4500) |
пиррол |
210 ( 15000) |
350 ( 300) |
пиридин |
195 ( 7500) |
250 ( 2000) |
Таким образом, достаточно очевидны как преимущества метода (доказательство наличия в исследуемом веществе группировок-хромофоров сопряженной диеновой, полиеновой и ароматической систем, а также карбонильной группы или их отсутствия; в простейших случаях возможность определения типа хромофора, длины цепи сопряжения, числа алкильных групп при хромофоре; количественный анализ, включая регистрацию изменения концентраций растворов во времени), так и ограничения метода (ограниченность рамок применения, так как многие типы органических соединений не имеют максимума поглощения в исследуемой области; сравнительно малые возможности при решении структурно-аналитических задач; в ряде случаев сильное влияние природы растворителя на характер спектра и возможность отклонений от закона Бугера- Ламберта-Бера; фотохимическая изомеризация веществ в процессе работы (например, цис-транс- изомеризация в диеновых и полиеновых системах).
Колебательная спектроскопия
Типы колебаний
Энергия, необходимая для возбуждения колебаний атомов в молекуле, соответствует энергии квантов света с длиной волны 1-15 мкм или волновым числом 400÷4000 см-1, т. е. электромагнитному излучению средней инфракрасной (ИК) области. Колебательные уровни молекул квантованы, энергия переходов между ними и, следовательно, частоты колебаний могут иметь только строго определенные значения. Поглощая квант света, молекула может переходить на более высокий колебательный уровень, обычно из основного колебательного состояния в возбужденное. Поглощенная энергия передается затем на возбуждение вращательных уровней или преобразуется в кинетическую энергию молекул. Колебания молекул проявляются в двух типах спектров: спектры поглощения в инфракрасной области (ИК-спектры) и спектры комбинационного рассеяния света (спектры КР).
Математическая модель колебаний многоатомных молекул сложна. Расчеты проведены только для простейших двухатомных молекул. Колебательная спектроскопия носит в основном эмпирический характер, т.е. основные частоты колебаний получены при сопоставлении спектров многих соединений одного класса. Это, однако, не умаляет ценности метода.
Основными типами колебаний являются валентные и деформационные.
Валентными колебаниями называются колебания ядер атомов вдоль линии связи, они обозначаются буквой (C=C, C=O и т. д.).
Приближенной механической моделью валентных колебаний может служить система из двух шаров, связанных жесткой пружиной (здесь шары изображают атомы, а пружина - химическую связь) (см. рис., А).
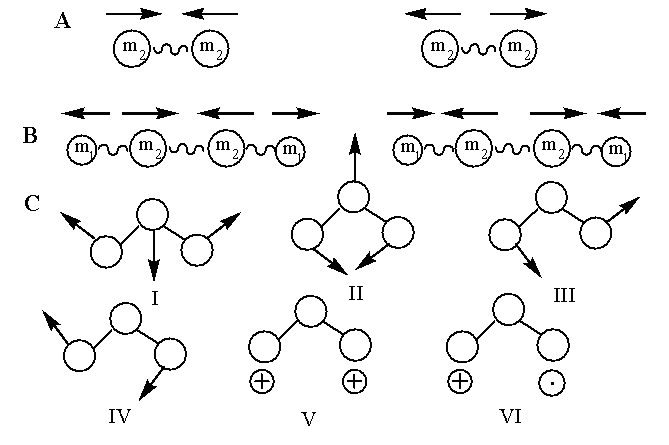
А, В – валентные колебания в молекулах;
С – деформационные колебания: I,II – ножничные; III,IV – маятниковые; V – веерные; VI – крутильные.
При растяжении или сжатии пружины шары начнут колебаться вокруг положения равновесия, т. е. будет осуществляться гармоническое колебание, описываемое уравнением
![]()
где - частота колебания; F- силовая постоянная, характеризующая прочность связи, или силу, возвращающую шары в положение равновесия; mr - приведенная масса шаров (атомов), вычисляемая по формулам
![]()
Частоты валентных колебаний определяются массой атомов и прочностью (энергией) связи. Чем масса больше, тем меньше частота, например:
C-C 1000 см-1; C-Н 3000 см-1
Чем связь прочнее, тем выше частота колебаний, например:
C-C 1000 см-1 |
C-О 1100 см-1 |
C-N 1050 см-1 |
C=C 1600 см-1 |
C=О 1700 см-1 |
C=N 1650 см-1 |
Возможно появление обертонов - колебаний, частота которых больше в целое число раз, чем у основных (2, 3 и т. д.). Обычно интенсивность обертонов много меньше: для первого обертона она составляет 1—10 % от интенсивности основного колебания; третий обертон обнаружить обычно не удается.
В системе из трех или четырех атомов возможны два типа валентных колебаний - синфазное (в одной фазе, или симметричное, s) и антифазное (в разных фазах, или антисимметричное, as) (рис. В), хотя термины полностью применимы и для симметричных молекул. Частота антифазного колебания всегда выше, чем синфазного.
Деформационные колебания связаны с изменением валентного угла, образованного связями у общего атома; они обозначаются буквой . Виды некоторых деформационных колебаний показаны на рис., С. Для возбуждения деформационных колебаний требуется меньшая энергия, чем в случае валентных колебаний, и, следовательно, они имеют меньшую частоту.
С увеличением числа атомов в молекуле число возможных колебаний быстро растет. В реальной молекуле колебания атомов тесно связаны друг с другом и взаимодействуют между собой. Спектры молекул представляют собой сложный набор различных колебаний, каждое из которых проявляется в узком интервале частот.
Интенсивность поглощения определяется, как и в УФ спектроскопии, молярным коэффициентом поглощения, однако в этом случае точность измерения существенно меньше. Обычно интенсивность полос выражают как поглощение (А) или пропускание (Т) светового потока в процентах. Полосы также оценивают по интенсивности как сильные (с.), средние (ср.) и слабые (сл.).