

18. Понятие о гомогенном катализе. Механизм действия катализатора. Энергетические диаграммы для некаталитической и каталитической реакции.
Катализ –
явление изменения скорости термодинамически
возможной ХР под влиянием катализаторов
–в-в, учавствующих в реакции, но остающихся
в неизменном количестве и составе после
ее завершения. Все кат. проц. подразделяют
на 2 группы.Гомогенный
катализ –
процессы,В которых реагирующие в-ва и
катализатор находятся в 1 фазе и обр.
гомогенную систему (напр., окисление
оксида серы кислородом в присутствии
оксида азота). при гетерогенном
катализе катализатор
представляет собой самостоятельную
фазу, граничащую с фазой реагентов. В
этом случае каталитическая р-ция
протекает на пов-ти раздела фаз (газ-тв.
тело, жидкость-тв. тело, жидкость-газ).
Наибольшее практическое значение имеют
КР, когда катализатор находится в ТВ.
фазе, а реагенты – в жидкой или
газообразной. ( Пример ГК – окисление
оксида серы кислородом в присутствии
оксида ванадия). В особую группу выделяют
ферментативные каталитические реакции,
что связано с происхождением катализаторов,
называемых ферментами.Ферменты –
специф-е белки, выполняющие каталитич.
ф-ии в живых системах. (высокая активность
и уникальная селективность).
Закономерности
КР: 1. К не влияет на положение равновесия.
Не входит в состав исх. в-в и продуктов
реакции, не может изм. DG. Катализатор
образует с реаг. в-вами промежуточный
комплекс, который затем разрушается с
обр. продуктов реакции. Скорость
каталитич. реакции ~ концентрации
катализатора(гомогенный), площади пов.
катализатора (гетерогенный). ОСНОВНОЙ
причиной увеличения скорости реакции
в результате катализа является
значительное уменьшение Еа. В некоторых
случаях ускорение реакции обусловлено
увеличением предэкспоненциального
множителя ko. Катализатор селективен
(увеличивает скорость преимущественно
одной из возм. р-й, и не вл. заметно на
скор. др.). Селективность К м.б.
охарактеризована долей реагента,
превратившегося в целевой продукт(интегральная
селективность), или отношением скорости
образования целев. прод. к сумме скоростей
хим. прев. реагентов по всем возм.
направлениям (диффер. селективность).
Наиб. сел. обл. ферменты и некот. гомог.
кат. гетерог. <=70%. В образовании хим.
связи с реаг. участвуют отд. группы
атомов кат. – каталитические или активные
центры. В гомогенном К кажд. мол. можно
рассм. как активный центр. Акт. центры
гетер. кат. нах. на пов. ТВ. тела.
2
концепции –
слитная (одностадийная) схкма катализа,
стадийная (раздельная) схема катализа.
Согласно слитной, р-я типа А+В->Р в
присутствии К: ![]() .
АВК – активированный комплекс исх. в-в
и катализатора, r=kCaCbCk.Энергетическая
диаг.
гомог. реак. в виде двух холмов, где холм
с К – ниже… (вершина - АК). В соответствии
со стадийной схемой в-ва реагенты
послед-но взаим., образуя на кажд. стадии
соотв. АК: 1)
.
АВК – активированный комплекс исх. в-в
и катализатора, r=kCaCbCk.Энергетическая
диаг.
гомог. реак. в виде двух холмов, где холм
с К – ниже… (вершина - АК). В соответствии
со стадийной схемой в-ва реагенты
послед-но взаим., образуя на кажд. стадии
соотв. АК: 1)![]() ,
2)
,
2)![]() .
(эн. диаг. для кат-х р. с 2-мя максимумами(2-й
ниже) ).
КР цикличны.
Активность
кат-ра. Мера К активности – число оборотов
К – число циклов, сов-х на 1 активном
центре за ед. времени. Для гомог число
оборотов:
.
(эн. диаг. для кат-х р. с 2-мя максимумами(2-й
ниже) ).
КР цикличны.
Активность
кат-ра. Мера К активности – число оборотов
К – число циклов, сов-х на 1 активном
центре за ед. времени. Для гомог число
оборотов:  ,
скорость реакции, конц. катализатора.
Для гетерог. n не удается опред, поэтому
активность хар-ют скоростью р-ции,
отнесенной к ед. площади пов-ти S
катализатора:
,
скорость реакции, конц. катализатора.
Для гетерог. n не удается опред, поэтому
активность хар-ют скоростью р-ции,
отнесенной к ед. площади пов-ти S
катализатора: 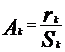 .
Одним
из важных отличий гетерогенных кат. от
гомогенных является зависимость их
св-в от условий приготовления и высокая
чувствительность к действию небольших
количеств посторонних в-в. Доб-ие нек.
ката-ки неакт. в-в иногда увел. эфф-ть К
(промоторы, модификаторы). В-ва, способные
понижать или полн. подавл. акт. кат. –
каталитические яды (отравление
катализатора).
ГОМОГЕННЫЙ
КАТАЛИЗ:
2 группы кат. пр. (как для гомог, так и для
гетеро) – гр., осуществляющиеся по
кислотно-основному механизму, по
окислительно-восстановительному.
Кислотно-основный
катализ:
Многие гомог. р-ции в р-рах катализируются
кислотами и основаниями (кислота –
в-во, склонное отдавать ион водорода,
основание – присоединять его –
сопряженное основание). Рассм. в-во В,
кот. вступает в реак. с кисл, осн. или с
ними обоими. Механизм кислотного взаим.:
1)В+НА<=>BH(+)+A(-); 2)BH(+)->P1+p2+H(+);
3)A(-)+H(+)<=>HA. Скорость КР:
.
Одним
из важных отличий гетерогенных кат. от
гомогенных является зависимость их
св-в от условий приготовления и высокая
чувствительность к действию небольших
количеств посторонних в-в. Доб-ие нек.
ката-ки неакт. в-в иногда увел. эфф-ть К
(промоторы, модификаторы). В-ва, способные
понижать или полн. подавл. акт. кат. –
каталитические яды (отравление
катализатора).
ГОМОГЕННЫЙ
КАТАЛИЗ:
2 группы кат. пр. (как для гомог, так и для
гетеро) – гр., осуществляющиеся по
кислотно-основному механизму, по
окислительно-восстановительному.
Кислотно-основный
катализ:
Многие гомог. р-ции в р-рах катализируются
кислотами и основаниями (кислота –
в-во, склонное отдавать ион водорода,
основание – присоединять его –
сопряженное основание). Рассм. в-во В,
кот. вступает в реак. с кисл, осн. или с
ними обоими. Механизм кислотного взаим.:
1)В+НА<=>BH(+)+A(-); 2)BH(+)->P1+p2+H(+);
3)A(-)+H(+)<=>HA. Скорость КР: ![]() ,
где k – константа скорости кат. реакции:
,
где k – константа скорости кат. реакции: ![]() ,
где kr-константа скорости некат. р., все
ост-е – константы скор. кат. реак. с
участием соотв. частиц
Выделяют 2
типа кислотно-осн. К – специфический и
общий кислотно-основной. Специфический
к-о К – скор. р-ции ~ конц. H+ или OH-. В этом
случае kH+ или kOH- велики по ср. с kHA и kA-.
(из Ур-ия соотв. множители убираем).. В
сильно основном р-ре – только ОН- (
,
где kr-константа скорости некат. р., все
ост-е – константы скор. кат. реак. с
участием соотв. частиц
Выделяют 2
типа кислотно-осн. К – специфический и
общий кислотно-основной. Специфический
к-о К – скор. р-ции ~ конц. H+ или OH-. В этом
случае kH+ или kOH- велики по ср. с kHA и kA-.
(из Ур-ия соотв. множители убираем).. В
сильно основном р-ре – только ОН- (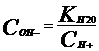 ),
КН20-ионное произведение воды. В области
промеж. знач. pH lgk не зависит от конц-й
ионов. В этом случае константа скорости
КР зависит только от kr. Реакции,
катализируемые всеми кислотами и осн.,
отн-ся к случаю общего кислотно-осн. К.
Если примен. такие условия, когда конц.
ионов Н+ и ОН- не влияют на скор., т.е.
соотв. конст. можно пренебречь, скорость
многих реакций зависит от концентраций
недиссоциированной кислоты и сопр.
основания А-. Считая, сто вклад. некат.
р-ции мал, получаем
:
),
КН20-ионное произведение воды. В области
промеж. знач. pH lgk не зависит от конц-й
ионов. В этом случае константа скорости
КР зависит только от kr. Реакции,
катализируемые всеми кислотами и осн.,
отн-ся к случаю общего кислотно-осн. К.
Если примен. такие условия, когда конц.
ионов Н+ и ОН- не влияют на скор., т.е.
соотв. конст. можно пренебречь, скорость
многих реакций зависит от концентраций
недиссоциированной кислоты и сопр.
основания А-. Считая, сто вклад. некат.
р-ции мал, получаем
: ![]() .
Окислительно-восстановительный
катализ.
Определяющей стадией многих ОВР является
перенос е от 1 реагента к др. Этот пр. в
ряде случаев протекает медленно. Тогда
применяют К, ускоряющие стадию переноса
е. (разложение пероксида водорода,
гидрирование, окисление орг. соед.
молек-м кислор.). Скорость реакции в
целом определяется скоростью лимитирующей
стадии последовательных реакций.
Автокатализ.
Явление ускорения р под действием
продукта самой ХР. В начале скор. реак.
очень низка, затем она резко увеличивается
и лишь на закл. этапе уменьшается. Нач.
пром., когда скор. низк – индукционный
период. (реакция йодирования ацетона,
кат-я в водн. р-рах ионами водорода).
Зависимость скорости от времени –
достиг. максимума, потом сниж (за счет
уменьш. конц. исх. реаг.).
.
Окислительно-восстановительный
катализ.
Определяющей стадией многих ОВР является
перенос е от 1 реагента к др. Этот пр. в
ряде случаев протекает медленно. Тогда
применяют К, ускоряющие стадию переноса
е. (разложение пероксида водорода,
гидрирование, окисление орг. соед.
молек-м кислор.). Скорость реакции в
целом определяется скоростью лимитирующей
стадии последовательных реакций.
Автокатализ.
Явление ускорения р под действием
продукта самой ХР. В начале скор. реак.
очень низка, затем она резко увеличивается
и лишь на закл. этапе уменьшается. Нач.
пром., когда скор. низк – индукционный
период. (реакция йодирования ацетона,
кат-я в водн. р-рах ионами водорода).
Зависимость скорости от времени –
достиг. максимума, потом сниж (за счет
уменьш. конц. исх. реаг.).
Катализом называют явление изменения скорости термодинамически возможной химической реакции под влиянием катализаторов – веществ, участвующих в реакции, но остающихся в неизменном количестве и составе после ее завершения. Все каталитические процессы подразделяют на две группы. К гомогенному катализу относятся процессы, в которых реагирующие вещества и катализатор находятся в одной фазе и образуют гомогенную систему, например некоторые газофазные каталитические реакции, многие каталитические реакции в растворах. Примером гомогенной каталитической реакции является окисление оксида серы (IV) кислородом в присутствии оксида азота (VI) Основным положением теории каталитических процессов является представление об образовании неустойчивых промежуточных соединений катализатора с реагирующими веществами. Для описания механизма протекания каталитических реакций в настоящее время используют две концепции. Одна из них соответствует слитной (одностадийной) схеме катализа, другая – стадийной (раздельной) схеме катализа. Согласно слитной теории катализа, некаталитическая реакция типа А + В = Р В присутствии катализатора К проходит по схеме А + В + К D (АВК) ¹ ® Р + К Здесь (АВК) ¹ -активированный комплекс исходных веществ и катализатора. Этот процесс в присутствии катализатора проходит в одну стадию и его скорость описывается кинетическим уравнением: r = k СА СВ СК Энергетическая диаграмма каталитической для некаталитической и каталитической реакции. В присутствии катализатора процесс идет с меньшей энергией активации. Чаще протекают каталитические реакции, осуществляемые по стадийной схеме. В соответствии с этой схемой вещества-реагенты последовательно взаимодействуют, образуя на каждой стадии соответствующий активированный комплекс: 1)А + К <> (АК) ->АК 2)АК + В <> (АКВ) -> Р + К согласно записанным уравнениям, промежуточное соединение АК может превратиться по обратной реакции с константой скорости k—1 в исходное вещество и катализатор или в продукты реакции Р и катализатор по второй реакции (константа скорости k2.
Каталитические реакции протекают по слитной и стадийной схемам, обычно в разных температурных интервалах. По слитной схеме часто осуществляются гомогенные каталитические реакции при температурах 300…400К и ферментативные реакции. По стадийной схеме протекают, как правило гетерогенные каталитические реакции при температурах 600…800К. Обе приведенные схемы отражают общую и характерную особенность механизма протекания каталитических реакций, а именноцикличность . в каталитических реакциях один и тот же активный центр или молекула катализатора может многократно (103-1011раз) вступать в химическое взаимодействие с молекулами реагента.
19) Понятие о гетерогенном катализе. Стадии гетерогенной каталитической реакции. Роль адсорбции в гетерогенном катализе. Энергетическая диаграмма гетерогенной каталитической реакции.
Гетерогенный
катализ.
В гетерог. К реаг-е в-ва и катализатор
находятся в разных агр. сост. Наиб. часто
катализатор предст. собой ТВ. в-во, а
реаг. являются газами или жид. Р прот.
на пов. К. => св-ва пов. сущ-но влияют на
активность К. Гетер К проц. оч сложны.
Если К имеет дост-но развитую пов-ть,
можно выд. 7 стадий кат процесса: 1)
диффузия реаг. в-в к пов. К (внешняя диф.)
2)диффузия в поры кат-ра (внутр. диф.)
3)адсорбция реагентов на пов-ти К 4)Хим.
превр.на пов-ти кат-ра(может происх. в
неск. стадий) 5) десорбция реаг. с пов. К
6)диф. продуктов р-ции в порах к внешней
пов-ти гранул катализатора. 7) диф. прод.
от внешн. пов. К
В ГК значит. роль
играет адсорбция (К р-ция происх. в
пов-ном слое).
Энерг.
диагр.
– отличается от гомог. кат. наличием
энтальпий адс. и дес. Мол. прод. должны
быть оторв. от пов-ти, на это затр. Е
(энтальпия дес.). Только некот. особ.
сочет. Еа, Ндес. и адс. будет спос. ускорению
К проц.
Кинетика
гетерог КР:
Для ГК пр. роль одного из реагентов
играют адсорбционные центры ТВ. пов-ти,
кот. следует включать в соотв. Ур-ия.
Элем. ХР между адс-ми частицами: ![]() .
Если прод. реак. заним. на пов. больше
адс. центров, чем исх. в-ва на вел. Dn, то
превр-ие в адсорбционном слое окажется
возможным при наличии недост. числоа
свободн. центров Q адс. Поэт. стех. Ур.
можно запис:
.
Если прод. реак. заним. на пов. больше
адс. центров, чем исх. в-ва на вел. Dn, то
превр-ие в адсорбционном слое окажется
возможным при наличии недост. числоа
свободн. центров Q адс. Поэт. стех. Ур.
можно запис:
![]() .
Если этот процесс протек в 1 стадию, то
скорость:
.
Если этот процесс протек в 1 стадию, то
скорость: ![]() .
(Dn>0). Если в элем. акте. прин. уч-ие мол.
в-ва А, не занимающие адс. центров на
пов. К (пост-ие из газовой фазы), то Ур-ие
прин. вид. – вместо а просто А(г). а выр-ие
скорости
.
(Dn>0). Если в элем. акте. прин. уч-ие мол.
в-ва А, не занимающие адс. центров на
пов. К (пост-ие из газовой фазы), то Ур-ие
прин. вид. – вместо а просто А(г). а выр-ие
скорости ![]() .
Существенное
отличие гетерог. проц. от гомогенного
сост в том, что для дан. элем. р-ции
порядок. и эффект. Еа зависят от
адсорбционной спос-ти всех присутств.
в сист. в-в. Если идет превр-ие адс. молекул
Аадс->Вадс+Dадс. Записываем Ур-ие
изотермы. Допускаем, что B и D адс-ся
слабо. Тогда, опять, 3 случая : А адс. слабо
(р-ция следует кинетике 1 пор). В-во А адс.
со средн. силой: r=kpa*m, где обычно m<1
(кинетика дробного порядка). 3. В-во А
адс-ся сильно. r=k.
.
Существенное
отличие гетерог. проц. от гомогенного
сост в том, что для дан. элем. р-ции
порядок. и эффект. Еа зависят от
адсорбционной спос-ти всех присутств.
в сист. в-в. Если идет превр-ие адс. молекул
Аадс->Вадс+Dадс. Записываем Ур-ие
изотермы. Допускаем, что B и D адс-ся
слабо. Тогда, опять, 3 случая : А адс. слабо
(р-ция следует кинетике 1 пор). В-во А адс.
со средн. силой: r=kpa*m, где обычно m<1
(кинетика дробного порядка). 3. В-во А
адс-ся сильно. r=k.
В гетерогенном катализе реагирующие вещества и катализатор находятся в разных агрегатных состояниях. Наиболее часто катализатор представляет собой твердое вещество, а реагенты являются газами или жидкостями. Реакция протекает на поверхности катализатора. Следовательно свойства поверхности (площадь, химический состав поверхностного слоя, его структура) существенно влияют на активность катализатора. Гетерогенные каталитические процессы очень сложны. Если катализатор имеет достаточно развитую поверхность, то можно выделить семь стадий каталитического процесса: -диффузия реагирующих веществ к поверхности катализатора (внешняя диффузия); -диффузия в поры катализатора (внутренняя диффузия); -адсорбция реагента на поверхности катализатора; -химическое превращение на поверхности катализатора (которое может происходить в несколько стадий); -десорбция продуктов с поверхности катализатора; -диффузия продуктов реакции в порах к внешней поверхности гранул катализатора; -диффузия продуктов от внешней поверхности катализатора. В гетерогенном катализе значительную роль играет адсорбция, так как каталитическая реакция протекает в поверхностном слое. Адсорбированные молекулы определенным образом ориентированы к поверхности, причем уменьшение энергии активации происходит за счет перераспределения связей и повышения энтропии активации. Энергетические диаграммы реакции, проходящей с участием гетерогенного катализатора и без него (стр 290 рис) отличаются от аналогичных диаграмм с участием гомогенного катализатора наличием энтальпии адсорбции и десорбции. Очень сильное адсорбционное взаимодействие может препятствовать прохождению химической реакции. Молекулы продуктов должны быть десорбированы (оторваны) от поверхности на это затрачивается энергия (энтальпия десорбции dНД>0) Слишком большое значение энтальпии десорбции также может затруднять протекание процесса. Только некоторое оптимальное сочетание энергии активации, энтальпии адсорбции и десорбции будет способствовать ускорению каталитического процесса.
20 Растворы. Современные представления о физико-химических процессах образования растворов. Энергетика процесса растворения. Ненасыщенный, насыщенный и пересыщенный растворы. Растворимость твердых веществ, зависимость растворимости от температуры, давления. Закон Генри-Дальтона. Способы выражения концентрации растворов.
![]()



![]()
![]()
![]()
![]()

![]()


![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()

![]()
![]()
![]()
В развитии физической химии исключительно важную роль сыграло изучение природы растворов, которые имеют огромное значение в жизни человека, животных и растений. Многие процессы, протекающие в земной коре и на ее поверхности, связаны с растворами, ни одно промышленное производство не обходится без их участия. Ученые многих поколений интересовались растворами. Еще Менделеев писал о взаимодействии растворителя и растворенного вещества. Растворами называются гомогенные (однородные) системы, состоящие из растворенных веществ, растворителей и продуктов их взаимодействия.Растворы могут представлять собой системы, находящиеся в газообразном, жидком или твердом состоянии (сплавы). Растворами называют однородные смеси нескольких компонентов. Содержание каждого компонента может непрерывно изменяться. Особенно часто в практике встречаются жидкие растворы (водные). Растворенные вещества могут быть твердыми, жидкими и газообразными. Свойства растворов определяются характером взаимодействия компонентов. Это взаимодействие зависит не только от вида сил, действующих между частицами, но и от формы и размеров частиц. Общий признак растворов – стремление к смешению. Различают растворы истинные и коллоидные. В истинных растворах вещества находятся в виде ионов или молекул, а в коллоидных в виде групп молекул (агрегатов определенных размеров). Важной характеристикой растворов является концентрация, показывающая в каком соотношении (весовом или объемном) взяты растворитель и растворенное вещество. Однородность растворов делает их очень сходными с химическими соединениями. Отличие их в том, что состав растворов может изменяться. Непостоянство растворов приближает их к механическим смесям, но от них они резко отличаются своей однородностью. Вывод. Растворы занимают промежуточное положение между механическими смесями и химическими соединениями. В настоящем растворе протекают одновременно два процесса: растворение и кристаллизация. Под влиянием растворителя от поверхности твердого вещества постоянно отрываются отдельные молекулы или ионы (растворимость твердых веществ) и равномерно распределяются по всему объему раствора. По мере увеличения концентрации раствора скорость растворения уменьшается, т.к. при этом протекает обратный процесс – процесс кристаллизации (молекулы, ионы сталкиваются с кристаллами и кристаллизуются). Через некоторое время наступает процесс его насыщения. В насыщенном растворе одновременно протекают два процесса: растворение и кристаллизация. При постоянной температуре скорость растворения и скорость кристаллизации равны, а поэтому концентрация насыщенного раствора при постоянной температуре постоянна. Следовательно, насыщенным называется раствор, который находится в динамическом равновесии с избытком растворенного вещества. Если концентрация какого-либо раствора будет меньше концентрации насыщенного раствора при данной температуре, то он называется ненасыщенным. Возможно дополнительное растворение до предела насыщения. Раствор, концентрация которого больше концентрации насыщенного при данных условиях, называют пересыщенным. Такие растворы получаются при осторожном охлаждении ненасыщенных растворов, концентрация которых близка к концентрации насыщенных. Растворимость понижается и содержание вещества окажется большим, чем в его насыщенном при такой же температуре, т.е. происходит пересыщение. Эти растворы очень неустойчивы. Встряхивание, попадание пыли и сразу же начинается кристаллизация. Растворимость твердых веществ всегда ограниченна. Абсолютно нерастворимых веществ в природе нет, хотя есть практически нерастворимые ВаSО4, Са СО3, Си(ОН)2, и др. Малорастворимые Са(ОН)2, Рв Сl2. самопроизвольное растворение идет медленно, его увеличивают измельчением в порошок, перемешиванием, повышением температуры. Влияние температуры разное у разных веществ. Зависимость растворимости твердых тел от температуры выражают в виде кривых (таблицы растворимости). Некоторые вещества при повышении температуры резко повышают растворимость, у других она падает. Растворимость газов падает. Понятие «растворимость» имеет качественный смысл. В качественном смысле растворимостью вещества называется его способность образовывать однородную систему с другим веществом, выполняющим функцию растворителя. Эта способность определяется характером взаимодействия между молекулами растворитель – растворитель, растворенное вещество – растворенное вещество, растворитель – растворенное вещество. Наибольшая взаимная растворимость достигается тогда, когда все эти силы имеют подобный характер. Неполярные или малополярные соединения хорошо растворимы в неполярных или малополярных растворителях и менее растворимы в высокополярных растворителях.. Так оксид углерода СО – малополярное соединение (дипольный момент 0,4×10-30Кл×м) – хорошо растворим в бензоле, молекулы которого неполярны (=0), и ограниченно растворим в воде – соединении с сильно выраженным дипольным характером (дипольный момент 6,11×10-30Кл×м). Вода является хорошим растворителем полярных соединений, например аммиака или этилового спирта, не только потому, что их молекулы обладают значительной полярностью (соответственно 4,94×10-30Кл×м и 5,66×10-30Кл×м), но и потому, что при этом сохраняется характер связей, существовавших в исходных компонентах. Вместо водородных связей между молекулами каждого компонента – воды, аммиака и спирта возникают подобные вязи между растворителем и растворенным веществом. Схема гидратации хлороводорода стр 422. Растворимость газов в жидкостях определяется не только природой компонентов, но и изменяется в широких пределах в зависимости от давления и температуры. С увеличением давления растворимость газов в жидкостях увеличивается. Эта закономерность выражается законом Генри: рi = Кi Хi где Хi – молярная доля i –го газа в растворе; рi – парциальное давление газа над раствором; Кi – постоянная Генри для i –го компонента. Закон Генри не выполняется, если растворение газа в жидкости сопровождается образованием новых химических веществ. Например, растворение в воде аммиака приводит к образованию гидроксида аммония, а растворение углекислого газа - к образованию угольной кислоты. Влияние температуры на растворимость количественно выражается уравнением: (dlnХi /dТ)р = dН*интi/RТ2 где dsdН*интi – энтальпия перехода 1 моль i –го компонента из чистого состояния в состояние насыщенного раствора. Если dsН*интi > 0, то растворимость увеличивается с ростом температуры, если dsН*интi <0 – уменьшается.Растворение газов в жидкостях в основном экзотермический процесс, поэтому растворимость большинства газов уменьшается с ростом температуры. Например, при Т=0оС в 100 г воды растворяется 5,56 мл СН4, а при Т=100оС – 1,7 мл Термодинамическим условием самопроизвольного образования раствора является уменьшение энергии Гиббса системы dsG<0, определяемой по уравнению: dsG = dsН -Тds S, где dsН – энтальпия (теплота) растворения; ds S энтропия растворения. Образование растворов протекает как с поглощением (dsН>0), так и с выделением (dsН <0) теплоты, поскольку сопровождается эндотермическими процессами разрушения исходных связей растворителя и растворенного вещества и экзотермическим процессом образования новых связей (сольватацией). Состав раствора может быть задан несколькими способами: Концентрацией растворенного вещества называют отношение количества растворенного вещества или его массы к объему раствора (моль/л, г/л). Концентрация – это отношение неоднотипных величин. Те величины, которые являются отношением однотипных величин, например отношение массы растворенного вещества к массе раствора, и формально не имеют своей единицы, называются долями. Таким образом, состав раствора может быть задан как концентрацией, так и долей растворенного вещества. Массовая доля растворенного вещества w –это отношение массы растворенного вещества к массе раствора. Объемная доля растворенного вещества v – это отношение объема растворенного вещества к объему раствора. Мольрность раствора – величина, численно равна отношению молярной массы растворенного вещества к объему раствора (моль⁄л).
21Р-р
называется идеальным, если химический
потенциал каждого компонента линейно
зависит от логарифма мольной доли этого
компонента.
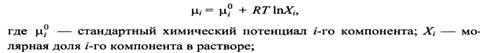
![]()
![]() Сл-я:
Т(кип)ра-ра выше Т(кип)раств-ля и
Т(крис)рас-ра ниже Т(крист)раств-ля.
Сл-я:
Т(кип)ра-ра выше Т(кип)раств-ля и
Т(крис)рас-ра ниже Т(крист)раств-ля.

![]()
![]()

![]()
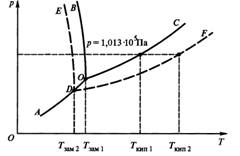
Существует несколько классификаций растворов. С точки зрения термодинамики целесообразно различать растворы идеальные и неидеальные или реальные. В идеальных растворах внутренняя энергия каждого компонента не зависит от концентрации, и молярный объем не изменяется при растворении. Компоненты при этом смешиваются как идеальные газы, и увеличение энтропии можно рассчитать по уравнениям, характерным для идеальных газов. Здесь сил взаимодействия между частицами нет, и вещества смешиваются без выделения или поглощения теплоты. При исследовании растворов широко пользуются методом моделей. Простейшей моделью является идеальный раствор. Идеальные растворы делят обычно на две группы: разбавленные (более точно бесконечно разбавленные) и совершенные. Образование идеального раствора не сопровождается изменением объема, тепловым эффектом, химическим взаимодействием. Такие растворы образуются в результате простого физического смешения (d Нсм = 0; dVсм = 0). Такие растворы получаются смешиванием неполярных жидкостей, характеризующихся близкими по силе молекулярными полями. В таких смесях отсутствуют явления сольватации, а отсюда нет и теплового эффекта растворения, нет и концентрации системы. Объем раствора равен сумме объемов смешиваемых компонентов. Это и есть идеальные или совершенные растворы. Роль их в теории растворов аналогична роли идеальных газов (потому их и называют идеальными растворами). Идеальные растворы довольно распространены. Пример: смешивание изомеров углеводорода (октаны и др.). Бензин, керосин, смесь бензола и толуола –идеальные растворы, представляют собой смесь различных углеводородов (жидких). Идеальные растворы имеют характер простых молекулярных смесей. К ним подходит «физическая» теория растворов. Физическая теория растворов предложена Вант-Гоффом и Аррениусом в 19 в. Согласно этой теории растворитель рассматривается как среда, в которой при растворении вещества его молекулы равномерно размещаются по всему объему раствора, межмолекулярные взаимодействия отсутствуют. Законы Рауля. Важнейшей характеристикой вещества, находящегося в жидком состоянии или твердом является давление насыщенного пара, это давление – константа вещества, определяющая равновесие жидкость ó пар, твердое веществоó пар. Само равновесие достигается, когда процессы испарения компенсируется процессами конденсации. При нагревании давление пара возрастает. Для разбавленных растворов относительное понижение давления пара растворителя (А) численно равно мольной доле растворенного вещества (В) (первый закон Рауля).
РА0 - РА = ХВ РА0 |
Так как для любого раствора сумма мольных долей Х А + Х В = 1, то формула будет
1 - РА = ХВ = 1 – Х А . Отсюда РА = РА0 × ХА РА0 |
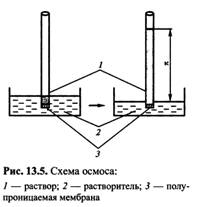
Где РА0 - давление пара чистого растворителя; РА – давление пара растворителя над раствором; ХА – мольная доля растворителя; Х В – мольная доля растворенного вещества. Таким образом, для разбавленных растворов давление пара растворителя пропорционально его мольной доле в растворе. Из закона Рауля возникает два следствия. Согласно одному из них температура кипения раствора выше температуры кипения растворителя. Это обусловлено тем, что давление насыщенного пара растворителя над раствором становится равным атмосферному давлению (условие кипения жидкости) при более высокой температуре, чем в случае чистого растворителя. Повышение температуры кипения d Ткип пропорционально моляльности раствора: сm. d Ткип= Кэ сm где Кэ – эбулиоскопическая постоянная растворителя Согласно второму следствию из закона Рауля температура замерзания (кристаллизации) раствора ниже температуры замерзания (кристаллизации) чистого растворителя. Это обусловлено более низким давлением пара растворителя над раствором, чем над растворителем. Понижение температуры замерзания (кристаллизации) d Тзам пропорционально моляльности раствора: d Тзам= Кк сm где Кк - криоскопическая постоянная раствора. Диаграмма состояния представляет собой графическое изображение зависимости между различными величинами, характеризующими состояние системы. Для однокомпонентных систем обычно используют диаграммы состояния, показывающие зависимость между давлением и температурой. Они называются фазовыми диаграммами состояния. Диаграмма показывает те состояния воды, которые термодинамически устойчивы при определенных значениях температуры и давления. Она состоит из трех кривых, разграничивающих все возможные температуры и давления на три области, отвечающие льду, жидкости и пару. Все три кривые пересекаются одной точке О. координаты этой точки – это единственная пара значений температуры и давления, при которых в равновесии могут находиться все три фазы: лед, жидкая вода и пар. Она носит название тройной точки. Кривая плавления исследована до весьма высоких температур. Кривая кипения оканчивается в критической точке. При температуре, отвечающей этой точке, - критической температуре – величины, характеризующие физические свойства жидкости и пара, становятся одинаковыми, так, что различие между жидким и парообразным сосоянием исчезает. Одной из особенностей воды, отличающих ее от других веществ, является понижение температуры плавления льда с ростом давления. Это обстоятельство отражается на диаграмме. Кривая плавления на диаграмме идет вверх влево, тогда как почти для всех других веществ она идет вверх вправо. Температура замерзания раствора ниже температуры замерзания воды, а температура кипения раствора выше температуры кипения воды. Самопроизвольный переход растворителя через полупроницаемую мембрану, разделяющую раствор и растворитель или два раствора с различной концентрацией растворенного вещества, называется осмосом. Осмос обусловлен диффузией молекул растворителя через полупроницаемую перегородку, которая пропускает только молекулы растворителя. Молекулы растворителя диффундируют из растворителя в раствор или из менее концентрированного раствора в более концентрированный, поэтому концентрированный раствор разбавляется (химический потенциал растворителя в растворе меньше химического потенциала чистого растворителя), при этом увеличивается и высота его столба. Количественно осмос характеризуется осмотическим давлением, равным силе, приходящейся на единицу площади поверхности, и заставляющей молекулы растворителя проникать через полупроницаемую перегородку. Оно равно давлению столба раствора в осмометре высотой h. При равновесии внешнее давление уравновешивает осмотическое давление. В этом случае скорости прямого и обратного переходов молекул через полупроницаемую перегородку становится одинаковыми. Если внешнее давление, приложенное к более концентрированному раствору, выше осмотического p, т.е. р>p, то скорость перехода молекул растворителя из концентрированного раствора будет больше, и растворитель будет переходить в разбавленный раствор (или чистый растворитель). Этот процесс, называемый обратным осмосом, используется для природных и сточных вод, для получения питьевой воды из морской. Осмотическое давление возрастает с увеличением концентрации растворенного вещества и температуры. Вант-Гофф предположил, что для осмотического давления можно применить уравнение состояния идеального газа: pV = nRТ или p = (n/V ) RТ откуда p = с RТ , где p - осмотическое давление (кПа), с – молярная концентрация раствора. Осмотическое давление прямо пропорционально молярной концентрации растворенного вещества и температуре. Осмос играет очень важную роль в биологических процессах, обеспечивая поступление воды в клетки и другие структуры. Растворы с одинаковым осмотическим давлением называются изотоническими. Если осмотическое давление выше внутриклеточного, то оно называется гипертоническим, если ниже внутриклеточного - гипотоническим.
22 Растворы электролитов. Теория электролитической диссоциации Аррениуса: степень диссоциации, константа диссоциации. Факторы, влияющие на них. Закон разбавления Оствальда.
![]()

![]()
![]()
![]()
![]()


По способности веществ распадаться или не распадаться в расплаве или растворе на катионы и анионы различают электролиты и неэлектролиты. Электролиты – вещества, которые подвергаются электролитической диссоциации, и вследствие чего их расплавы или растворы проводят электрический ток. К электролитам принадлежат все соли, а также кислотные, основные и амфотерные гидроксиды. Раствор электролита представляет собой смесь молекул растворителя и сольватированных (ионы растворенного вещества, окруженные соответственно ориентированными диполями растворителя) молекул и ионов растворенного вещества. Относительное количество молекул, распавшихся на ионы, характеризующее степень диссоциации электролита α , зависит от природы растворителя, природы и концентрации электролита, температуры, давления и наличия других электролитов в растворе. Процесс распада полярного вещества в растворе на ионы называют электролитической диссоциацией (ионного – ионизацией). По способности к электролитической диссоциации электролиты обычно подразделяют на сильные и слабые. К сильным электролитам обычно относят вещества, которые в растворе практически полностью диссоциированы на ионы. Слабыми электролитами считают вещества, степень диссоциации, которых невелика. Понятие степень диссоциации электролита α как величины, равной отношению числа распавшихся (диссоциированных) молекул Nдисс к общему числу молекул N0 электролита, α = Nдисс /Nо было введеноАррениусом – создателем первой количественной теории растворов электролитов. Теория электролитической диссоциации и основанная на ней классификация кислот и оснований в полной мере применимы лишь к водным растворам. Процесс электролитической диссоциации возникающий в результате сольватации, обратим, т.е. наряду с равпадом молекул растворенного вещества происходит их образование из ионов: Кm Аn D mКZ1+ + nАZ2- где Кm Аn – молекула электролита; КZ1+- катион; АZ2- - анион; Z1 и Z2 - заряд аниона и катиона соответственно; n и m – стехиометрические коэффициенты. Равновесие между ионами и молекулами электролита подчиняется закону действия масс. Поэтому важной характеристикой процесса диссоциации является константа диссоциации (константа ионизации) Кd (С) , вычисленная по равновесным концентрациям молекул и ионов: Кd (С) = [КZ1+] m [АZ2-]n ⁄[К mАn] , где [КZ1+], [АZ2-] –равновесные молярные концентрации катионов и анионов соответственно; [К mАn] - равновесная молярная концентрация недиссоциированных молекул электролита. Константу равновесия процесса диссоциации принято обозначать Ка в случае слабых кислот и Кb для слабых оснований. Пример диссоциации слабого основания (гидроксид аммония) NН4ОН D NН4+ + ОН- Кb = [NН4+] [ОН-] /[NН4ОН] = 1,8 ×10-5 Многоосновные кислоты и многоосновные основания диссоциируют ступенчато. Константа диссоциации характеризует процесс диссоциации данного электролита в данном растворителе, но не зависит от концентрации электролита и при постоянной температуре Кd (С) =соnst. Очевидно, что степень диссоциации α тем больше, чем ниже концентрация, т.е. чем сильнее разбавлен раствор. В состоянии равновесия концентрации катионов КZ+ и анионов АZ- будут равны [КZ+] = [АZ-] = α С, а концентрация недиссоциированных молекул [КА] = (С – α С) = С ( 1- α) подставляя эти выражения в уравнение для константы диссоциации получим: Кd (С) = α2 × С 1- α - это выражение описывает закон разбавления (разведения) Оствальда для слабых электролитов. В случае когда степень диссоциации электролита α<<1, что имеет место при С /Кd (С) >100, величиной α по сравнению с 1 можно пренебречь и закон разбавления Оствальда записать в упрощенном варианте Кd (С) = α2С. Из закона разбавления Оствальда следует, что степень диссоциации уменьшается с увеличением концентрации слабого электролита.
23 ) Закон Рауля для растворов электролитов. Коэффициент диссоциации (i) и его связь со степенью диссоциации. Элементы современной теории сильных электролитов (теория Дебая-Хюккеля). Ионная сила, активность и коэффициент активности.

![]()

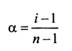

![]()
![]()
![]()

![]()
![]() на
на
Понижение давления пара растворителя над раствором определяется в основном количеством растворенных частиц. Однако количество растворенных частиц в растворах электролитов, в отличие от растворов неэлектролитов, определяется не только концентрацией раствора, но и степенью диссоциации электролита, поскольку все молекулы или часть молекул электролита в растворе распадаются на ионы. Применяя закон Рауля к растворам электролитов Вант-Гофф ввел поправочный коэффициент i в уравнение для осмотического давления Р = С R Т. Коэффициент i , учитывает увеличение числа частиц в растворе в результате электролитической диссоциации: dр/р0i = i Х2 (Р = i С R Т). Коэффициент диссоциации i показывает, во сколько раз число частиц в растворе электролита больше числа частиц в растворе неэлектролита той же концентрации, (для растворов неэлектролитов i=1, а для растворов электролитов i> 1). При диссоциации уксусной кислоты количество образовавшихся ионов n=2. (СН3СООН DСН3СОО- + Н+). Число ионов в 1л раствора Nион = anСNА , А число недиссоциированных молекул растворенного вещества Nнедисс = (1-a)СNА , Где a = Nдис/N0 - степень диссоциации электролита (N0 = Nдисс + Nнедисс), с – молярная концентрация раствора (моль/л). Таким образом, коэффициент диссоциации i связан со степенью диссоциации a электролита соотношением: a = (i - 1) /(n-1).и, значит по относительному изменению давления пара растворителя над раствором известной концентрации можно определить степень диссоциации электролита. Растворы сильных электролитов обнаруживают особенности в поведении, не соответствующие их полной диссоциации на ионы. Так, реальная концентрация ионов оказывается значительно меньше концентрации, задаваемой при приготовлении раствора. Кажущаяся (определяемая экспериментально) степень диссоциации сильных электролитов в соответствии с опытными данными меньше 1 даже в разбавленных растворах. Это связано с тем, что в растворах электролитов наблюдается некоторая степень упорядоченности взаимного расположения ионов, вызванная электростатическим взаимодействием катионов и анионов. На небольших расстояниях от каждого иона преимущественно располагаются ионы противоположного знака, т.е. вокруг каждого иона в растворе создается ионная атмосфера. Таким образом, для процессов диссоциации и химических реакций, протекающих в растворах с участием сильных электролитов, а также в концентрированных растворах слабых электролитов, нельзя рассчитывать константы равновесия на основании концентраций свободных ионов, которых нет в реальных системах. Кроме того, различная степень сольватации веществ, участвующих в реакции, по-разному изменяет скорости прямой и обратной реакций, что также приводит к зависимости константы равновесия от общего содержания ионов в растворе. Поэтому для описания свойств реальных растворов, как и других реальных систем используют метод активностей Льюиса, в котором для учета межионных и межмолекулярных взаимодействий введено понятие эффективной концентрации или активности. Подстановка активности вместо концентрации в термодинамические соотношения, справедливые для идеальных растворов, позволяет применять их для описания любых систем. Активность a электролита суммарно отражает все эффекты взаимодействия ионов между собой и с молекулами растворителя: a = g Сm, где Сm – моляльная концентрация электролита; g - коэффициент активности, который можно рассматривать как меру различия поведения электролита в данном растворе и в растворе, который принимают за идеальный. Для идеальных растворов g = 1. Бесконечно разбавленные растворы по своим сойствам приближаются к идеальным, поэтому в таких растворах полагают g»1. Коэффициенты активности и, следовательно, сами активности определяют экспериментально, измеряя различные свойства раствора, например, давление пара растворителя, температуру кипения или кристаллизации раствора и др. Электростатическая теория сильных электролитов, развитая в трудах Дебая, Хюккеля, позволяет вычислить средний коэффициент активности g± -сильного бинарного электролита в разбавленных растворах. Сила электростатического взаимодействия ионов с их окружением (ионной атмосферой) определяется плотностью заряда в этом окружении, а плотность заряда, в свою очередь, зависит от того, сколько ионов находится в единице объема раствора, т.е. от их концентрации, и от того, какой заряд несут эти ионы. Мерой этого взаимодействия является ионная сила раствора I, рассчитываемая по формуле: I = 0,5 S Сm,iZi 2 где Сm,i - моляльная концентрация i –иона; Zi – зарядовое число i –го компонента. В очень разбавленных растворах (I<0,1) средний коэффициент активности электролита зависит только от ионной силы раствора и не зависит от природы присутствующих в растворе ионов.
24) Электрическая проводимость растворов электролитов: удельная и молярная. Зависимость электропроводности от концентрации растворов сильных и слабых электролитов. Закон Кольрауша. Коэффициент электропроводности и степень диссоциации.
![]()
![]()
![]()
![]()
![]()



![]()

![]()
![]()
![]()
![]()
24) Равновесия в водных растворах слабых электролитов. Диссоциация воды. Ионное произведение воды. Водородный и гидроксидный показатели.
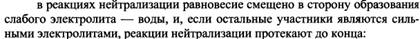
![]()

![]()

![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
![]()
![]()
![]()
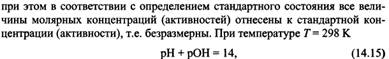
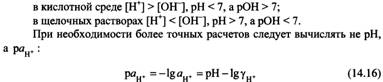
26) Равновесие в системе труднорастворимый электролит - его насыщенный раствор. Произведение растворимости (ПР). Расчет растворимости соединения по значению ПР. Условия растворения и образования осадка.
![]()
![]()
![]()
![]()

![]()

![]()
![]()
![]()
![]()
27) Направление протекания реакций с участием электролитов (образование осадка, газа, слабого электролита). Гидролиз солей. Типы реакций гидролиза. Степень и константа гидролиза.
(дополнение)

![]()
![]()
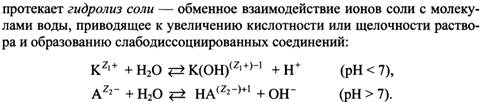

![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
![]()
![]()


![]()
 Если
гидролиз протекает либо по катиону,
либо по аниону:
Если
гидролиз протекает либо по катиону,
либо по аниону:
![]()
![]()
 Если
и по катиону и по аниону, то
Если
и по катиону и по аниону, то
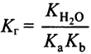
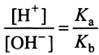
28) Возникновение двойного электрического слоя и скачка потенциала на границе металл - раствор электролита. Стандартный электродный потенциал. Понятие о стандартном водородном электроде. Уравнение Нернста для расчета электродного потенциала. Ряд напряжений металлов.

![]()
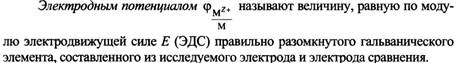


![]() уравнение
Нернста
уравнение
Нернста


29) Классификация электродов (1,2 рода). Металлические электроды. Газовые электроды: водородный, кислородный. Зависимость потенциалов водородного и кислородного электродов от рН.

![]()
![]()
![]()

![]()
Уравнение
электродного процесса ![]()

![]()
![]()
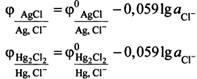
Система металл, погруженный в раствор электролита, называется электродом. Электроды подразделяются на обратимые и необратимые. Если изменить направление электрического тока во внешней цепи на противоположное, то на обратимом электроде протекает тот же самый процесс в обратном направлении, а на необратимом – другой процесс. Серебряная пластинка, находящаяся в растворе нитрата серебра, представляет собой обратимый электрод. Электродный процесс Аg+ +е DАg Протекает в прямом и обратном направлениях. Серебряная пластинка, находящаяся в растворе кислоты, служит примером необратимого электрода. В зависимости от направления тока во внешней цепи на электроде происходит восстановление катионов водорода 2Н+ + 2е ® Н2# Или окисление атомов серебра Аg ® Аg+ +е. В зависимости от свойств веществ и заряженных частиц, участвующих в электрохимических процессах, и характера равновесия обратимые электроды классифицируют на электроды первого второго рода, окислительно-восстановительные и ионообменные. Электрод первого рода представляет пластинку, изготовленную из простого вещества (металла или полупроводника) и погруженную в раствор, содержащий его ионы. В качестве примера можно привести серебряный и селеновый электроды. Аg + | Аg : Sе2- | Sе Для их электродных процессов характерно участие только одного вида ионов: Аg + + е D Аg Sе + 2е D Sе2- Электроды второго рода представляют собой металл, покрытый слоем его малорастворимого соединения (соли, оксиды, гидроксиды) и погруженный в раствор, содержащий анионы, одноименные с анионами труднорастворимого соединения. Условная запись электрода второго рода АZ- | МА, М. в качестве примера можно привести хлоридсеребряный Сl | Аg Сl, Аg процесс протекающий на электроде Аg Сlт + е D Аgт + Сl-р. В электродах второго рода окисленной формой является малорастворимое соединение (МА), восстановленной – атом металла (М) и анион раствора (АZ-). Среди электродов первого рода в отдельную группу выделяют газовые электроды, к которым относятся водородный, кислородный электроды и др. водородный электрод обратим относительно катиона, кислородный относительно аниона. Все газовые электроды конструктивно устроены одинаково. Они представляют собой инертный металл (Рt) с развитой поверхностью, хорошо проводящей электрический ток и обладающий каталитическими свойствами по отношению к электродному процессу. Платиновая пластинка электролитически покрывается слоем мелкодисперсной платины с целью увеличения адсорбции газа поверхностью металла. Платина одновременно контактирует с газом и раствором, содержащим соответствующие ионы. В стандартном кислородном электроде платиновая пластинка погружена в раствор щелочи (NаОН, КОН), с активностью гидроксид ионов равной 1 моль/л. Давление чистого кислорода (или его парциальное давление в смеси газов над раствором) составляет 101,3 кПа. В щелочной среде кислородному электроду Н20, ОН- | О2 Рt соответствует уравнение электродного процесса О2 г + 2Н2О р + 4е D 4ОН-р схема кислородного электрода в кислотной среде Н2О, Н+ | О2 Рt схема водородного электрода в щелочной и нейтральной средах: Н2О, ОН- | Н2, Рt уравнения электродных процессов: 2Н2Ор+ 2е D Н2 г + 2ОН- р О2 г + 4Н+р +4е D 2Н2Ор Зависимость электродного потенциала водородного электрода от рН j Н+/Н2 = - 0,059 рН - 0,0295 lg рН2 при рН2 =1 j Н+/Н2 = - 0,059 рН (для чистой воды рН = 7 электродный потенциал равен -0,414В) Зависимость электродного потенциала кислородного электрода от рН jО2,Н2О/ОН- = 1,229 – 0,059 рН + 0,0147 lg рО2 при рО2 = 1 jО2,Н2О/ОН- = 1,229 – 0,059 рН Анализируя уравнение электродного потенциала для водородного электрода, можно сделать вывод, что потенциал водородного электрода линейно увеличивается с уменьшением водородного показателя рН (ростом кислотности) среды и уменьшением парциального давления газообразного водорода над раствором. Потенциал кислородного электрода линейно увеличивается с уменьшением рН раствора и увеличением парциального давления газообразного кислорода над ним.
30) Гальванические элементы и их классификация. Процессы, протекающие при работе ГЭ. Расчет ЭДС и работы ГЭ. Окислительно-восстановительные и концентрационные ГЭ. Определение рН раствора.

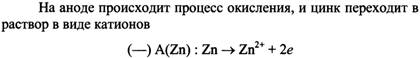
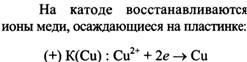
![]()

![]()
![]()

![]()
![]()

![]()
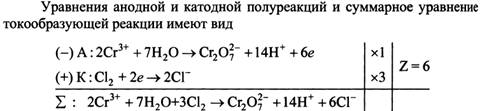

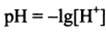
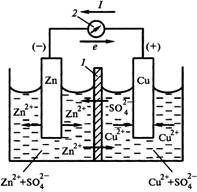
Если два различных металла погрузить в раствор электролита, то между ними возникает электрическое напряжение (разность потенциалов). Такая схема: Металл 1 ⁄ Раствор электролита ⁄ Металл 2 называется гальваническим элементом, или гальванической цепью. Вместо металлов в гальванической цепи можно использовать и другие вещества с металлической проводимость, например графит (угольный электрод). Возникновение разности потенциалов между обоими металлическими электродами объясняется различной склонностью металлов отдавать катионы в раствор электролита. У поверхности каждого из электродов возникает двойной электрический слой, который оказывает противодействие дальнейшему переходу катионов в раствор. Если оба металла соединить металлическим проводником (обладающим электронной проводимостью), то вследствие электропроводимости раствора электролита (ионной проводимости) получается замкнутая электрическая цепь. В этой цепи поток электронов будет перемещаться от менее благородного металла через внешний участок цепи (металлический проводник) к более благородному металлу. При этом в растворе электролита катионы будут двигаться к благородному металлу и разряжаться под действием имеющихся на нем электронов. В результате в замкнутой гальванической цепи возникает электрический ток. Каждый гальванический элемент состоит из двух электродов (окислительно-восстановительных пар), один из которых является поставщиком электронов, а другой их принимает. При этом на одном электроде возникает избыток электронов, а на другом - недостаток. Электрод с избытком электронов называют отрицательным полюсом гальванического элемента, или анодом, а электрод с недостатком электронов - положительным полюсом, или катодом. Отрицательным полюсом гальванического элемента является менее благородный металл, на котором имеется избыток электронов. Положительным полюсом гальванического элемента является более благородный металл, на котором имеется недостаток электронов. Электроны по внешнему участку цепи (по металлическому проводнику) переходит от отрицательного к положительному полюсу гальванического элемента. При условиях примерно равных концентраций электронов в растворах, в которые погружены электроды гальванического элемента, металл с меньшим значением стандартного потенциала будет отрицательным полюсом, а металл с большим значением стандартного потенциала – положительным полюсом. (в гальваническом элементе с цинковым и свинцовым электродами отрицательным полюсом будет цинк (φ0=- 0,763В), а положительным полюсом свинец (φ0=- 0,126В). (стандартный потенциал свинца более положителен, чем цинка) На аноде происходит процесс окисления, и цинк переходит в раствор в виде катионов. Масса цинковой пластинки уменьшается, остающиеся на ней электроны сообщают ей отрицательный заряд. Напряжение гальванического элемента тем выше, чем больше отличаются между собой значения стандартного потенциала электродов. Напряжение, которое показывает вольтметр, подключенный к полюсам гальванического элемента, называется напряжением на клеммах. Это напряжение вследствие наличия внутреннего сопротивления источника напряжения меньше действительного напряжения, называемого электродвижущей силой, сокращенно э.д.с. (обозначение U). Действительное напряжение гальванического элемента равно разности между стандартным потенциалом положительного полюса и стандартным потенциалом отрицательного полюса. U = φ0 пол. п. - φ0 отр . Если на электродах испытывает превращение один моль вещества, то по закону Фарадея через систему протекает количество электричества равное ΖF, где Ζ – число молей эквивалентов в одном моле вещества. Таким образом, максимальная электрическая работа гальванического элемента при превращении одного моля вещества Wмэ = ΖFЕ, где Е – эдс гальванического элемента. В то же время максимальная полезная работа Wм.р которую может совершить система при протекании реакции при постоянном давлении, равна энергии Гиббса Wм.р = - ΔG Частным случаем химических гальванических элементов являются окислительно-восстановительные элементы. Из двух электродов хотя бы один должен быть окислительно-восстановительным. Если в качестве второго электрода использовать стандартный водородный, то ЭДС элемента (-) Р t, Н2 | Н+ | | Fе3+ , Fе2+ | Р t РН2 = ОН+ = 1 Равна электродному потенциалу оислительно-восстановительной системы. Е = j Fе3+ / Fе2+ - j0 Н+/Н2 = j0 Fе3+ / Fе2+ + (RТ/F) ln (аFе3+/ аFе2+), где j0 Fе3+ / Fе2+ - стандартный потенциал окислительно-востановительной системы, равный ее потенциалу при аFе3+ = аFе2+ Электродные и токообразующие процессы в таком элементе описываются уравнениями (-) А : Н2 г → 2Н+р + 2е 1 (+) К : Fе3+р + е → Fе2+р 2 ∑ : 2Fе3+р + Н2 г → 2 Fе 2+р + 2Н+р Концентрационные гальванические элементы состоят из двух одинаковых электродов, у которых различаются активности одного или нескольких участников электродного процессов. Они генерируют электрическую энергию за счет выравнивания химических потенциалов веществ в растворах. Существуют следующие концентрационные гальванические элементы: - элементы с различной активностью иона в растворах электролита катодного и анодного пространств, например никелевый концентрационный гальванический элемент (-)Ni | Ni2+ | | Ni 2+ | Ni (+) а1Ni2+ < а2Ni2+ уравнение Нернста для расчета ЭДС такого элемента имеет вид Е = (RТ / 2F) ln (а2Ni2+ /а1Ni2+ ) , где а1Ni2+ и а2Ni2+ активности катионов никеля в анодном и катодном пространствах соответственно. Уравнения электродных процессов (-) А : Ni → Ni2+ + 2е (+) К : Ni2+ +2е →Ni - элементы с одним раствором электролита, у которого различаются активности металла в составе сплавов катода и анода или давление газа в газовых полуэлементах, например амальгамный концентрационный элемент: (-) Сd (Нg) | Сd SО4 | Сd (Нg) (+) а1 Сd (Нg) > а2 Сd (Нg) и водородный концентрационный элемент (-) Рt, Н2 | Н+ | Н2, Рt (+) р1Н2 > р 2Н2 уравнение Нернста для расчета ЭДС такого элемента имеет вид Е = (RТ / 2F) ln (а1Сd (Нg) / а2 Сd (Нg) ) , где а1Сd (Нg) и а2 Сd (Нg) активности кадмия в амальгамах анода и катода Анодами в концентрационных гальванических элементах всегда являются электроды с меньшими значениями активностей окисленной формы аВф = соnst (первый пример) или с большими значениями активностей восстановленной формы при аОф = соnst (второй пример) стандартная ЭДС для концентрационных гальванических элементов равна нулю. Водородный показатель рН –это взятый с обратным знаком десятичный логарифм концентрации ионов Н3О+ рН = - lg [Н3О] + =7.нейтральным будет раствор, у которого рН = 7. у кислых растворов рН<7, у щелочных рН >7. Величина водородного показателя играет огромное значение в некоторых явлениях природы. От него зависит урожайность сельскохозяйственных культур. Кислые почвы известкуют. На основании знания рН почв разрабатываются методы улучшения почв. Для измерения рН существуют различные методы. Приближенно можно определить реакцию раствора с помощью индикаторов, окраска которых изменяется в зависимости от концентрации ионов Н+ или ОН-. Лакмус краснеет в кислых средах – реакция кислая. Фенолфталеин в кислых - бесцветный (рН<8), а в щелочных - фиолетовый. Рн крови человека и животных величина постоянная. Коррозионные свойства воды тоже зависят от рН. Для многих растворов существуют таблицы рН
31) Электролиз. Последовательность разряда ионов в расплавах и водных растворах при электролизе. Вторичные процессы при электролизе. Электролиз с растворимым и нерастворимым анодами. Потенциал разложения. Законы Фарадея. Выход по току; электрохимический эквивалент. Применение электролиза в технике. Расчет толщины металлического покрытия, наносимого методом электролиза.\

![]()

![]()
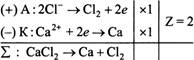 В
водных растворах электролитов появляется
второе вещество – вода. И при рН<>7,
соли могут подвергаться гидролизу.
В
водных растворах электролитов появляется
второе вещество – вода. И при рН<>7,
соли могут подвергаться гидролизу. 

![]()


![]()
![]()
![]()

![]()

32) Гальванические элементы и их классификация. Процессы, протекающие при работе ГЭ. Расчет ЭДС и работы ГЭ. Окислительно-восстановительные и концентрационные ГЭ. Определение рН раствора.
Если два различных металла погрузить в раствор электролита, то между ними возникает электрическое напряжение (разность потенциалов). Такая схема: Металл 1 ⁄ Раствор электролита ⁄ Металл 2 называется гальваническим элементом, или гальванической цепью. Вместо металлов в гальванической цепи можно использовать и другие вещества с металлической проводимость, например графит (угольный электрод). Возникновение разности потенциалов между обоими металлическими электродами объясняется различной склонностью металлов отдавать катионы в раствор электролита. У поверхности каждого из электродов возникает двойной электрический слой, который оказывает противодействие дальнейшему переходу катионов в раствор. Если оба металла соединить металлическим проводником (обладающим электронной проводимостью), то вследствие электропроводимости раствора электролита (ионной проводимости) получается замкнутая электрическая цепь. В этой цепи поток электронов будет перемещаться от менее благородного металла через внешний участок цепи (металлический проводник) к более благородному металлу. При этом в растворе электролита катионы будут двигаться к благородному металлу и разряжаться под действием имеющихся на нем электронов. В результате в замкнутой гальванической цепи возникает электрический ток. Каждый гальванический элемент состоит из двух электродов (окислительно-восстановительных пар), один из которых является поставщиком электронов, а другой их принимает. При этом на одном электроде возникает избыток электронов, а на другом - недостаток. Электрод с избытком электронов называют отрицательным полюсом гальванического элемента, или анодом, а электрод с недостатком электронов - положительным полюсом, или катодом. Отрицательным полюсом гальванического элемента является менее благородный металл, на котором имеется избыток электронов. Положительным полюсом гальванического элемента является более благородный металл, на котором имеется недостаток электронов. Электроны по внешнему участку цепи (по металлическому проводнику) переходит от отрицательного к положительному полюсу гальванического элемента. При условиях примерно равных концентраций электронов в растворах, в которые погружены электроды гальванического элемента, металл с меньшим значением стандартного потенциала будет отрицательным полюсом, а металл с большим значением стандартного потенциала – положительным полюсом. (в гальваническом элементе с цинковым и свинцовым электродами отрицательным полюсом будет цинк (φ0=- 0,763В), а положительным полюсом свинец (φ0=- 0,126В). (стандартный потенциал свинца более положителен, чем цинка) На аноде происходит процесс окисления, и цинк переходит в раствор в виде катионов. Масса цинковой пластинки уменьшается, остающиеся на ней электроны сообщают ей отрицательный заряд. Напряжение гальванического элемента тем выше, чем больше отличаются между собой значения стандартного потенциала электродов. Напряжение, которое показывает вольтметр, подключенный к полюсам гальванического элемента, называется напряжением на клеммах. Это напряжение вследствие наличия внутреннего сопротивления источника напряжения меньше действительного напряжения, называемого электродвижущей силой, сокращенно э.д.с. (обозначение U). Действительное напряжение гальванического элемента равно разности между стандартным потенциалом положительного полюса и стандартным потенциалом отрицательного полюса. U = φ0 пол. п. - φ0 отр . Если на электродах испытывает превращение один моль вещества, то по закону Фарадея через систему протекает количество электричества равное ΖF, где Ζ – число молей эквивалентов в одном моле вещества. Таким образом,максимальная электрическая работа гальванического элемента при превращении одного моля вещества Wмэ = ΖFЕ, где Е – эдс гальванического элемента. В то же время максимальная полезная работа Wм.р которую может совершить система при протекании реакции при постоянном давлении, равна энергии Гиббса Wм.р = - ΔG Частным случаем химических гальванических элементов являются окислительно-восстановительные элементы. Из двух электродов хотя бы один должен быть окислительно-восстановительным. Если в качестве второго электрода использовать стандартный водородный, то ЭДС элемента (-) Р t, Н2 | Н+ | | Fе3+ , Fе2+ | Р t РН2 = ОН+ = 1 Равна электродному потенциалу оислительно-восстановительной системы. Е = j Fе3+ / Fе2+ - j0 Н+/Н2 = j0 Fе3+ / Fе2+ + (RТ/F) ln (аFе3+/ аFе2+), где j0 Fе3+ / Fе2+ - стандартный потенциал окислительно-востановительной системы, равный ее потенциалу при аFе3+ = аFе2+ Электродные и токообразующие процессы в таком элементе описываются уравнениями (-) А : Н2 г → 2Н+р + 2е 1 (+) К : Fе3+р + е → Fе2+р 2 ∑ : 2Fе3+р + Н2 г → 2 Fе 2+р + 2Н+р Концентрационные гальванические элементы состоят из двух одинаковых электродов, у которых различаются активности одного или нескольких участников электродного процессов. Они генерируют электрическую энергию за счет выравнивания химических потенциалов веществ в растворах. Существуют следующие концентрационные гальванические элементы: - элементы с различной активностью иона в растворах электролита катодного и анодного пространств, например никелевый концентрационный гальванический элемент (-)Ni | Ni2+ | | Ni 2+ | Ni (+) а1Ni2+ < а2Ni2+ уравнение Нернста для расчета ЭДС такого элемента имеет вид Е = (RТ / 2F) ln (а2Ni2+ /а1Ni2+ ) , где а1Ni2+ и а2Ni2+ активности катионов никеля в анодном и катодном пространствах соответственно. Уравнения электродных процессов (-) А : Ni → Ni2+ + 2е (+) К : Ni2+ +2е →Ni - элементы с одним раствором электролита, у которого различаются активности металла в составе сплавов катода и анода или давление газа в газовых полуэлементах, например амальгамный концентрационный элемент: (-) Сd (Нg) | Сd SО4 | Сd (Нg) (+) а1 Сd (Нg) > а2 Сd (Нg) и водородный концентрационный элемент (-) Рt, Н2 | Н+ | Н2, Рt (+) р1Н2 > р 2Н2 уравнение Нернста для расчета ЭДС такого элемента имеет вид Е = (RТ / 2F) ln (а1Сd (Нg) / а2 Сd (Нg) ) , где а1Сd (Нg) и а2 Сd (Нg) активности кадмия в амальгамах анода и катода Анодами в концентрационных гальванических элементах всегда являются электроды с меньшими значениями активностей окисленной формы аВф = соnst (первый пример) или с большими значениями активностей восстановленной формы при аОф = соnst (второй пример) стандартная ЭДС для концентрационных гальванических элементов равна нулю. Водородный показатель рН –это взятый с обратным знаком десятичный логарифм концентрации ионов Н3О+ рН = - lg [Н3О] + =7.нейтральным будет раствор, у которого рН = 7. у кислых растворов рН<7, у щелочных рН >7. Величина водородного показателя играет огромное значение в некоторых явлениях природы. От него зависит урожайность сельскохозяйственных культур. Кислые почвы известкуют. На основании знания рН почв разрабатываются методы улучшения почв. Для измерения рН существуют различные методы. Приближенно можно определить реакцию раствора с помощью индикаторов, окраска которых изменяется в зависимости от концентрации ионов Н+ или ОН-. Лакмус краснеет в кислых средах – реакция кислая. Фенолфталеин в кислых - бесцветный (рН<8), а в щелочных - фиолетовый. Рн крови человека и животных величина постоянная. Коррозионные свойства воды тоже зависят от рН. Для многих растворов существуют таблицы рН
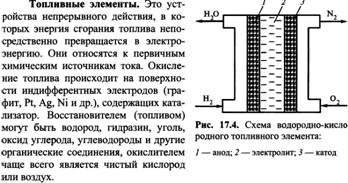
33) Практическое применение электрохимических процессов. Химические источники тока. Аккумуляторы. Свинцовый аккумулятор. Топливные элементы. Водородно-кислородный топливный элемент.



![]()
![]()
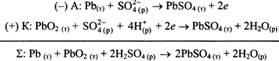
![]()

34) Коррозия. Классификация коррозионных процессов по характеру разрушений, по виду агрессивной среды, по механизму протекания. Скорость равномерной коррозии.
Коррозия
- самопроизвольное разрушение материалов
вследствие их физико-хим. взаимодействия
с окружающей средой. Если разрушение
произошло из-за мех. причины - эрозия.
Процесс коррозии железа - ржавление



![]()
![]()
![]()
![]()
![]()
![]()
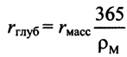
![]()
Коррозия – это разрушение конструкций и изделий из металлических материалов (металлов и сплавов), происходящее вследствие их физико-химического взаимодействия с окружающей средой, которую называют коррозионной, а образовавшиеся химические соединения – продуктами коррозии. Коррозия сопровождается выделением энергии и рассеиванием продуктов коррозии в окружающей среде. Процесс коррозии железа и его сплавов называют ржавлением. Коррозионные среды бывают жидкими и газообразными, токопроводящими и неэлектролитами, естественными и искусственно созданными. К газообразным относятся природная атмосфера и газы, образующиеся при сгорании топлива или выделяющиеся в различных химических производствах. Жидкие – это жидкости-электролиты (водные растворы солей, кислот, щелочей. Морская вода) и жидкости-неэлетролиты (сернистая нефть, бензин, керосин). Естественными. Кроме атмосферы, являются вода и почва, искусственными – многие химические вещества. По характеру разрушения поверхности различают коррозию: - сплошную (общую), при которой поражается вся поверхность изделия. Она бывает равномерной и неравномерной; -локальную (местную), при которой поражаются лишь отдельные участки поверхности. Она проявляется в виде пятен, язв и питтинга (точечного разрушения на большую глубину). Перечисленные коррозионные разрушения являются макроскопическими дефектами. К микроскопическим дефектам относятся разрушения, происходящие при коррозии: -селективной (избирательной), т.е. в случае преимущественного разрушения одного или нескольких компонентов сплавов, например обесцинкование латуни; -межкристаллитной (интеркристаллитной), т.е. при разрушении по границам кристаллитов (или зерен), приводящем к ослаблению связи между ними, например окисление чугунов при переменном нагреве и охлаждении. Кристаллитами (или зернами) называют кристаллы поликристаллического тела, имеющие неправильную форму в отличие от правильно ограненных кристаллов; -транскристаллитной, т.е. при разрушении, возникающем под действием механических напряжений и сопровождающемся появлением глубоких транскристаллитных трещин. По механизму протекания коррозию подразделяют на химическую и электрохимическую. Причина коррозии металлов и сплавов состоит в их термодинамической неустойчивости, поэтому коррозионные процессы протекают самопроизвольно и сопровождаются убылью энергии Гиббса. Химическая и электрохимическая коррозия относится к гетерогенным окислительно-восстановительным процессам, протекающим на поверхности металлов и сплавов (на границе раздела фаз материал - коррозионная среда. При этом разрушаемый материал являющийся восстановителем, непосредственно взаимодействует с окислителем коррозионной среды. Изменение температуры может ускорять или замедлять процесс электрохимической коррозии. Это связано с ее различным влиянием на скорость любой стадии сопряженных реакций. По влиянию кислотности раствора (рН среды) на процесс электрохимической коррозии все металлы подразделяют на пять групп, каждая из которых имеет свой вид зависимости: -металлы с высокой коррозионной стойкостью в кислых, нейтральных и щелочных растворах, такие как Аg, Аи, Рt и др. скорость их коррозии не зависит от рН раствора; - металлы, устойчивые в кислотных растворах, но нестойкие в щелочных – Мо, Та, W и др; -металлы малостойкие в кислотных растворах, но устойчивые в щелочных – Ni, Сd, - металлы, устойчивые в растворах близких к нейтральным, но разрушающиеся в щелочных и кислотных из-за амфотерности – Zn, Аl, Sn, Pb; - металлы малостойкие в кислотных растворах, в интервале значений рН 4…8,5 имеют постоянную скорость коррозии, которая при рН>10 резко уменьшается следствие образования на их поверхности малорастворимых гидроксидов – Fе, Мg, Сu, Мn. Влияние давления на скорость электрохимической коррозии связано, главным образом с изменением растворимости газа, участвующего в катодном процессе. Скорость движения электролита в большей степени влияет на коррозию, протекающую с кислородной деполяризацией, чем на коррозию с водородной деполяризацией. Влияние состава (вида, числа, концентрации компонентов) электролитной среды также имеет сложный характер. С возрастанием концентрации раствора скорость коррозии начале увеличивается, а затем падает. Такая зависимость типична для коррозии металлов в нейтральных растворах солей (Nа2 SО4, NаСl, КСl), протекающей с кислородной деполяризацией. Электрохимическая коррозия значительно ускоряется в присутствии небольшого количества веществ, называемых в соответствии с характером их действия активаторами (ускорителями или стимуляторами). К их числу галогенид –ионы Сl-, Вr-, I-, которые адсорбируясь на поверхности защитной пленки, вытесняют из нее кислород. При этом образуются растворимые галогениды металлов, которые, облегчая доступ коррозионной среды, способствуют началу и дальнейшему усилению коррозии. Особенно велико влияние хлорид-ионов на растворение таких металлов, кА железо, никель, алюминий.
35) Химическая коррозия. Высокотемпературная газовая коррозия металлов. Законы рост оксидных пленок. Факторы, определяющие защитные свойства пленки. Фактор Пиллинга-Бедвордса. Коррозия в среде.



![]() Важное
условие при образовании оксидной пленки:
ориентационное соответствие металлу,
т.е. максимальное сходство кристаллических
решеток металла и образующегося оксида
при минимальном смещении атомов.
Важное
условие при образовании оксидной пленки:
ориентационное соответствие металлу,
т.е. максимальное сходство кристаллических
решеток металла и образующегося оксида
при минимальном смещении атомов.
![]()
![]()
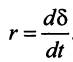
![]()

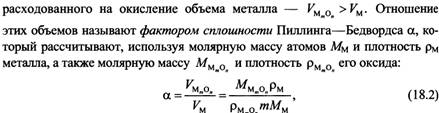

![]()
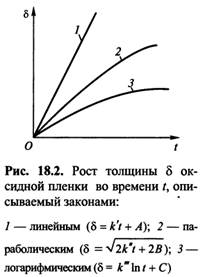
Обычно скорость r газовой коррозии, т.е. процесса окисления, выражают через скорость роста толщины δ оксидной пленки во времени t: r = dδ ⁄ dt. Рост толщины пленки, т.е. окисление поверхности металла, может проходить в соответствии с различными кинетическими зависимостями, или законами: линейным, параболическим, логарифмическим. Согласно линейному закону, скорость процесса окисления постоянна во времени. Этот закон выполняется как при полном отсутствии оксидной пленки на поверхности, так и при наличии тонкой или незащитной (пористой, несплошной) оксидной пленки, у которой α <1. Во всех этих случаях доступ кислорода к поверхности свободен и лимитирующей стадией процесса является поверхностная химическая реакция, протекающая с постоянной скоростью, т.е. окисление осуществляется в кинетическом режиме. По линейному закону происходит окисление щелочных и щелочноземельных металлов, а также ванадия, вольфрама и молибдена при высоких
температурах. У первых оно обусловлено их разогревом из-за плохого отвода теплоты, вызванного образованием на поверхности рыхлых оксидных пленок, препятствующих Ии оттоку, у вторых – летучестью их оксидов при высоких температурах. Дифференциальное уравнение скорости процесса окисления: d δ ⁄ d t=k! Для линейного закона. В соответствии с параболическим законом скорость процесса окисления обратно пропорциональна толщине оксидной пленки. Этот закон соблюдается, когда на поверхности металла при его окислении образуется пленка, обладающая защитными свойствами, т.е. сплошная и непористая, для которой α >1. согласно параболическому закону окисляются вольфрам, кобальт, никель, а также медь в интервале температур 300…10000С и железо – 500…1000оС Дифференциальное уравнение скорости процесса окисления: d δ ⁄ d t=k!! ⁄ δ для параболического закона. Логарифмический закон имеет место, когда происходит либо уплотнение защитной оксидной пленки, либо появление в ней дефектов в виде пузырей или расслоений, тормозящих процессы встречной диффузии ионов кислорода и металла. При этом наблюдается сильное затухание процесса окисления, и рост толщины оксидной пленки осуществляется медленнее, чем по параболическому закону. В соответствии с логарифмическим законом окисляются медь при температуре ниже 100оС, тантал – ниже 150оС, железо – ниже 400оС, а также алюминий, цинк и никель ниже 150оС. Скорость процесса окисления в этом случае обратно пропорциональна времени его протекания. Дифференциальное уравнение скорости процесса окисления: d δ ⁄ d t=k!!! ⁄ t для логарифмического закона. Из большого числа внутренних факторов существенное влияние на скорость окисления при газовой коррозии оказывает состояние поверхности металла, связанное с образованием, устойчивостью и разрушением оксидных пленок. Их защитные свойства в значительной степени определяются природой и составом сплавов. Так, хром, алюминий, кремний и углерод очень сильно замедляют процесс окисления стали вследствие образования пленок с высокими защитными свойствами. Аналогичная картина наблюдается и при легировании меди бериллием, оловом алюминием и цинком. Наоборот, элементы, образующие легкоплавкие и летучие оксиды, например ванадий, вольфрам, молибден ускоряют процесс окисления. Внешние факторы, такие как вид, состав, давление, температура и скорость движения газовой среды, время ее контакта, режим нагрева оказывают большее влияние на скорость коррозии, чем внутренние. При повышении температуры, с одной стороны, понижается термодинамическая возможность газовой коррозии, с другой, - увеличивается константа скорости химической реакции и коэффициент диффузии, а также изменяются защитные свойства оксидной пленки. В целом с ростом температуры скорость коррозии увеличивается в соответствии с зависимостью, близкой к экспоненциальной. Колебания температуры, особенно попеременные нагрев и охлаждение, вызывают быстрое разрушение защитной пленки из-за возникновения больших внутренних напряжений. С повышением парциального давления кислорода скорость окисления ряда металлов при высокой температуре увеличивается, а затем при достижении некоторого критического значения уменьшается и остается достаточно низкой в широком интервале давления. Наблюдаемое явление получило название высокотемпературной пассивацией. Пассивное состояние металла обусловлено образованием на его поверхности совершенной по структуре оксидной пленки. В оксидных пленках определенной толщины и совершенной структуры (без трещин, пор, вакансий и др.) процессы встречной диффузии прекращаются. Такие пленки и являются защитными. Чтобы обладать защитными свойствами, оксидная пленка должна удовлетворять следующими требованиям: быть сплошной, беспористой, химически инертной к агрессивной среде, иметь высокие твердость, износостойкость, адгезию (прилипаемость к металлу) и близкий к металлу коэффициент термического расширения. Главным требованием является условие сплошности Пиллинга-Бедвордса, согласно которому объем образовавшегося оксида должен быть больше израсходованного на окисление объема металла VМmОn>VМ. Отношение этих объемов называют фактором сплошности Пиллинга-Бедвордса α , который рассчитывают, используя молярную массу атомов Мм и плотность ρМ металла, а также молярную массу ММmОn и плотность ρМmОn его оксида: α = VМmОn ⁄VМ = ММmОnρМ ⁄ρМmОn mММ где m – число атомов металла в молекуле оксида. Если α>1, то формирование и рост толщины пленки при окислении происходит в условиях сжатия, поэтому она является сплошной и может обладать защитными свойствами. Если α < 1, то пленка в процессе своего формирования и роста испытывает растяжения, которые способствуют ее разрушению и появлению трещин, различных дефектов, вследствие чего кислород свободно проникает к поверхности металла. Химическая коррозия характерна для сред, преимущественно не проводящих электрический ток. В зависимости от вида этих сред различают: химическую коррозию в жидкостях-неэлектролитах; химическую газовую коррозию (газовую). Они сопровождаются возникновением электрического тока и протекают по механизмам гетерогенных окислительно-восстановительных реакций.. Жидкости-неэлектролиты – это неэлектропроводные жидкие коррозионные среды неорганического (жидкий бром, расплавленная сера и др.) и органического (нефть, керосин, бензол и др.) происхождения. В чистом виде они малоагрессивны, однако присутствие в них даже малых количеств примесей (меркаптанов, сероводорода, воды, кислорода и др.) резко увеличивает их химическую активность. Так, меркаптаны (R-SН) и сероводород (Н2S), содержащиеся в сырой нефти, вызывают коррозию Fе, Сu, Ni, Аg, Рb, Sn с образованием соответственно их меркаптидов (М (SR)z) и сульфидов (МmSn). Fе0 → Fе+2 + 2е 2Н+1 + 2е → Н2 ↑ Fе + Н2S→FеS +Н2↑
36) Электрохимическая коррозия. Случаи образования коррозионных гальванопар. Электродные процессы в различных средах. Привести пример. Факторы, приводящие к электрохимической коррозии. Скорость электрохимической коррозии: рН и состав среды, температура и концентрация окислителя. Стимуляторы и ингибиторы коррозии. Коррозия малоуглеродистой стали в различных средах.




![]()
![]()
![]()

![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

 Причина
коррозий - термодинамическая
неустойчивость.
Электрохимическая
коррозия протекает в средах с хорошей
ионной проводимостью. Причины, создающие
неоднородность в системе металл-электролит
(возникновение коррозионных микроэлементов):
1. неоднородность металлической фазы.
2. неоднородность жидкой фазы. 3.
неоднородность наложения внешних
условий.
Причина
коррозий - термодинамическая
неустойчивость.
Электрохимическая
коррозия протекает в средах с хорошей
ионной проводимостью. Причины, создающие
неоднородность в системе металл-электролит
(возникновение коррозионных микроэлементов):
1. неоднородность металлической фазы.
2. неоднородность жидкой фазы. 3.
неоднородность наложения внешних
условий.
![]()


![]()




Абсолютно коррозионно-стойких металлов нет. Золото –стойкое в обычных условиях – растворяется в растворах цианидов калия или натрия вследствие образования устойчивых комплексных ионов. В результате коррозии металлические изделия теряют свои ценные технические свойства, поэтому важное значение имеет антикоррозионная защита металлов и сплавов. Коррозия – это разрушение конструкций изделий из металлических материалов (металлов и сплавов), происходящее вследствие их физико-химического взаимодействия с окружающей средой, которую называют коррозионной (или агрессивной), а образовавшиеся химические соединения –продуктами коррозии. По механизму процесса коррозию подразделяют на химическую и электрохимическую. Причина коррозии металлов и сплавов состоит в их термодинамической неустойчивости, поэтому коррозионные процессы протекают самопроизвольно и сопровождаются убыль энергии Гиббса: (drG)р,Т<0. Чем меньше (более отрицательно) значение (drG 0)р,Т коррозионного процесса, тем выше термодинамическая возможность его протекания. В случае электрохимической коррозии (drG 0)р,Т связана со стандартной ЭДС Е0 коррозионного гальванического элемента уравнением: ZFЕ0 = -drGТ0 . Химическая и электрохимическая коррозия относятся к гетерогенным окислительно-восстановительным процессам, протекающим на поверхности металлов и сплавов. При этом разрушаемый материал, являющийся восстановителем, непосредственно взаимодействует с окислителем коррозионной среды. На практике чаще всего приходится иметь дело с электрохимической коррозией. Она, в отличие от химической, сопровождается возникновением электрического тока и протекает, как правило, в средах с хорошей ионной проводимостью. По условиям осуществления различают: коррозию в электролитах, атмосферную коррозию, электрокоррозию, коррозию под напряжением и др. причинами возникновения электрохимической коррозии служат различные виды неоднородностей как самой поверхности металла или сплава, так и коррозионной среды. В результате вся поверхность, соприкасающаяся с токопроводящей коррозионной средой, разделяется на катодные и анодные участки, которые имеют очень малые размеры и чередуются друг с другом. В такой среде они представляют собой совокупность огромного числа короткозамкнутых гальванических микроэлементов, вследствие чего электрохимическую коррозию называют гальванической коррозией. Обычно имеют место следующие основные случаи неоднородностей в системе металл – электролит, которые приводят к возникновению и функционированию коррозионных микроэлементов: - неоднородность металлической фазы, обусловленная наличием загрязнений, примесей в металле, пленок на его поверхности, различным химическим и фазовым состоянием сплава; - неоднородность жидкой фазы, обусловленная различной концентрацией в электролите (коррозионной среде) ионов данного металла и растворенного кислорода на отдельных участках контакта фаз, неодинаковым значением рН отдельных зон объема электролита и др.; - неоднородность наложения внешних условий: неодинаковая температура отдельных участков поверхности металла (более нагретые являются анодами); различный уровень механических напряжений в одной и той же детали; неравномерное наложение внешнего электрического поля и др. в системах возможно возникновение коррозионных не только микро-, но и макроэлементов, например, при контакте с электролитом двух соприкасающихся деталей, изготовленных из металлов различной активности (так называемая контактная коррозия). Механизм электрохимической коррозии сводится к возникновению и функционированию коррозионных гальванических макро- и микроэлементов, поэтому ее процессы аналогичны процессам, протекающим в химических источниках тока: гальванических и топливных элементах, аккумуляторах основное отличие коррозионных процессов – отсутствие внешней цепи. Электроны в процессе коррозии не выходят из корродирующего металла, а перемещаются внутри него от анодных участков к катодным. Процесс электрохимической коррозии представляет собой совокупность двух сопряженных (взаимосвязанных) полуреакций, одновременно протекающих на поверхности металла: - анодной, сопровождающейся окислением атомов металла на анодных участках поверхности (в оцинкованном железе в водном растворе электроны переходят от цинка к железу): (-) А: Zn -> Zn2+ + 2е - катодной, сопровождающейся восстановлением окислителя коррозионной среды на катодных участках поверхности (на поверхности железа разряжаются ионы водорода): (+) К: 2 Н2О +2е -> Н2 + 2ОН- Zn2+ + 2ОН- = Zn(ОН)2 повреждение защитного слоя цинка в оцинкованном железе приводит к разрушению этого слоя, поскольку он состоит из более активного металла – цинка. Окислители электрохимической коррозии называют деполяризаторами. К наиболее часто встречающимся деполяризаторам относятся молекулы кислорода О2, воды Н2О и ионы водорода Н+. Основными катодными реакциями с их участием при электрохимической коррозии являются: 1. в аэрированных (насыщенных кислородом ) коррозионных средах: - нейтральных и щелочных (рН³7) (+) К: О2 + 2Н2О + 4е -> 4ОН- - кислотных (рН<7) (+) К: О2 + 4Н+ + 4е -> 2Н2О 2. в деаэрированных (несодержащих растворенный кислород) коррозионных средах: - нейтральных и щелочных (рН³7) (+) К: 2Н2О + 2е -> Н2 +ОН- - кислотных (рН<7) (+) К: 2Н+ +2е -> Н2 Ингибиторы коррозии: изменение состава и свойств коррозионной среды, т.е. уменьшение ее агрессивности, осуществляют либо введением в нее специальных веществ – ингибиторов коррозии, либо соответствующей ее обработкой. Защиту ингибиторами применяют в системах с постоянным или мало обновляемым объемом коррозионной среды. (в котлах, цистернах, химических аппаратах) ингибиторами называются химические соединения или композиции на их основе, введение которых в небольших количествах (до 1% масс) в коррозионную среду резко снижает скорость коррозии. По химическому составу различают органические и неорганические ингибиторы, по условиям применения – жидкофазные (ингибиторы для растворов) и летучие ингибиторы, дающие защитный эффект в условиях атмосферной коррозии, по механизму своего действия на процесс электрохимической коррозии – катодные, анодные и экранирующие (изолирующие) ингибиторы. В свою очередь, жидкофазные ингибиторы подразделяют на кислотные, щелочные и нейтральные, в соответствии с тем, в какой среде они применяются. Механизм защитного действия большинства ингибиторов заключается в адсорбции (концентрировании) их на корродирующей поверхности и и последующем торможении анодных (анодные ингибиторы) и катодных (катодные ингибиторы) процессов элетрохимической коррозии, а также в образовании защитных и пассивирующих пленок (экранирующие ингибиторы). К анодным ингибиторам относят гидроксид и бензоат натрия, различные фосфаты, силикаты, тетрабораты и др. Анодные и катодные ингибиторы, являясь преимущественно неорганическими веществами, оказывают эффективное защитное действие только в нейтральных и щелочных растворах. В сильнокислых растворах используют экранирующие ингибиторы-органические соединения, содержащие в своем составе атомы серы, кислорода, азота (альдегиды, фенолы, меркаптаны, амины, соли ароматических карб. кислот
Коррозия малоуглеродистой стали в различных средах. Малоуглеродистая сталь содержит до 0,2% углерода, основное – железо. 1) в аэрированных (насыщенных кислородом) коррозионных средах: -нейтральных и щелочных (рН => 7) (-) А: Fе " Fе2+ + 2е 2 (+) К: О2 + 2Н2О + 4е " 4 ОН 1 Fе + О2 + 2Н2О " 2 Fе (ОН)2$ -в кислой среде (рН<7) (-) А: Fе " Fе2+ + 2е 2 (+) К: О2 + 4 Н+ + 4е " 2Н2О 1 2 Fе + О2 + 4НСN "2 Fе(СN)2 $ + 2Н2О 2) в деаэрированной (несодержащей растворенного кислорода ) коррозионной среде: -нейтральных и щелочных (рН=>7) (-) А: Fе " Fе2+ + 2е (+) К: 2Н2О + 2е " Н2# + 2ОН- Fе + 2Н2О → Н2# + Fе (ОН)2$ -в кислой среде (рН<7) (-) А: Fе " Fе2+ + 2е (+) К: 2Н+ + 2е → Н2# Fе + 2НСN → Н2# + Fе(СN)2 $
37) Защита металлов от газовой коррозии. Методы защиты металлов от электрохимической коррозии: легирование, обработка среды, неорганические (в том числе металлические) и органические покрытия; электрохимическая защита (протекторная, катодная, анодная).

![]()
![]()
![]()

 Катодная
защита: подключение защищаемой конструкции
к отрицательному полюсу внешнего
источника постоянного тока, присоединение
к защищемой конструкции электрода,
изготовленногоиз более активного
металла.
Анодная защита - андоная
поляризация: потенциал защищаемого
металла смещают в положительную сторону
до значений, лежащих в пассивной области
андоной поляризационной кривой, путсем
присоединения защищаемой конструкции
к положительному полюсу внешего источника
тока, а вспомогательного электрода к
отрицательному.
Катодная
защита: подключение защищаемой конструкции
к отрицательному полюсу внешнего
источника постоянного тока, присоединение
к защищемой конструкции электрода,
изготовленногоиз более активного
металла.
Анодная защита - андоная
поляризация: потенциал защищаемого
металла смещают в положительную сторону
до значений, лежащих в пассивной области
андоной поляризационной кривой, путсем
присоединения защищаемой конструкции
к положительному полюсу внешего источника
тока, а вспомогательного электрода к
отрицательному. 
![]()
![]() Защитные
покрытия, спобосы: металлизация,
плакирование, темродиффузионный метод,
газофазный метод. Силикатные эмали,
конверсионные покрытия, лакокрасочные
покрытия.
Защитные
покрытия, спобосы: металлизация,
плакирование, темродиффузионный метод,
газофазный метод. Силикатные эмали,
конверсионные покрытия, лакокрасочные
покрытия.
Защита от коррозии – это комплекс мероприятий, направленных на предотвращение и замедление коррозионных процессов, сохранение и поддержание в работоспособном состоянии узлов и агрегатов машин, оборудования и сооружений в требуемый период их эксплуатации. Способы защиты от коррозии выбирают на стадии конструирования и осуществляют в процессе изготовления и эксплуатации объектов. Для защиты металлов от химической коррозии возможно применять: -легирование поверхностное или объемное другими металлами, например хром или никель, добавленные к стали в качестве легирующих компонентов, при высокой температуре диффундируют к поверхности и образуют устойчивый оксидный слой. В качестве легирующих добавок используют Сr, Ni, Мо, Сu, и др. в результате получают сплавы с более высокой коррозионной стойкостью, чем исходные материалы -металлические изделия смазывают неокисляющимися маслами, последние хорошо смачивают металл при повышенной температуре в жидком виде и при застывании образуют на поверхности слой, изолирующий металл от окружающей среды. Для более надежной защиты металлов от коррозии в состав смазок вводят ингибиторы (замедлители); -на поверхности металла наносят раствор высокомолекулярных соединений в низкокипящем растворителе. После испарения растворителя на поверхности металла остается полимерная пленка, преграждающая доступ окислителя к металлу; -металлическую поверхность окрашивают раствором пигмента (красящего вещества) в олифе (частично окисленном растительном масле). Олифа в тонком слое быстро окисляется кислородом воздуха и образует плотную пленку на металле; -наносят металлические защитные покрытия. Используют металлы, на поверхности которых при действии воздуха или других окислителей образуется устойчивая защитная пленка (Аl, Zn, Sn, Сr, Рb, Ni), или наносят металлы пассивные в химическом отношении (Аu, Аg, Сu) Электрохимическая коррозия развивается при контакте металлов с раствором электролита. При электрохимической коррозии на металле протекают одновременно два процесса: окисление металла – анодный процесс Мо -> Мn+ + n е; И восстановление окислителя например кислорода воздуха: О2 + Н2О + 4е -> 4ОН- (кислородная коррозия). Существуют различные способы защиты от коррозии, основанные на снижении агрессивности коррозионной среды, нанесение защитных покрытий и применение электрохимических методов – электрохимическая защита. Один из видов электрохимической защиты – катодная защита заключается в том, что защищаемый металл соединяют с отрицательным полюсом внешнего источника постоянного тока, а положительный полюс – с вспомогательным материалом, который будет окисляться и тем самым защищать основной металл. В настоящее время катодную защиту широко применяют для защиты трубопроводов, кабельных установок и металлоконструкций в воде (морской, речной), грунте и др средах. В основе анодной защиты лежит анодная поляризация: смещение потенциала металла защищаемой конструкции в положительную сторону до значений, лежащих в пассивной области анодной поляризационной кривой. Ее осуществляют присоединением защищаемой конструкции к положительному полюсу внешнего источника тока, а вспомогательного электрода – к отрицательному, при этом конструкция – анод, а электрод –катод. Анодная защита в отличие от катодной применима только к легкопассивируемым (покрывающимся пассивной пленкой оксида). Это большинство переходных металлов и сплавов на их основе, включая нержавеющие и углеродистые стали. При анодной защите ток противоположен по направлению току при катодной защите. Достоинства анодной защиты: высокая рассеивающая способность –возможность защит на большом удалении от катода; невысокая защитная плотность тока, а следовательно и небольшие потребления энергии. К недостаткам следует отнести ограничение области применения (анодная защита не осущетсвима для Мg, Сd, Аg, Сu и медных сплавов), а также то. Что скорость коррозии при анодной защите, в отличие от катодной, никогда не падает до нуля. Другой вид электрохимической защиты – протекторная защита осуществляется путем присоединения к защищаемому металлу протектора – более активного (т.е. менее благородного металла), который легче окисляется и, тем самым предохраняет основной металл от коррозии. Так, для защиты от коррозии изделий из железа и его сплавов в качестве протектора обычно применяют магний. Электрохимическая защита используется в широких масштабах в судостроении.
38)
39)
40) Сплавы. Понятие о физико-химическом анализе. Термический анализ. Кривые охлаждения. Диаграмма состояния (плавкости) бинарной металлической системыс образованием эвтетики. Анализ диаграммы по правилу фаз Гиббса-Коновалова.
Твёрдые
многокомпонентные металлические системы
получили название сплавов.
Диаграммы
состояния систем твердое тело – жидкость
строят на основании результатов термического
анализа.
Сущность метода состоит в следующем.
Берут вещество или смесь веществ и
нагревают выше температуры плавления
– получается гомогенная система. В
жидкость погружают высокотемпературный
термометр (или термопару), нагревание
прекращают и через определенные
промежутки времени фиксируют температуру.
Строят график изменения температуры
со временем, называемый кривой
охлаждения.
Смеси, образующие простую эвтектику
Диаграммы
такого типа встречаются при изучении
равновесий в системах: олово – свинец,
цинк – кадмий, бензол – нафталин, хлорид
натрия – вода. В таких системах компоненты
неограниченно растворимы друг в друге
в жидком состоянии и совершенно не
растворяются в твердом состоянии.
Диаграмма такого типа изображена на
рис. 1.
По оси абсцисс отложена массовая
доля одного из компонентов В, по оси
ординат – температура. Поскольку
вещества находятся в конденсированном
состоянии, давление на фазовые равновесия
практически не влияет. Область на
диаграмме, расположенная выше кривой
СЕD, отвечает гомогенной жидкой системе.
Точки С и D – это температуры кристаллизации
(или плавления) чистых компонентов А и
В. Если кчистому компоненту А добавить
некоторое количество компонента В, то
температура начала кристалллизации
понизится. Мы знаем, что растворы начинают
замерзать при более низкой температуре,
чем чистый растворитель (следствие из
закона Рауля). Линия СЕ называется
линией ликвидуса (или
линией жидкости). Точно так же, если
взять чистый компонент В и прибавлять
к нему компонент А, температура начала
кристаллизации будет понижаться
(кривая ДЕ). Точка пересечения кривых
ликвидуса Е называется эвтектической
точкой,
а соответствующая смесь – эвтектической
смесью или
просто эвтектикой (слово
«эвтектика» происходит от греческого
«легкоплавкий»).
Таким образом, эвтектика
представляет собой смесь веществ,
имеющая минимальную температуру
замерзания
Линия
MN на диаграмме называется линией
солидуса,
то есть твердого тела. Ниже этой линии
существуют только твердые фазы.
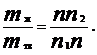 Теперь
возьмем надиаграмме точку n. Эта точка
соответствует гетерогенной системе,
состоящей из твердой и жидкой фаз. Для
того, чтобы определить состав фаз,
проведем через точку n горизонталь.
Точка пересечения с линией ликвидуса
n 1 покажет состав жидкой фазы. В точке
n 2 горизонталь пересекается с осью
ординат, отвечающей w В = 100 %. Следовательно,
твердая фаза представляет чистый
компонент В. Относительные количества
твердой и жидкой фаз можно найти по
правилу рычага:
Теперь
возьмем надиаграмме точку n. Эта точка
соответствует гетерогенной системе,
состоящей из твердой и жидкой фаз. Для
того, чтобы определить состав фаз,
проведем через точку n горизонталь.
Точка пересечения с линией ликвидуса
n 1 покажет состав жидкой фазы. В точке
n 2 горизонталь пересекается с осью
ординат, отвечающей w В = 100 %. Следовательно,
твердая фаза представляет чистый
компонент В. Относительные количества
твердой и жидкой фаз можно найти по
правилу рычага:
41) Диаграмма состояния бинарной металлической системы с неограниченной растворимостью компонентов. Понятие о твердом растворе. Условия образования твердого раствора замещения. Анализ диаграммы по правилу фаз Гиббса-Коновалова.
значение имеют исследования зависимости температур начала и конца кристаллизации твердых веществ от состава системы. Графики, выражающие эту зависимость, называются фазовыми диаграммами или диаграммами плавкости.
Системы, компоненты которых неограниченно растворимы в жидком и твердом состояниях
В таких системах при кристаллизации выделяются одновременно оба компонента, входящие в одну кристаллическую фазу – твердый раствор. Во время кристаллизации состав твердой фазы непрерывно меняется и эвтектика отсутствует. эвтектика представляет собой смесь веществ, имеющая минимальную температуру замерзания. Область I – гомогенная система – жидкий раствор (расплав); II – гетерогенная система, состоящая из жидкого раствора и твердого раствора; III – гомогенная система – твердый раствор. Линия CSm 1 D соответствует температурам начала кристаллизации, а кривая Cr 1 PD – температурам плавления твердых растворов. При охлаждении жидкого расплава, отвечающего точке m до температуры m 1, начнется кристаллизация твердого раствора. Для того, чтобы найти состав его первой порции, необходимо из точки m 1 провести горизонталь до линии плавления. Точкп пересечения Р отвечает составу твердого раствора Наоборот, если состояние системы определяется точкой r , она представляет собой твердый раствор. При нагревании его до температуры, соответствующей точке r 1, начнется плавление. Чтобы найти состав начинающей образовываться жидкой фазы, проведем горизонталь до линии CSm 1 D - точка пересечения S отвечает составу жидкого раствора. Если состояние системы характеризуется точкой, лежащей в области II, то состав равновесных фаз и их относительные количества можно найти так, как это было описано при рассмотрении предыдущих диаграмм.
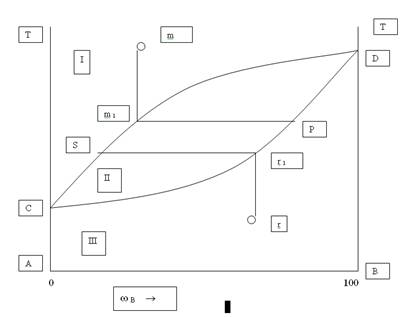
42) Диаграмма плавкости бинарной металлической системы с образованием химического соединения. Анализ диаграммы по правилу фаз Гиббса-Коновалова. Понятие об интерметаллидах.
Сущность
метода состоит в следующем. Берут
вещество или смесь веществ и нагревают
выше температуры плавления – получается
гомогенная система. В жидкость погружают
высокотемпературный термометр (или
термопару), нагревание прекращают и
через определенные промежутки времени
фиксируют температуру. Строят график
изменения температуры со временем,
называемый кривой охлаждения.
Если
это соединение термически устойчиво,
то есть при плавлении не разлагается,
то кривая его охлаждения подобна кривой
охлаждения чистого вещества (рис. 2, а).
Диаграмма состояния такой системы
представлена на рис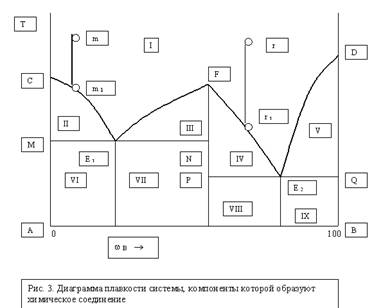 Точка
F на диаграмме соответсвует температуре
плавления химического соединения AmBn.
Диаграмма состоит как бы из двух
эвтектических диаграмм, подобных
изображенной на рис. 1. Каждая из частей
диаграммы имеет свою точку эвтектики.
Точка E 1 отвечает эвтектической смеси,
состоящей из кристаллов вещества А и
химического соединения, а точка E 2
- эвтектической смеси химического
соединения и компонента B. На диаграмме
можно выделить следующие области:
I
– гомогенная система, содержащая оба
компонента в жидкой фазе;
II –
гетерогенная система, состоящая из
кристаллов А и жидкого раствора обоих
компонентов;
III, IV – гетерогенные
системы, состоящие из кристаллов
химического соединения и жидкого
раствора;
V – гетерогенная система
– кристаллы компонента В и раствор;
VI
– твердая эвтектическая смесь, содержащая
кристаллы А и химического соединения
и избыток кристаллов А;
VII - твердая
эвтектическая смесь AmBn с А и избыток
кристаллов AmBn ;
VIII - твердая
эвтектическая смесь, содержащая кристаллы
химического соединения и компонента В
и избыток кристаллов химического
соединения;IX – та же смесь и избыток
кристаллов компонента В.При охлаждении
жидкого раствора, состав которого
определяется точкой m, при температуре,
определяемой точкой m 1 , начнется
кристаллизация вещества А. Если же взять
смесь r, то при понижении температуры
до r 1 начнут выпадать кристаллы химического
соединения.
Число степеней свободы
в точке F равно нулю, так как для этой
точки существует математическое
уравнение, связывающее концентрации
компонентов, а именно: на m моль А
приходится n моль В, и число независимых
компонентов равно 1. Тогда: с = 1 – 2 + 1 =
0. Таким образом, на данной диаграмме
имеется 5 точек, соответсвующих
нонвариантным системам: это точки C, D,
E 1, E 2, F.
Точка
F на диаграмме соответсвует температуре
плавления химического соединения AmBn.
Диаграмма состоит как бы из двух
эвтектических диаграмм, подобных
изображенной на рис. 1. Каждая из частей
диаграммы имеет свою точку эвтектики.
Точка E 1 отвечает эвтектической смеси,
состоящей из кристаллов вещества А и
химического соединения, а точка E 2
- эвтектической смеси химического
соединения и компонента B. На диаграмме
можно выделить следующие области:
I
– гомогенная система, содержащая оба
компонента в жидкой фазе;
II –
гетерогенная система, состоящая из
кристаллов А и жидкого раствора обоих
компонентов;
III, IV – гетерогенные
системы, состоящие из кристаллов
химического соединения и жидкого
раствора;
V – гетерогенная система
– кристаллы компонента В и раствор;
VI
– твердая эвтектическая смесь, содержащая
кристаллы А и химического соединения
и избыток кристаллов А;
VII - твердая
эвтектическая смесь AmBn с А и избыток
кристаллов AmBn ;
VIII - твердая
эвтектическая смесь, содержащая кристаллы
химического соединения и компонента В
и избыток кристаллов химического
соединения;IX – та же смесь и избыток
кристаллов компонента В.При охлаждении
жидкого раствора, состав которого
определяется точкой m, при температуре,
определяемой точкой m 1 , начнется
кристаллизация вещества А. Если же взять
смесь r, то при понижении температуры
до r 1 начнут выпадать кристаллы химического
соединения.
Число степеней свободы
в точке F равно нулю, так как для этой
точки существует математическое
уравнение, связывающее концентрации
компонентов, а именно: на m моль А
приходится n моль В, и число независимых
компонентов равно 1. Тогда: с = 1 – 2 + 1 =
0. Таким образом, на данной диаграмме
имеется 5 точек, соответсвующих
нонвариантным системам: это точки C, D,
E 1, E 2, F.
При благоприятных условиях в металлических системах образуются фазы с определённым стехиометрическим соотношением компонентов. Они получили название интерметаллических соединений
Состав таких соединений весьма разнообразен, но определяется он в основном указанными выше двумя причинами - размером атомов, характером и числом электронов в зоне проводимости. Например если радиус атома А в 1,26 раза больше радиуса атома В, то объём атома А будет в 2 раза больше, чем В. В этом случае в кристаллической решётке металла атом В займёт одно место, а атом А - два. Соотношение атомов будет АВ 2. Фазы, образующиеся по такому принципу, называются фазами Лавеса. Типичным примером такой фазы может служить соединение MgCu 2.
43-44) Общая характеристика, получение, физические и химические свойства s-металлов IA и IIA групп: взаимодействие s-металлов с простыми (кислородом, водородом и др.) и сложными веществами. Применение в технике.
Жесткость воды. Виды жесткости. Единицы измерения жесткости. Методы устранения жесткости воды.
Жесткость воды - совокупность химических и физических свойств воды, связанных с содержанием в ней растворенных солей щелочноземельных металлов, кальция и магния.
Виды жесткости: карбонатная (временная) и некарбонатная (остаточная/постоянная).
Единицы измерения жесткости: 1 ммоль/л или 1 моль/м3 эквивалентов ионов Ca(2+), Mg(2+), Fe(2+).
Способы устранения карбонатной жесткости: 1) кипячение; 2) известкование - добавление негашеной CaO и гашеной Ca(OH)2 извести.
Способы устранения некарбонатной жесткости: 1) добавление рассчитаного количества соды (Na2CO3); 2) добавление фосфатов натрия.
Дополнение:
Карбонатная жесткость обусловлена присутствием в воде гидрокарбонатов кальция, магния и железа.
Уравнения реакций с помощью которых может быть устранена карбонатная жесткость:
1) Ca(HCO3)2 =(t)> CaCo3 \/ +CO2 /\ + H2O
2) Ca(HCO3)2 + CaO = 2CaCO3 \/ + H2O
Некарбонатная жесткость обусловлена наличием в воде сульфатов, а также хлоридов и нитратов ионов кальция, магния, железа.
Уравнения реакций с помощью которых может быть устранена некарбонатная жесткость:
1) CaSO4 + Na2CO3 = CaCO3 \/ + Na2SO4
2) CaCl2 + 2Na3PO4 = Ca3(PO4)2 \/ + 6NaCl
Уравнение для расчета общей жесткости воды: Hобщ=Нкарб+Ннекарб
Формула для расчета объемной емкости ионита (катионита, анионита): ε
=Нобщ*V(Н2О)/m
Молярная концентрация раствора (С) - количество в-ва в одном литре раствора, моль/л.
Молярная концентрация эквивалента раствора (Сэ или N) - кол-во в-ва эквивалента в одном литре раствора, моль экв/л (при числовых значениях указывают размерность, 1N=1н)
Формула для расчета количества вещества эквивалента: nэкв=m/Мэкв
Химический эквивалент - условная частица, соответствующая (эквивалентная) одному иону водорода в обменных реакциях или одному электрону в ОВР.
Возможные формулы для решения задач:
Hкарб=Нвр=(N(HCl)*V(HCl)/V(H2O))*1000=.. [моль/м3]
Нобщ=(N(тр.Б)*V(тр.Б)/V(H2O))*1000=.. [моль/м3]
45) d-Металлы. Положение d-металлов в периодической таблице. Зависимость изменения физических свойств d-металлов от их электронного строения. Общие химические свойства d-металлов: взаимодествие с кислородом, водой, кислотами и щелочами. Зависимость кислотно-основных и окислительно-восстановительных свойств соединений d-металлов от степени окисления d-металла. Понятие о металлообразных соединения. их применение.
Св-ва. Большое число степеней окисления. Образование комплексных соединений. Образование окрашенных соединений. Парамагнетизм. Способность катализировать реакции. Все d – элементы являются металлами. Как правило, они отличаются высокой твердостью, тугоплавкостью, значительной электропроводностью. Для каждой декады d – элементов наиболее устойчивы конфигурации d 0 (Sc, Y, La), d 5 (Mn, Tc, Re), d 10 (Zn, Cd, Hg). Примеры: Ti 4 + (d 0), Fe 3 + (d 5), Zn 2 + (d 10) - устойчивы; Cr 2 + (d 4), Mn 3 + (d 4) – нестабильны. особенности d – элементов: большой выбор их валентных состояний и, как правило, широкие пределы изменения окислительно-восстановительных и кислотно-основных свойств. Полярность связей в соединениях с ростом степени окисления уменьшается. Для степени окисления (1 и 2) связь близка к ионной, для максимальной степени окисления она приближается к ковалентной. Поэтому, например, низшие оксиды и гидроксиды являются основными, высшие – кислотными, многие низшие галогениды – ионные кристаллы (хорошо растворимые соли), высшие галогениды – легкоплавкие, легколетучие вещества, подвергающиеся гидролизу. Хим. Св-ва (на примере Mn): Mn + 2 HCl = Mn Cl 2 + H 2 (j 0 (Mn 2 + / Mn) = - 1, 19 B). Mn + 8 HNO 3 = 3 Mn (NO 3 ) 2 + 2 NO 2 + 4 H 2 O, Mn + 2 H 2 SO 4 (конц.) = MnSO 4 + SO 2 + 2 H 2 O. Mn + 2 H2O = Mn(OH) 2 ¯ + H 2 (при нагревании), Химические св-ва d-металлов определяются степенью заполнения электронами подуровня d, а также возможностью возбуждения электронов подуровня д и подуровня с для образования валентных связей. Д-металлы, обладающие небольшим кол-вом электронов в подуровне Д, способны вступать в химические реакции с элементарными или сложными окислителями как восстановители и в первую очередь будут устанавливать химические связи электроны подуровня Д. Д-металлы, обладающие электронными парами в подуровне Д, имеют меньшие степени возбуждения и их окислительные числа тоже меньше, так как не все электроны подуровня Д могут принимать участие в хим. реакциях. Д-металлы III, IV, V, VI, VII групп в высших степенях окисления проявляются св-ва, подобные св-вам р-элементов соответствующих групп. Д-металлы VIII, I, II групп, за исключением железа и его аналогов, обладают малыми степенями окисления и соединения их проявляют только металлические св-ва. Соединениям высшей степени окисления св-ны ковалентно-полярные связи, приближающие эти соединения к соединениям р-элементов. Соединения Д-металлов высшей степени окисления в химических реакциях могут выступать как окислители, так как их устойчивость уменьшается, а следовательно, окислительная активность увлечивается от подгруппы титана к подгруппе железа.
46|)Общая характеристика фимических и физических свойств p-элементов IIIA группы. Бор, аллюминий. Их получение, химические свойства (взаимодествие с кислородом, галогенами, растворами кислот и щелочей). Применение в технике.
В основном состоянии атомы элементов 3 - ей группы имеют конфигурацию ns2np1 с одним неспаренным электроном и двумя вакантными р - орбиталями. В соединениях, как правило, атомы находятся в состоянии sp 2 - или sp3 – гибридизации с одной вакантной р – или sp3 – гибридной орбиталью. Участие этой орбитали в донорно-акцепторном взаимодействии позволяет увеличиваться координационному числу (КЧ) атомов до четырех. Начиная с Al , в атомах появляются d – орбитали. С их участием КЧ может повышаться до 6, что особенно характерно для тяжелых элементов Ga, In, Tl. Координационная ненасыщенность атомов, образующих 3 связи, и наличие низких по энергии вакантных орбиталей приводит к тому, что, за исключением бора, в простых веществах элементов осуществляется металлическая связь. ФИЗ-ХИМ св-ва: Бор Кристаллы бора черного цвета, тугоплавки ( Т пл. = 2300 0 С), диамагнитны, обладают полупроводниковыми свойствами ( ширина запрещенной зоны D Е = 1,55 эВ). При комнатной температуре бор химически инертен и взаимодействует непосредственно только с фтором: 2B + 3F2 = 2 B F3 . При нагревании бор окисляется хлором, кислородом и некоторыми другими неметаллами:4 B + 3 O2 = 2 B2 O3 (700 0 C) , Df H = - 1264 к Дж/ моль , 2 B + 3 Cl2 = 2 B Cl3 . Оксид бора B2 O3 – кристаллическое вещество, О = В – О – В = О, угол ВОВ = 95 0 . При взаимодействии с водой переходит медленно в борную кислоту: B2 O3 + 3 Н2 О = 2 Н3 В О3. Отношение к галогенам. 2 В + 3 Г2 = 2 В Г3 , У бора есть вакантная 2 р – орбиталь, поэтому бор может образовывать комплексные соединения: B F3 + H F = H [BF4], Отношение к воде. 2 В + 6 Н2 О (пар) = 2 Н3 В О3 + 3 Н2 ( температура красного каления). Отношение к кислотам и щелочам. 2 B (аморфный) + 2 NaOH + 2 H2 O = 2 Na B O2 + 3 H2 Концентрированные кислоты H2 SO4 и HNO3 окисляют В до Н3 В О3 : В + 3 HNO3 (конц.) = Н3 В О3 + 3 NO2 , 2 В + 3 H2 SO4 (конц.) = 2 Н3 В О3 + 3 SO2 . Отношение к водороду. С водородом бор не реагирует Получение бораАморфный бор образуется по реакциям: В2 О3 + 3 Mg = Mg O + 2 B , K[B F4] + 3 Na = K F +3 Na F + B Алюминий Легкоплавкий серебристый металл малой плотности, обладает высокой электропроводностью и пластичностью. От взаимодействия с кислородом, парами воды и углекислым газом атмосферы алюминий защищен плотной оксидной пленкой Al2 O3 . 2 Al + 1,5 O2 = Al 2 O3 , Df H 0 = - 1669,4 к Дж / моль Алюминий химически активный металл. При комнатной температуре он реагирует с хлором, бромом, а при нагревании – с фтором, иодом, серой, азотом, углеродом с образованием соответствующих бинарных соединений: Al Cl3, Al Br3, Al2 S3, Al N, Al 4 C3 Лишенный оксидной пленки алюминий реагирует с водой 2 Al + 6 H2 O = 2 Al (OH)3 + 3 H2 . Алюминий растворяется в щелочах с образованием комплексных гидроксоалюминатов: 2 Al + 2 Na OH + 6 H2 O = 2 Na [Al (OH)4] + 3 H2 . Алюминий легко растворяется в разбавленных растворах неокислительных кислот: 2 Al + 6 H Cl = 2 Al Cl 3 (Al 2 O3 + 6 H Cl = 2 Al Cl 3 + 3 H2 O ). Малый радиус и большой положительный заряд катиона Al 3+ служат причиной того, что он является активным комплексообразователем (к. ч. = 6 ): [Al (H2 O)6 ] 3+ ; [Al F6 ] 3 - ; Получение алюминия Промышленный метод получения алюминия – электролиз расплава Na 3 [Al F6 ] Анод (уголь) (+) : Al 2 O3 – 6 е = 2 Al 3+ + 1,5 O2 , Катод (уголь) (-): 2 Al 3+ + 6 e = 2 Al (ж). Применение Алюминий – основа легких сплавов, его применяют для производства различных емкостей и аппаратов, фольги, проволоки, восстановителем в алюмотермии, для производства кабелей.
47) Общая характеристика элементов 4А группы. Олово, свинец. Их получение; взаимодействие с кислородом, галогенами, растворами кислот и щелочей. Применение в технике.
У атомов элементов 4-ой группы в основном состоянии есть вакантные орбитали на внешнем уровне. В основном состоянии они имеют конфигурацию n s2 np2 . Свойства соединений элементов 4-ой группы определяются тем, что в них нет ни реакционноспособных неподеленных пар, ни вакантных р – орбиталей. Свинец представляет типичный металл с плотной кубической упаковкой частиц в структуре ( КЧ = 12). Для свинца характерна низшая степень окисления +2. Большинство солей Pb 2+ (PbCl2, PbI 2, PbSO4) малорастворимы, поэтому Pb плохо растворяется в соляной и серной кислотах и при стандартных условиях взаимодействует только с азотной кислотой. При нагревании растворяется в растворах щелочей: Pb + 2 КОН + 2 Н2 О = К2 [Pb (OH) 4] + H 2 Олово. При низких температурах устойчиво a - Sn (серое олово), имеющее алмазоподобную решетку. При 13,2 0С происходит переход a - Sn Û b - Sn (белое олово), d20 (a - Sn) = 5,75 г/см3 ; d20 (b - Sn) = 7,31 г/см3 . Обычное белое олово должно превращаться в серое при температурах (- 30) – (- 50) 0С. Из-за большой разницы в плотностях во время перехода металл рассыпается порошок. Для олова уже в примерно равной степени характерны соединения, в которых оно находится в степенях окисления +2 и + 4. В химическом отношении олово ведет себя как малоактивный амфотерный металл. На холоду оно реагирует с галогенами, но не окисляется кислородом. Олово медленно растворяется в разбавленных кислотах с образованием солей Sn 2+ и выделением H2. Концентрированные серная и азотная кислоты окисляют олово до SnО 2. При нагревании олово растворяется в щелочах с образованием станнитов – гидроксокомплексов Sn (II), которые в присутствии окислителей переходят в соединения Sn (IV): Sn + 2 КОН + 2 Н2 О = К2 [Sn (OH) 4] + H 2 ; Sn + 2KOH +2 H2 O2 = K2 [Sn (OH) 6]; К2 [Sn (OH) 4] + H2 O2 = K2 [Sn (OH) 6]. И на последок =): 3 Pb + 8 HNO3 (разб.) = Pb (N O3) 2 + 2 NO + 4 H2 O. Sn + 4 HNO3 (конц.) = Sn O 2 . n H2 O¯ + 4 NO2 + 2 H2 O; , но при реакции с разбавленной HNO3, олово ведет себя как металл и дает катион Sn 2+. Свинец при любых условиях реагирует с образованием нитрата свинца (II). В состав группы входят 5 элементов: два неметалла – углерод и кремний, находящиеся в во втором и третьем периодах системы Менделеева и 3 металла –германий (промежуточный между неметаллами и металлами, олово и свинец, находящиеся в конце больших периодов-IV, V, VI. Для всех этих элементов характерно то, сто они имеют на внешнем энергетическом уровне 4 электрона. И поэтому могут проявлять степень окисления от +4 до -4. эти элементы образуют газообразные соединения с водородом : СН4, Si Н4, Sn Н4, PbН4. при нагревании на воздухе соединяются с элементами подгруппы кислорода, серы и с галогенами. Степень окисления +4 получается при переходе 1s –электрона на свободную р- орбиталь. С увеличением радиуса атома уменьшается прочность связи наружных электронов с ядром. Неметаллические свойства уменьшаются, а металлические нарастают. (снижаются температура плавления и кипения и т.д) Олово не принадлежит к числу широко распространенных металлов (содержание в земной коре 0,04%) но оно легко выплавляется из руд и поэтому стало изветсно человеку в виде его сплавов с медью (бронза) со времен глубокой древности. Встречается в виде кислородного соединения SnО2 (оловянного камня, из которого получается посредством восстановления углем). В свободном состоянии олово – серебристо-белый мягкий металл. При сгибании палочки олова слышится характерный треск, обусловленный трением отдельных кристаллов друг о друга. Олово обладает мягкостью и тягучестью и легко может быть прокатано в тонкие листы, называемые оловянной фольгой или станиолем. Кроме обычного белого олова, кристаллизующегося в тетрагональной системе, существует другое видоизменение олова –серое олово кристаллизующееся в кубической системе и имеющее меньшую плотность. Белое олово устойчиво при температурах выше 14оС, а серое – при температурах ниже 14оС, поэтому при охлаждении белое олово превращается в серое. В связи со значительным изменением плотности металл при этом рассыпается в серый порошок. Это явление получило название оловянной чумы. Быстрее всего превращение белого олова происходит при температурах около 30оС; оно ускоряется в присутствии зародышей кристаллов серого олова. Сплавы олова с сурьмой и медью применяются для изготовления подшипников. Эти сплавы (оловянные баббиты) обладают высокими антифрикционными свойствами. сплавы олова со свинцом – припой – широко применяются для пайки. В качестве легирующего компонента олово входит в некоторые сплавы меди. На воздухе олово при комнатной температуре не окисляется , но нагретое выше температуры плавления постепенно превращается в диоксид олова SnО2 вода не действует на олово. Разбавленная соляная и серная кислота действуют очень медленно. Концентрированные эти кислоты, при нагревании растворяют олово. Sn + 2 НСl = SnСl 2 + Н2/\ Sn + 4Н2SО4 = Sn( SО4)2 + 2 SО2/\ + 4Н2О Чем концентрированнее азотная кислота, тем интенсивнее идет реакция 4Sn + 10 НNО3 = 4Sn(NО3)2 + NН4NО3 + 3Н2О Sn + 4 НNО3 = Н2SnО3\/ + 4NО2/\ +Н2О в концентрированной Концентрированные щелочи реагируют Sn + 2NаОН = Nа2SnО2 +Н2/\ Nа2SnО2 станиит натрия. В растворах соли гидратированы (окружены молекулами воды). На воздухе олово покрывается защитной пленкой. Олово образует комплексные соединения Nа2[Sn(ОН)4] 40% олова идет на производство консервных банок. Железо покрывают оловом. Олово образует устойчивые соединения со степенью окисления +2,+4. SnО (II) –желтый порошок, получается при разложении Sn(ОН)2 –амфотерный гидроксид Sn(ОН)2+ 2NаОН = Nа[Sn(ОН)4] Диоксид олова встречается в природе. Образуется при сжигании Sn на воздухе. Оловянная кислота Н2SnО3 нерастворима (белый порошок) взаимодействует со щелочами Н2SnО3 + 2 NаОН +Н2О = Nа2 [Sn(ОН)4] Хлорид олова растворим SnСl2, гидрид олова SnН4 –бесцветный, очень ядовитый газ. Свинец. Руда. Из которой получают свинец называется свинцовый блеск. PbS + 3О2 = 2PbО + 2SО2 –обжиг. PbО плавят вместе с коксом и получают свинец, который потом очищают. Свинец – голубовато- белый тяжелый металл. Он мягок и режется ножом. Свинец широко используется в технике. Наибольшее его количество расходуется на изготовление оболочек кабелей и пластин аккумуляторов. Свинец идет на изготовление боеприпасов и изготовление дроби. Он входит в состав многих сплавов (подшипники, типографский шрифт, припой). Свинец хорошо поглощает g - излучения и используется для защиты от них при работе с радиоактивными веществами. На воздухе быстро окисляется и покрывается защитной оксидной пленкой, защищающей от дальнейшего окисления. Вода не взаимодействует со свинцом, но в присутствии воздуха разрушает его. 2Pb + О2 + 2Н2О = 2Pb (ОН)2 При соприкосновении с жесткой водой покрывается защитной пленкой нерастворимых солей и не разрушается дальше. Разбавленная соляная и серная кислоты почти не реагируют со свинцом. С концентрированной серной кислотой, при нагревании получается Pb(НSО4)2. С разбавленной азотной кислотой реагирует быстрее, чем с концентрированной . взаимодействует со щелочами Pb + 4КОН +Н2О = К4[Pb (ОН)6] + Н2/\ гидроксоплюмбит калия.Все растворимые соединении свинца ядовиты Степени окисления характерны +2, +4 PbО =- желтый порошок после прокаливания (500оС) приобретает красновато-желтый цвет и называется глетом. Гидроксид свинца Pb(ОН)2 амфотерен. Pb(ОН)2 + 4 NаОН = 2 Nа[Pb (ОН)6] При сплавлении Pb(ОН)2 с сухими щелочами получаются соли плюмбиты: Pb(ОН)2 + 2 NаОН = Nа2 PbО2 + 2Н2О Ацетат свинца применяют при крашении Pb(СН3СОО)2 PbS – черного цвета. Бумажка, смоченная раствором соли свинца быстро темнеет, если в воздухе есть сероводород. Это качественная реакция на соли сероводорода. Соединения свинца (IV) –соли плюмбаты СаО + PbО = Са PbО3 большинство нерастворимы. Аккумуляторы состоят из решетчатых свинцовых пластин, одни заполнены диоксидом свинца, а другие – металлическим губчатым свинцом. Пластины погружены в раствор 35-40% Н2SО4 . при работе идет разряд: Pb + SО42- -> PbSО4\/ + 2е Металлический свинец окисляется , а диоксид свинца восстанавливается. PbО2 + SО42 - + 4Н+ -> Pb SО4\/+ Н2О Электроны, отдаваемые атомами свинца передаются по внешней цепи. Pb – анод, а PbО2-катод. В растворе Н2SО4 происходит перенос ионов. Ионы SО42- - движутся к аноду, Н+ - к катоду.
48) Физические свойства кремния и германия. Собственная и примесная проводимость элементарных полупроводников. Способы получения и методы очистки полупроводниковых материалов.
Кремний существует только в одной модификации: его кубические кристаллы построены по типу алмаза. В отличие от алмаза кристалл кремния непрозрачен и имеет отчетливый металлический блеск, что связано с уменьшением ширины запрещенной зоны между 3р – и 3d – орбиталями по сравнению с 2р – и 3s – орбиталями углерода. Хотя в атоме кремния имеются вакантные орбитали (3d) и его КЧ может быть равно 6, тетраэдрическое окружение и КЧ = 4 более характерны для кремния, особенно в соединениях с кислородом. В силу этого кристаллический кремний, где его КЧ = 4, уже валентно - и координационно насыщен и поэтому сравнительно инертен. При стандартных условиях он реагирует только с фтором. Взаимодействие с другими элементами, в том числе с кислородом и галогенами, протекает только при нагревании до 500 – 600 0 С. Si + O2 = Si O2, Df H 0 (298) = - 911 к Дж /моль (600 0С) При высоких температурах кремний проявляет сильные восстановительные свойства: Si (k) + 2 H2 O (г) = Si O2 (k) + 2 H2, D H 0 = - 427 к Дж, D S 0 = - 98 Дж / К. химически инертен, не растворим в воде, кислотах (кроме HF), растворяется в щелочах Si O2 + 2 NaOH = Na2 SiO3 + H2 O (сплав), Si O2 + 4 HF = SiF4 + 2 H2 O. H2Si O3 – очень слабая кислота, химическая формула условна (Si O2. nH2 O). Мелкокристаллический кремний, загрязненный различными примесями, получают из диоксида Si O2 восстановлением его магнием, алюминием или углеродом. Например, Si O2 + 2 Mg = Si + 2 Mg O. Почти все количество особо чистого кремния, применяемого в полупроводниковой технике, производят с использованием следующих реакций: Si I4 = Si + 2 I2 (1000 0C) ; Si H4 = Si + 2 H2 (700 0C); Германий – по свойствам похож на кремний. Единственная форма его простого вещества имеет кубическую алмазоподобную структуру с заметной делокализацией электронов по кристаллу, что делает германий типичным полупроводником. При стандартных условиях германий несколько более инертен, чем кремний: ни кислоты, ни щелочи не действуют на него. При комнатной температуре устойчив к воде и воздуху. Ge + O2 = Ge O2 (t > 1000 0C), Ge + H2 = не реагирует. Даже с концентрированными щелочами он реагирует лишь в присутствии сильных окислителей, образуя гидроксокомплексы: Ge + 2 NaOH + 2 H2 O2 = Na2 [Ge (OH) 6]. В отличие от кремния, германий способен при нагревании растворяться в концентрированных кислотах – окислителях: Ge + 4 H2 SO4 = Ge (SO4) 2 + 2 SO2 + 4 H2 O, Ge + 4 HNO3 (конц.) = H2 GeO3 + 4 NO2 + H2 O. Подобно олову Ge дает соединения, где он находится в степени окисления +2 (GeО, GeCl2 и другие). 28 14 Si имеет три стабильных изотопа и три радиоактивных. Существует в двух формах: аморфной (бурый порошок) и кристаллической (в виде кристаллов октаэдрической формы) – серый со стальным блеском. Обладает низкой электропроводностью. На внешнем энергетическом слое 2 s и 2р-электрона. В возбужденном состоянии s-электроны распариваются, и один электрон переходит в свободную р ячейку. становится 4 валентных электрона (SiО2). В этом сродство с углеродом. Известен гомологический ряд Sin Н 2n + 2 (как у предельных углеводородов, но их только 6). По распространенности в природе занимает 2 место после кислорода. Минералы – кремнезем, кварц, горный хрусталь, силикаты –соли кремневой кислоты, тальк, полевой шпат, глины. Проявляет сходство с бором. Он как и бор начинает серию неметаллов в IV периоде. Сходство по степени электропроводности. Кристаллический Si похож на аморфный. Но кристаллы кремния обладают еще и металлическим блеском. При обычных условиях взаимодействует только с F (фтор) Si + 2 F = Si F4. при повышении температурывзаимодействует со многими неметаллами. Si + 2Сl2 = SiСl4 ; Si + О2= SiО2; Si + С = SiС (карборунд) изготавливают шлифовальные круги, точильные бруски. реагирует с металлами Мg, Сu, Fе с образованием силицидов. Мg 2Si . в воде нерастворим.а в водных растворах щелочей: Si + 2NаОН + Н2О = Nа2SiО3 +2Н2/\ Кислоты не действуют на кремний за исключением НF (плавиковая) Si + 4НF = SiF4 + 2Н2/\ Применяется кремний главным образом в металлургии и в полупроводниковой технике. В металлургии для удаления кислорода из расплавов металлов и служит составной частью сплавов (железа, меди, алюминия) В полупроводниковой технике для изготовления фотоэлементов, усилителей, выпрямителей. Полупроводниковые элементы на основе кремния выдерживают нагрев до 250оС. Соединение кремния SiО2 – твердое вещество очень тугоплавкое, широко распространено, встречается в виде двух модификаций – кристаллического (кварц, песок) и аморфного. С водой не реагирует. Раствор Nа2SiО3 и К2SiО3 – называют жидким стеклом (изготавливают клей, водонепроницаемые ткани)силикаты входят в состав горных пород (асбест, полевой шпат). Силикаты, содержащие алюминий называют алюмосиликатами. 7232Gе 1 s2 2 s2 2р63 s23d10 3р64 s2 4р2 на внешнем энергетическом уровне 4 s2 4р2 в нормальном состоянии имеет 2 спаренных s –элекрона и 2 неспаренных р электрона. В возбужденном состоянии s электроны распариваются и один занимает свободную р орбиталь. Получается 4 валентных электрона. Германий был одним из 3 –х элементов, предсказанных еще Менделеевым. Это типичный рассеянный элемент и добывают его из отходов цветной металлургии и из пыли, осаждающейся в дымоходах газогенераторов. Он содержится в каменном угле. Это хрупкий металловидный неметалл, четырехвалентный почти во всех соединениях. При обычных условиях он устойчив. Образует очень непрочные газообразные соединения с водородом. GеnН2 n +2 , окисел GеО2, ангидрид слабой и неустойчивой кислоты.Н2GеО3, соли которой называются германатами. Основной потребитель монокристаллического германия – радиотехника. Германиевый диод размером с кукурузное зернышко представляет собой пластинку из германия, в которую упирается металлический усик. Он действует как выпрямитель, заменяющий вакуумную диодную лампу, превосходя ее надежностью работы, долговечностью и компактностью. Транзисторы из германия имеют те же самые преимущества перед ламповыми триодами. Здесь играют роль особые свойства полупроводников. Как и кремний германий кристаллизуется подобно решетке алмаза. Каждый атом связан с 4 другими и связь осуществляется двумя электронами, по одному от каждого атома. Но в алмазе все валентные электроны закреплены прочно, а в кремнии и германии так слабо, что за счет энергии теплового движения отрываются от связываемых ими атомов и становятся свободными. Постоянное присутствие в кристаллах кремния и германия свободных электронов сообщает этим веществам электронную проводимость. При переходе каждого электрона из связанного в свободное состояние в ковалентной связи возникает пустое место ( или «дырка»). Такие «дырки» могут заполняться путем перемещения в них валентных электронов из ковалентных связей. Если ввести элементV группы – сурьму, то ее атомы заменят часть атомов германия , то увеличится «дырочная» проводимость, увеличится значение электропроводности. Например кристалл с поверхностью всего в 100см3 достаточен для выпрямления переменного тока для электровоза в 400 лошадиных сил. На том же свойстве основано использование полупроводников в радиотехнике. При комнатной температуре германий устойчив к действию воздуха, кислорода, воды, соляной и разбавленной серной кислот азотная и концентрированная серная кислота окисляют его до диоксида GеО2, особенно при нагревании: Gе + 2 Н2SО4 = GеО2\/ + 2 SО2/\ + 2Н2О Германий взаимодействует со щелочами в присутствии водорода . при этом образуются соли германиевой кислоты – германаты. Gе + 2NаОН +2Н2О = Nа2GеО3 + 2Н2О. Соединения германия (ll) малоустойчивы. Для использования германия в производстве полупроводников его тщательно очищают. Для придания ему электрических свойств в него вводят очень небольшие количества прuмесей (элементы lll и V групп мышьяк, сурьма, алюминий, галлий.