

Билеты химия.
1) Развитие представлений о строении атома. Уравнение волны де Бройля. Принцип неопределенности Гейзенберга.
Достижения экспериментальной физики к концу 19 в. со всей убедительностью доказали неправомерность представлений о неделимости атома. Французский физик Беккерель в 1986 г обнаружил самопроизвольное испускание урановыми рудами ранее неизвестного вида излучен6ия, проникающего через вещества. Несколько позднее то же явление было обнаружено и основательно изучено французскими учеными П. Кюри и М Склодовской-Кюри, которые объяснили наблюдаемое излучение естественной радиоактивностью. Они открыли (1898г) в урановых рудах два новых и более мощных источника излучения, чем сам уран. Ими оказались радиоактивные элементы полоний и радий. Было найдено, что радий претерпевает многоступенчатый спонтанный распад, который заканчивается образованием стабильного свинца. Поскольку атомы свинца качественно отличаются от атомов радия, такое превращение элементов можно объяснить только тем, что атомы обоих элементов построены из одинаковых, более мелких, чем сами атомы частиц. Это послужило основанием для глубокого теоретического и экспериментального изучения строения атома. Следующий этап в становлении квантовой теории строения атома начался с теоретического обоснования французским ученым де Бройлем двойственной природы материальных частиц, в частности электрона. Распространив идею Эйнштейна о двойственной природе света на вещество, де Бройль постулировал (1924г), что поток электронов наряду с корпускулярным характером обладает и волновыми свойствами. Длина волны любого движущегося объекта l=h¤mn. Где m – масса частицы; n - скорость движения частицы; h – постоянная Планка; l - длина волны объекта, называемая длиной волны де Бройля. Волновые свойства микрочастиц выражаются также в ограниченности применения к ним некоторых понятий классической механики, а именно координаты и импульса. Например, один из способов наблюдения за объектом – воздействие на него электромагнитного излучения (свет, радиоволны) и регистрации отраженного сигнала, что широко используется в радиолокации, эхолокации. Причем, чем сильнее воздействие, тем сильнее отраженный сигнал. Если ведется наблюдение за макрообъектами. То действие на них электромагнитного излучения не изменяет ни их положения, ни скорости. В случае наблюдения за объектами микромира (например электронами) ситуация выглядит иначе. При воздействии кванта света (фотона) на микрочастицу ее скорость не остается без изменения. Зная положение микрочастицы в какой-то момент времени, нельзя в это же мгновение определить ее скорость, поскольку она уже изменилась. Гейзенберг в 1927 г предложил соотношения, которые получили название принципа неопределенности. Согласно этому принципу, невозможно одновременно точно определить координаты частицы и ее импульс.
Наличие
в атоме массивного, но малого по размерам
(по сравнению с атомом) положительного
электрического заряда – ядра.( ![]() см,
размер атома
см,
размер атома ![]() см).
Исходя из этого Резерфорд построил
планетарную модель атома (в центре атома
находится положительно заряженное
массивное ядро, а легкие отрицательные
заряды (электроны) вращаются по различным
орбиталям вокруг этого ядра.)
см).
Исходя из этого Резерфорд построил
планетарную модель атома (в центре атома
находится положительно заряженное
массивное ядро, а легкие отрицательные
заряды (электроны) вращаются по различным
орбиталям вокруг этого ядра.)
Первый постулат Бора: атомная система может находиться только в особых стационарных или квантовых состояниях, каждому из которых соответствует определенная энергия. В стационарных состояниях атом не излучает. Второй постулат: При переходе атома из одного стационарного состояния в другое, излучается или поглощается квант с энергией, равной разности энергий этих состояний.
Де
Бройль предположил, что двойственной
природой обладает не только свет, но и
любой мат. объект. Длина волны любого
движущегося объекта: ![]() .
В случае наблюдения за объектами
микромира: воздействие на них фотона
(для определения координаты), ее скорость
меняется.
.
В случае наблюдения за объектами
микромира: воздействие на них фотона
(для определения координаты), ее скорость
меняется.
Принцип
Гейзенберга:
Невозможно одновременно точно определить
координаты частицы и ее импульс. ![]() (аналогично
y, z). Где дельты – погрешность определения
координат, погрешность определения
проекций импульса на оси координат. Для
волн Де Бройля:
(аналогично
y, z). Где дельты – погрешность определения
координат, погрешность определения
проекций импульса на оси координат. Для
волн Де Бройля: ![]() ,
где вторая пси-амплитуда волн Де Бройля
(координатная волновая функция).
,
где вторая пси-амплитуда волн Де Бройля
(координатная волновая функция).
2) 2. Квантовомеханическая теория строения атома. Уравнение Шредингера. Волновая функция. Радиальная и угловая составляющие. Квантовые числа. Атомные уровни, подуровни и орбитали. Формы s-, p-, d- атомных орбиталей.
Современные квантово-механические представления о строении электронной оболочки атома исходят из того, что движение электрона в атоме нельзя описать определенной траекторией. Можно рассматривать лишь некоторый объем пространства. В котором находится электрон. Поскольку электрон обладает одновременно свойствами частицы и волны, то подходом к объяснению строения электронной оболочки может быть как корпускулярная, так и волновая теория; обе они приводят к одинаковому наглядному представлению, сформулированному как орбитальная модель атома. Атомная орбиталь – это геометрический образ, отвечающий объему пространства вокруг атомного ядра, который соответствует 90%-ой вероятности нахождения в этом объеме электрона (как частицы) и одновременно 90%-ой плотности заряда электрона (как волны). Собственной характеристикой каждого электрона в атоме является спин. Два электрона, находящиеся в одной атомной орбитали, различаются по спину. В квантовой механике каждая атомная орбиталь определяется тремя квантовыми числами. Главное квантовое число n – может принимать целочисленные значения от 1 до ¥. В Периодической системе элементов максимальному значению главного квантового числа соответствует номер периода. Орбитальное квантовое число l – определяем орбитальный момент количества движения (импульс) электрона, точное значение его энергии и форму орбитали. Может принимать значения 0,1,2,3,…(n-1). Орбитальное квантовое число определяет форму атомной орбитали. При l=0 это сфера, при l=1 – объемная восьмерка (гантель), при l =2 четырехлепестковая розетка. Магнитное квантовое число ml определяет возможные значения проекции орбитального момента количества движения электрона на фиксированное направление в пространстве (например ось Z) движение электрона вокруг ядра можно сравнить с движением тока по замкнутому контуру. При этом возникает магнитное поле, вектор напряженности Н которого направлен перпендикулярно плоскости движения электрона. Если атом находится во внешнем магнитном поле, то, согласно квантово-механическим представлениям, его электроны должны располагаться так, чтобы проекции их магнитных моментов на направление этого поля были целочисленными. При этом они могут принимать как отрицательные, так и положительные значения, включая нулевое. В общем случае магнитное кантовое число характеризует ориентацию атомной орбитали в пространстве относительно внешней силы. Магнитное квантовое число определяет ориентацию орбитального углового момента относительно некоторого фиксированного направления. Орбитальному квантовому числу l =0 отвечает единственное значение магнитного квантового числа ml =0. Эти значения l и ml характеризуют все s –орбитали, которые имеют форму сферы. Так как в этом случае магнитное квантовое число принимает только одно значение, то каждый s подуровень состоит только из одной орбитали. Рассмотрим р-подуровень. При l =1 орбитали имеют форму гантелей, магнитное квантовое число принимает следующие значения ml = -1, 0, +1. Следовательно, р-подуровень состоит из трех атомных орбиталей, которые располагаются вдоль осей координат, их обозначают рx, рy, рz Законы движения частиц в квантовой механике выражаются уравнением Шредингера, которое играет в ней ту же роль, что и законы Ньютона в классической механике. Уравнение Шредингера представляет собой дифференциальное уравнение в частных производных. Э. Шредингер в 1926 г. предложил использовать волновое уравнение в качестве модели для описания поведения электрона в атоме – уравнение, связывающее энергию системы с ее волновым движением. Стационарное уравнение Шредингера для одной частицы можно записать в следующей форме: - h2__ (d2y + d2y + d2y) + Uy = Ey 8p2m (dx2 dy2 dz2 ) где U – потенциальная энергия частицы; E – ее полная энергия; x, y, z – декартовы координаты; переменная величина y называется волновой функцией. Эта функция описывает все свойства системы в стационарном состоянии – состоянии, которое изменяется во времени. Функция y зависит от координат частиц и может зависеть от времени. Каждая частица (или набор частиц) характеризуется квантово-механической волновой функцией, которая описывает состояние данной системы. Функцию y можно рассматривать как амплитуду волнового процесса, следовательно, она может быть больше и меньше нуля, а также мнимой величиной. В соответствии с физическим смыслом волновая функция конечная, непрерывная и однозначная, а также обращается в ноль там, где частица не может находиться. Уравнение Шредингера можно решить точно только для очень простых систем (атом водорода). Решая уравнение Шредингера в сферических координатах, получают ynlml = R®rnlΘ(θ)lmlФ(φ)ml где R® - радиальная составляющая волновой функции; Θ(θ) и Ф(φ) – угловые составляющие волновой функции. Следовательно, волновые функции зависят от трех целочисленных параметров n, l, ml - , которые называют квантовыми числами. Волновую функцию описывающую состояние электрона, называют атомной орбиталью.
Квантово-механическая
модель атома не обладает наглядностью.
Законы движения частиц в квантовой
механике выражаются уравнением
Шредингера. Уравнение, связывающее
энергию системы с ее волновым движением.
Стационарное уравнение Шредингера для
одной частицы: 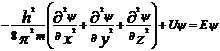 ,
где U – пот. энергия частицы, E – ее полная
энергия.
,
где U – пот. энергия частицы, E – ее полная
энергия. ![]() -вероятность
нахождения частицы в данном месте
пространства. Для волн Де Бройля:
,
где вторая пси-амплитуда волн Де Бройля
(координатная волновая функция).
В
квантовой механике каждая АО характеризуется
3-мя квантовыми числами. Главное квантовое
число n может принимать целочисленные
значения от 1 до бесконечности. Оно
определяет номер энергетического
уровня, интервал энергий электронов,
находящихся на данном уровне, размеры
орбиталей, число подуровней данного
энергетического уровня, в ПСЭ max значению
n соответствует номер периода. Орбитальное
квантовое число l определяет орбитальный
момент количества движения (импульс)
электрона, точное значение его энергии
и форму орбиталей. Может принимать
целочисленные значении от 0…n-1. Каждому
численному значению l соответствует
определенная геометрическая форма
орбиталей и приписывается буквенное
обозначение (s, p, d, f). Магнитное квантовое
число m определяет возможные значения
проекции орбитального момента количества
движения электрона на фиксированное
направление в пространстве. Принимает
отрицательные и положительные значения
l, включая 0. Общее число значений = 2l+1.
От значения m зависит взаимодействие
магнитного поля, создаваемого электроном,
с внешним магнитным полем. В общем случае
m характеризует ориентацию АО в
пространстве относительно внешней
силы. Общее число возможных значений
m соотв. числу способов расположения
орбиталей данного подуровня в пространстве,
т.е. общему числу орбиталей на данном
подуровне.
S-орбитали имеют форму
сферы(характеризуются l=0, m=0), P-орбитали
– гантели(объемные восьмерки) – l=1,
m=-1,0,1. (3 АО, расположенных вдоль осей
координат). d-подуровень – l=2, m=-2,-1,0,1,2.
5АО, dxy,dxz,dyz,dx*2-y*2, dz*2.
Собственный момент
импульса электрона – спин. Может
принимать только значение = ½. Проекция
вектора спина на опред. направление
внешнего поля наз. спиновым квантовым
числом, ms=+-1/2. Спин – проявление
релятивистских эффектов на микроскопическом
уровне.
-вероятность
нахождения частицы в данном месте
пространства. Для волн Де Бройля:
,
где вторая пси-амплитуда волн Де Бройля
(координатная волновая функция).
В
квантовой механике каждая АО характеризуется
3-мя квантовыми числами. Главное квантовое
число n может принимать целочисленные
значения от 1 до бесконечности. Оно
определяет номер энергетического
уровня, интервал энергий электронов,
находящихся на данном уровне, размеры
орбиталей, число подуровней данного
энергетического уровня, в ПСЭ max значению
n соответствует номер периода. Орбитальное
квантовое число l определяет орбитальный
момент количества движения (импульс)
электрона, точное значение его энергии
и форму орбиталей. Может принимать
целочисленные значении от 0…n-1. Каждому
численному значению l соответствует
определенная геометрическая форма
орбиталей и приписывается буквенное
обозначение (s, p, d, f). Магнитное квантовое
число m определяет возможные значения
проекции орбитального момента количества
движения электрона на фиксированное
направление в пространстве. Принимает
отрицательные и положительные значения
l, включая 0. Общее число значений = 2l+1.
От значения m зависит взаимодействие
магнитного поля, создаваемого электроном,
с внешним магнитным полем. В общем случае
m характеризует ориентацию АО в
пространстве относительно внешней
силы. Общее число возможных значений
m соотв. числу способов расположения
орбиталей данного подуровня в пространстве,
т.е. общему числу орбиталей на данном
подуровне.
S-орбитали имеют форму
сферы(характеризуются l=0, m=0), P-орбитали
– гантели(объемные восьмерки) – l=1,
m=-1,0,1. (3 АО, расположенных вдоль осей
координат). d-подуровень – l=2, m=-2,-1,0,1,2.
5АО, dxy,dxz,dyz,dx*2-y*2, dz*2.
Собственный момент
импульса электрона – спин. Может
принимать только значение = ½. Проекция
вектора спина на опред. направление
внешнего поля наз. спиновым квантовым
числом, ms=+-1/2. Спин – проявление
релятивистских эффектов на микроскопическом
уровне.
3) Строение многоэлектронных атомов. Принцип наименьшей энергии. Принцип Паули. Правило Хунда. Правило Клечковского. Электронные и электронно-графические формулы атомов элементов в основном и возбужденном состоянии.
В
многоэлектронных атомах, как и в атоме
водорода, состояние каждого электрона
можно характеризовать квантовыми
числами. Межэлектронное отталкивание
приводит к тому, что энергия электронов,
имеющих одно и то же значение n, но разные
значения l, становится различной.
Последовательность заполнения е
подуровней определяется принципом
наименьшей энергии, принципом Паули и
правилом Хунда.
Принцип
наименьшей энергии:
заполнение электронами АО происходит
в порядке возрастания их энергии.
Установлена энергетическая диаграмма
для различных АО в много-е нейтральных
атомов, находящихся в основном состоянии(с
наименьшей энергией). Правило
Клечковского:
энергия АО возрастает в соотв. с
увеличением n+l. При одинаковом значении
суммы энергия меньше у АО с меньшим
значением n.
Принцип
Паули:
в атоме не м.б. 2 е с одинаковым значением
4х квантовых чисел. Этот набор значений
полностью определяет энергетическое
состояние е. 2 е, находящихся на одной
АО называются спаренными. Общее число
орбиталей на эн. уроне со зн. n = n*2.
Следовательно, max электронная емкость
= 2n*2.
Правило
Хунда определяет
последовательность заполнения АО е в
пределах одного подуровня и гласит: При
данном значении l (в пределах 1 подуровня)
в основном состоянии электроны
располагаются т.о., что значение суммарного
спина атома max(на подуровне должно быть
max число неспаренных e).
Распределение
е по разл. АО называют е конфигурацией
атома. Эл.
конфигурация с
наименьшей энергией соответствует
основному состоянию атома, остальные
конфигурации относятся к возбужденным
состояниям. ЭК атома изображают 2мя
способами: в виде е формул и е-графических
диаграмм. При написании е формул
используют n и l. Подуровень обозначают
с помощью n и l(буквой). Число е на подуровне
характеризует верхний индекс. Например,
для основного состояния атома водорода: ![]() В
случае е-графических диаграмм распределение
е по подуровням представляют в виде
квантовых ячеек. Орбиталь принято
изображать квадратом, около кот.
проставлено обозн. подуровня. Подуровни
на каждом уровне д.б. немного смещены
по высоте (энергия различна). Электроны
изображаются против. стрелками в завис.
от значения спина.С учетом структуры
ЭК атомов все известные Эл. в соответствии
со значением орбитального квантового
числа последнего заполняемого подуровня
можно разбить на 4 группы: s, p, d и
f-элементы.
Отклонения от правила n+l
наблюдаются у нек. элементов – это
связано с тем, что с увеличением главного
квантового числа различия между энергиями
подуровней уменьшаются.
В
случае е-графических диаграмм распределение
е по подуровням представляют в виде
квантовых ячеек. Орбиталь принято
изображать квадратом, около кот.
проставлено обозн. подуровня. Подуровни
на каждом уровне д.б. немного смещены
по высоте (энергия различна). Электроны
изображаются против. стрелками в завис.
от значения спина.С учетом структуры
ЭК атомов все известные Эл. в соответствии
со значением орбитального квантового
числа последнего заполняемого подуровня
можно разбить на 4 группы: s, p, d и
f-элементы.
Отклонения от правила n+l
наблюдаются у нек. элементов – это
связано с тем, что с увеличением главного
квантового числа различия между энергиями
подуровней уменьшаются.
Число электронов, которые могут находиться на одном энергетическом уровне, определяется формулой 2n2, где n – номер уровня. Максимальное заполнение первых четырех энергетических уровней: для первого уровня – 2 электрона, для второго – 8, для третьего – 18, для четвертого – 32 электрона. Максимально возможное заполнение электронами более высоких энергетических уровней, в атомах известных элементов не достигнуто. Квантово-механические расчеты показывают, что в многоэлектронных энергия электронов одного уровня неодинакова; электроны заполняют атомные орбитали разных видов и имеют разную энергию. Каждый энергетический уровень, кроме первого, расщепляется на такое число энергетических подуровней, сколько видов орбиталей включает этот уровень. Второй энергетический уровень расщепляется на два подуровня (2s – и 2p-подуровни), третий энергетический уровень – на три подуровня (3s-, 3p- и 3d-подуровни). Каждый s-подуровень содержит одну s орбиталь, каждый р-подуровень – три р-орбитали, каждый d-подуровень семь f-орбиталей. Закономерность заполнения электронных оболочек атомов определяется принципом запрета, установленным в 1925 г швейцарским физиком Паули (принцип Паули): В атоме не могут одновременно находиться два электрона с одинаковым набором четырех квантовых квантовых чисел (заполнение электронами орбиталей происходит следующим образом: сначала на каждой орбитали располагается по одному электрону, затем, после заполнения всех орбиталей происходит распределение вторых электронов с противоположным спином). Используя понятия квантовые числа можно сказать, что: Каждый электрон в атоме однозначно характеризуется своим набором четырех квантовых чисел - главного n, орбитальногоl, магнитного ml, и спинового ms. Заселение электронами энергетических уровней, подуровней и атомных орбиталей подчиняется следующему правилу: В невозбужденном атоме все электроны обладают наименьшей энергией (принцип наименьшей энергии). Это означает, что каждый из электронов, заполняющих оболочку атома, занимает такую орбиталь, чтобы атом в целом имел минимальную энергию. Последовательно квантовое возрастание энергии подуровней происходит в следующем порядке: 1s - 2s -2р - 3s – 3р - 4s –3d - 4р - 5s -…. Такой порядок увеличения энергии подуровней определяет расположение эле Ментов в Периодической системе. Заполнение атомных орбиталей внутри одного энергетического подуровня происходит в соответствии с правилом, сформулированным немецким физиком Ф. Хундом (1927г) (правило Хунда): При данном значении квантового числа l (т.е. в пределах одного подуровня) в основном состоянии электроны располагаются таким образом, что значение суммарного спина атома максимально. Это означает, что на подуровне должно быть максимально возможное число неспаренных электронов. Порядок возрастания энергии атомной орбитали в сложных атомах описывается правилом Клечковского: энергия атомной орбитали возрастает в соответствии с увеличением n +l главного и орбитального квантовых чисел. При одинаковом значении суммы энергия меньше у атомной орбитали с меньшим значением главного квантового числа. Распределение электронов по различным атомным орбиталям называют электронной конфигурацией атома. Электронная конфигурация с наименьшей энергией соответствует основному состоянию атома, остальные конфигурации относятся к возбужденным состояниям. Электронную конфигурацию атома изображают двумя способами – в виде электронных формул и электронно-графических диаграмм. При написании электронных формул используют главное и орбитальное квантовые числа. Подуровень обозначают с помощью главного квантового числа (цифрой) и орбитального квантового числа (соответствующей буквой). Число электронов на подуровне характеризует верхний индекс. Например. Для основного состоянии атома водорода электронная формула: 1s1. Более полно строение электронных подуровней можно описать с помощью электронографических диаграмм, где распределение электронов по подуровням представляют в виде квантовых ячеек. Орбиталь в этом случае принято условно изображать квадратом, около которого проставлено обозначение подуровня. Подуровни на каждом уровне должны быть немного смещены по высоте, так как их энергия несколько различается. Электроны обозначают стрелками или ¯ в зависимости от знака спинового квантового числа. С учетом структуры электронных конфигураций атомов все известные элементы в соответствии со значением орбитального квантового числа последнего заполняемого подуровня можно разбить на четыре группы: s –элементы, р-элементы, d-элементы, f-элементы.
4) Периодический закон Д.И. Менделеева. Основные энергетические характеристики атома – энергия ионизации и сродства к электрону. Электроотрицательность. Закономерности их изменения по периодам и группам периодической таблицы.
В настоящее время ПЗ формулируется: свойства хим. элементов, а также форма и свойства образуемых ими соединений находятся в периодической зависимости от величины заряда ядер их атомов. Каждый из периодов (исключая первый) начинается типичным металлом (щелочной группы) и заканчивается инертным газом. В периоде, с увеличением заряда ядра наблюдается изменение св-в от металлических к типично неметаллическим, что связано с увеличением числа е на внешнем энергетическом уровне. В Группах объединены элементы, имеющие сходное е строение внешнего эн. уровня. Эл-ты аналоги (в 1 группе) проявляют схожие хим. св-ва. Т.о., при послед. увеличении зарядов атомных ядер периодически повторяется конфигурация ЭО и, как следствие, периодически повторяются хим. св-ва элементов. В этом заключается физ. смысл периодического закона. Номер группы, как правило, указывает на число е, способных участвовать в образовании хим. связей (валентные электроны). – физический смысл номера группы. Важнейшие характеристики атомов: размеры, энергия ионизации, сродство к электрону, электроотрицательность. Атомные радиусы - Орбитальные радиусы атомов изменяются периодически. В периодах, по мере роста заряда ядер ОР уменьшаются (при одинаковом числе эн. уров. в периоде возрастает заряд ядра, а след., и притяжение е к ядру.). В группах с ростом заряда ядер ОР атомов увеличиваются. В главных подгруппах такое увел. происх. в большей степени. Энергия ионизации – минимальная Е, которую требуется затратить на то, чтобы удалить данный е с АО невозбужденного атома на б.б. расстояние от ядра без сообщения ему кин. Е. (Э+Е->Э*+ + е), Е – в кДж/моль. Е хар-ет спос. ат. удерж. е – важная хар-ка его хим. акт-ти. Для много-е атомов можно рассматривать несколько Е, соотв-х Е отрыва 1,2, 3… е. (в периодах Е увел, в группах уменьшается). Потенциал ионизации –разность потенциалов, под воздействием которой е обретает Е, соотв. Е ионизации, измеряют в вольтах. Сродство к е атома – энергия, которая выделяется (или затрачивается) при присоединении в нейтральному атому е с образованием отрицательного иона: Э+е->Э*- + Е, в кДж/моль. Сродство к е считают положительным, если присоед. сопр. выделением Е и наоборот. Зависит от е стр-ры атома. Наибольшим сродством обладают Эл. гр. VIIA (галогены). В подгруппах сверху вниз уменьшается, но не всегда монотонно. Электроотрицательность. Это способность атома в молекуле или сложном ионе притягивать к себе е, учавствующие в образовании хим. связи. В периоде эо аозрастает с увеличением порядкового номера, а в группе, как правило, убывает по мере увеличения ядра. Т.о. наим эо – s-эл. 1-ой группы, наиб. – p-эл-ты 6, 7 групп.
Утверждение атомно-молекулярной теории на рубеже 18-19 в сопровождалось бурным ростом числа известных химических элементов. К 1830 г их число достигло 55. существование такого количества элементов, разнообразных по своим свойствам требовало их упорядочения и систематизации. Многие ученые занимались поиском закономерностей и даже открыли явление периодичности свойств (Мейер), но они не смогли объединить все элементы в единую периодическую систему, т.к. в их системах многие элементы не находили себе места. Периодический закон был открыт Менделеевым в 1869 г и сформулирован следующим образом: свойства простых тел, а также форма и свойства их соединений находятся в периодической зависимости от атомного веса элементов. Сейчас основываясь на теории строения атома периодический закон формулируется: свойства химических элементов, а также форма и свойства образуемых ими соединений находятся в периодической зависимости от величины заряда ядер их атомов. графическим изображением периодического закона является таблица Периодической системы элементов. В таблице элементы располагаются в порядке возрастания заряда ядра их атомов. Число электронов, находящихся на внешнем уровне в атомах элементов, расположенных в порядке увеличения порядкового номера периодически повторяются, а поэтому периодически изменяются и свойства (физические и химические) элементов и их соединений. По числу заполненных энергетических уровней элементы делят на семь периодов каждый период начинается со щелочного металла и заканчивается (инертным) газом. Внешний слой которого полностью заполнен электронами (их не более 8), а на первом уровне – 2. Внешние электронные оболочки у многих элементов сходны (Li, К, Nа..) (Ве, Мg, Са..) (F, Сl, Вr, I..). Каждая из этих групп оказывается в определенной главной подгруппе. У них заполняются s или р-подуровни внешнего слоя. Максимальное число электронов на этих подуровнях 2+6=8. Всего 8 главных подгрупп и 8 побочных подгрупп . В периодах слева направо радиусы атомов меняются незначительно. С увеличением заряда ядра атомов наблюдается постепенное изменение свойств от металлических к типично неметаллическим, что связано с увеличением числа электронов на внешнем энергетическом уровне. Первые три периода содержат только s и р элементы. Четвертый и последующие периоды включают в свой состав также элементы, у которых происходит заполнение d и f подуровней соответствующих внутренних энергетических уровней. При этом f элементы объединяются в семейства, называемые лантаноидами (4 f-элементы) и актиноидами 5 f-элементы. В вертикальных колонках, называемых группами, объединены элементы, имеющие сходное электронное строение внешнего энергетического уровня при различных значениях главного квантового числа и поэтому проявляют сходные химические свойства. Для химии особый интерес представляет энергетическое состояние электронов внешних уровней, так как именно они ответственны за образование химических связей. Энергия ионизации Е1 – минимальная энергия, которую требуется затратить на то, чтобы удалить данный электрон с атомной орбитали невозбужденного атома на бесконечно большое расстояние от ядра без сообщения ему кинетической энергии. Энергия ионизации соответствует следующему процессу: Э + ЕI → Э+ + е, где ЕI - кДж / моль. Энергия ионизации количественно характеризует способность атома удерживать электроны, что является важной характеристикой его химической активности. Энергии ионизации возрастает в периоде по мере увеличения порядкового номера элемента. Наименьшее ее значение имеют щелочные металлы, находящиеся в начале периода. Наибольшее значение энергии ионизации характерно для инертных газов, находящихся в конце периода. В группе элементов энергия ионизации уменьшается с повышением порядкового номера элемента. Это обусловлено увеличением размеров атомов и экранированием внешних электронов внутренними. Сродство к электрону атома ЕА – энергия, которая выделяется (или затрачивается) при присоединении к нейтральному атому электрона с образованием отрицательного иона: Э + е → Э- + ЕА где ЕА - кДж / моль. Сродство к электрону считают положительным, если присоединение электрона сопровождается выделением энергии (ЕА> 0). Если для присоединения электрона нужно затратить энергию, то сродство к электрону считается отрицательным (ЕА<0). Сродство к электрону зависит от электронной структуры атома. Наибольшим сродством к электрону обладают элементы подгрупп 7А (галогены) у большинства металлов и благородных газов сродство к электрону невелико или даже отрицательно. Наименьшее значение сродства к электрону у атомов с заполненными и наполовину заполненными s и р-подуровнями. В подгруппах сверху вниз сродство к электрону атомов уменьшается, но не всегда монотонно. Вследствие экспериментальных трудностей значение сродства к электрону известны не для всех атомов. Понятие электроотрицательности элементов ввел американский физикохимик Полинг. По определению Полинга электроотрицательность – это способность атома в молекуле или сложном ионе притягивать к себе электроны, участвующие в образовании химической связи. Электроотрицательность зависит от типа соединений, валентного состояния элемента. Поэтому такая характеристика имеет условный характер. Однако ее использование полезно для качественного объяснения типа химических связей и свойств соединений. В периоде электроотрицательность возрастает с увеличением порядкового номера элемента (слева направо), а в группе, как правило, убывает по мере увеличения заряда ядра (сверху вниз). Таким образом , наименьшее значение электроотрицательности имеют s-элементы 1 группы, а наибольшее р-элементы 6 и 7 групп.
5) Химическая связь. Зависимость потенциальной энергии от межъядерного расстояния в двухатомной молекуле. Виды химической связи. Основные характеристики химической связи: длина, энергия, кратность связи, валентный угол. Виды химических связей. Ионная связь. Металлическая связь. Межмолекулярные взаимодействия. Водородная связь.
Химическая связь – совокупность взаимодействий атомов, приводящая к образованию устойчивых систем (молекул, комплексов, кристаллов.). Она возникает, если в результате перекрывания е облаков атомов происходит уменьшение полной энергии системы. Мерой прочности служит энергия связи, которая определяется работой, нужной для разрушения данной связи. Виды хим. связи: ковалентная (полярная, неполярная, обменная и донорно-акцепторная), ионная, водородная и металлическая. Длина связи – расстояние между центрами атомов в молекуле. Энергия и длина связей зависят от характера распределения Эл. плотности между атомами. На распределение е плотности влияет пространственная направленность хим. связи. Если 2-х атомные молекулы всегда линейны, то формы многоатомных молекул м.б. различны. Угол между воображаемыми линиями, которые можно провести через центры связанных атомов называется валентным. Распределение е плотности так же зависит от размеров ат. и их эо. В гомоатомных Эл. плотность распределена равномерно. В гетероатомных смещена в том направлении, которое способствует уменьшению энергии системы.
Между
молекулами, валентно-насыщенными в
обычном понимании, на расстояниях,
превышающих размеры частиц, могут
проявляться электростатические силы
межмолекулярного притяжения, или так
называемые силы Ван-дер-Ваальса. Как
показывают квантово-механические
расчеты, Епр. определяется суммой
ориентационного, индукционного и
дисперсионного взаимодействий:
Епр=Еор+Еинд+Едисп. Ориентационное вз.
проявляется между полярными молекулами,
которые при приближении поворачиваются
(ориентируются) друг к другу разноименными
полюсами так, стобы пот Е сист. стала
мин. Индукционное взаимодействие связано
с процессами поляризации молекул
окружающими диполями.
Дисперсионное
взаимодействие возникает у любых
молекул, независимо от их строения и
полярности. Вследствие мгновенного
несовпадения центров тяжести зарядов
е облака и ядер образуется мгновенный
диполь, который индуцирует мгновенные
диполи в др.частицах.
Силы притяжения
Ван-дер-Ваальса – дальнодействующие.
На небольших расстояниях между молекулами
заметными становятся близкодействующие
силы отталкивания (силы Паули), которые
возрастают при сближении частиц. Для
неполярных молекул Е межмол. взаим
опис.: ![]() ,
где l-расст-е между мол., а и b – пост.,
завис от прир вещ-в. U0 – Е взаим. молек.
на равновесном расстоянии… <=1…5кДж/моль,
т.е. по сравнению с ковалентной связью
межмолек. взаим. оч слабое.
Промежуточный
характер между валентным и межмолекулярным
взаимодействием имеет водородная
связь.
Она хар-на для жидкостей, в состав молекул
которых (вода, спирты, кислоты) входит
положительно поляризованный атом
водорода. Малые размеры и отсутствие
внутренних е позволяют атому Н вступать
в доп. взаим. с ков-но с ним не связ.
отриц-но поляризованным атомом др.
молекулы. Такая специф. связь имеет
черты электростатич. и дон-акц. взаим.
и прив. к обр. ассоциатов молекул.
Е
водородных связей невелика (8…80 кДж/моль),
и в области выс. т-р эти связи практически
не существуют.
,
где l-расст-е между мол., а и b – пост.,
завис от прир вещ-в. U0 – Е взаим. молек.
на равновесном расстоянии… <=1…5кДж/моль,
т.е. по сравнению с ковалентной связью
межмолек. взаим. оч слабое.
Промежуточный
характер между валентным и межмолекулярным
взаимодействием имеет водородная
связь.
Она хар-на для жидкостей, в состав молекул
которых (вода, спирты, кислоты) входит
положительно поляризованный атом
водорода. Малые размеры и отсутствие
внутренних е позволяют атому Н вступать
в доп. взаим. с ков-но с ним не связ.
отриц-но поляризованным атомом др.
молекулы. Такая специф. связь имеет
черты электростатич. и дон-акц. взаим.
и прив. к обр. ассоциатов молекул.
Е
водородных связей невелика (8…80 кДж/моль),
и в области выс. т-р эти связи практически
не существуют.
6) Ковалентная связь. Механизм ее образования: обменные и донорно-акцептор-ный. Метод валентных связей. Гибридизация атомных орбиталей; виды гибридизации. Геометрическая конфигурация молекул. Свойства ковалентной связи: направленность, насыщаемость, полярность. Электрический (дипольный) момент связи и молекулы.
Ковалентная связь – связь, осуществляемая за счет образования е пар, принадлежащих обоим атомам. Различают полярную и неполярную. В случае полярной: в чистом виде может возникать только между одинаковыми атомами за счет объединения е с различными спинами в е пары. Ковалентная полярная связь возникает между атомами разных элементов, обладающих различной е-отрицательностью. При этом МО искажаются, т.к. е смещаются к более е-отр. Э (и при сохраняющейся е нейтральности молекулы, в ней появляются центры положительных и отрицательных зарядов, молекула становится диполем). . Способ образования ковалентной связи, когда каждый атом отдает по 1 е для образования общей е пары называется обменным. Донорно-акцепторный тип ковалентной связи – Один атом предоставляет пару е, другой – свободную орбиталь.(нередко превышает число неспаренных е в его атомах…). Донорно-акцепторный механизм образования связи отличается от обменного только происхождением общей е пары, во всем остальном оба эти механизма тождественны. Часто один и тот же атом может выступать как в роли донора, таки в роли акцептора е. Механизм образования связи между такими атомами называют дативным. МВС базируется на: каждая пара ат. в молекуле удерживается вместе при помощи одной или нескольких общих е пар; одинарная ков. связь 2-мя электронами с антипараллельными спинами, расп. на валентных орбиталях связывающихся атомов; при образовании связи происходит перекрывание волновых функций электронов, ведущее к увеличению е плотности между ат. и уменьшению общей Е системы; связь образуется в том направлении, при котором возможно максимальное перекрывание волновых функций; угол между связями в молекуле соответствует углу между образующими связь электронными облаками; из 2х орбиталей атома более прочную связь образует та, которая сильнее перекрывается орбиталью др. атома. Геометрическая форма молекул зависит от направленности хим. связи. Атомы, у которых валентные е расположены на s АО, способны образовывать одинаково прочные связи в любых направлениях. Общее е облако в таких случаях сосредоточено вдоль линии связи (s-связь). Для p-АО макс. перекрещивание возможно и по линии связи ядер, и по обе стороны от нее(p-связь). Для d-орбиталей возможно s, p, δ-связь. Гибридизация валентных орбиталей: при образовании хим. связей исходные атомные орбитали смешиваются, взаимно изменяются, образуя равноценные гибридные орбитали, которые отличаются от АО значительным увеличением электронной плотности в определенном направлении пространства. sp-ГО являются диагональными О, т.е. угол между ними = 180. sp2-ГО (тригональные) расположены под углом 120. При sp3 гибридизации образуется 4 тетрагональные ГО, направленные к вершинам тетраэдра. 109,28’.(109,5) sp-прямая линия, sp2, dp2, sd2 – треугольник, pd2-тригональная пирамида, sp3-тетраэдр, dsp2-квадрат, sp3dz*2 – тригональная бипирамида, sp3dx*2-y*2 – квадратная пирамида, sp3d2-октаэдр. Образование комплексных соединений: донорно-акцепторный механизм взаимодействия комплексообразователя и лигандов. Комплексообразователь выступает в роли акцептора, а лиганд – в роли донора е. Геом. форма комплексных частиц определяется типом гибридизации АО комплексообразователя. Прием *ГВО* основан на принципе максимального перекрывания е облаков и содержит условия устойчивой гибридизации орбиталей: в гибридизации участвуют орбитали с близкими значениями энергии, т.е. s- и p-АО внешнего и d-АО внешнего и предвнешнего ЭУ и с дост. выс. е плотн. ГО должны быть ориент. и выт. в пр-ве так, чтобы Е их отталк. была миним., а перекр. с орб. соседн. ат. наиб. полно. Учитывается, что ГО вследств. их асимметрии в образовании p-связей участвовать не могут. Теория отталкивания е пар валентных орбиталей: конфигурация связей многовал. ат. обуславливается числом связывающих и несвязывающих е пар в валентной оболочке центрального атома; ориентация облаков е пар вал. орб. опред. макс. взаимн. отталкиванием заполняющих их е. Теория ОЭПВО построена на след. допущ.: 1) неразличимости е, 2) действия кулоновских Сид, 3)действия сил отталквивания Паули.
Ковалентная связь - это связь, возникающая между атомами за счет образования общих электронных пар. В основе ее также лежит представление о приобретении атомами энергетически выгодной и устойчивой электронной конфигурации из 8 электронов (для атома водорода из 2). Такую конфигурацию атомы получают не путем отдачи или присоединения электронов как в ионной связи, а посредством образования общих электронных пар. Механизм образования такой связи может быть обменный или донорно-акцепторный. К обменному механизму относят случаи, когда в образовании электронной пары от каждого атома участвует по одному электрону. Например водород: Н2 Н. +Н. →Н:Н или Н-Н. Связь возникает благодаря образованию общей электронной пары за счет объединения неспаренных электронов. У каждого атома есть по одному s –электрону. Атомы Н равноценны и пары одинаково принадлежат обоим атомам. По этому же принципу происходит образование общих электронных пар (перекрывание р-электронных облаков) при образовании молекулы Сl2. При образовании молекулы N2 Образуются 3 общие электронные пары. Перекрываются р-орбитали. Связь называется неполярная. При образовании молекулы хлороводорода перекрывается орбиталь s-электрона водорода и орбиталь р-электрона хлора Н-Сl. Связывающая электронная пара смещена к атому хлора, в результате чего образуется диполь, который измеряется дипольным моментом. Связьназывается полярная. По донорно-акцепторному механизму происходит образование иона аммония. Донор (азот) имеет электронную пару, акцептор – (Н+) свободную орбиталь, которую пара электронная азота может занять. В ионе аммония три связи азота с водородом образованы по обменному механизму, а одна по донорно-акцепторному. Все 4 связи равноценны. Ковалентные связи классифицируют не только по механизму образования общих электронных пар, соединяющих атомы, но и по способу перекрывания электронных орбиталей , по числу общих пар, а также по смещению их. По способу перекрывания – σ (сигма s- s, s-р, р-р) π (р-р гантели перекрываются двумя местами). В молекуле азота между атомами существуют одна σ-связь и две π-связи, которые находятся в двух взаимно перпендикулярных плоскостях. По числу общих электронных пар различают: одинарные Н2, НСl; двойные С2Н4, СО2; тройные N2. По степени смещенности: полярные и неполярные. Связь между атомами с одинаковой электроотрицательностью – неполярная, с разной – полярная. Исследования ученых позволили сделать вывод, что химическая связь в молекуле водорода осуществляется путем образования пары электронов с противоположно направленными спинами. Каждый электрон занимает место в квантовых ячейках обоих атомов, т.е. движется в силовом поле, образованном двумя силовыми центрами – ядрами атомов водорода. Это представление о механизме образования химической связи было развито учеными Гейтлером и Лондоном на примере водорода.это было распространено и на более сложные молекулы. Разработанная на этой основе теория образования химической связи получила название метода валентных связей. Метод ВС дал теоретическое объяснение важнейших свойств ковалентной связи, позволил понять строение большого числа молекул. Хотя этот метод не оказался универсальным и в ряде случаев не в состоянии правильно описать структуру и свойства молекул – все же он сыграл большую роль в разработке квантово-механической теории химической связи и не потерял своего значение до настоящего времени. В основе метода ВС лежат следующие положения: - ковалентная связь образуется двумя электронами с противоположно направленными спинами, причем эта электронная пара принадлежит двум атомам. -ковалентная связь тем прочнее, чем в большей степени перекрываются взаимодействующие электронные облака. Геометрическая форма s –орбитали сферическая, от центра к краям размазанная (более плотная у ядра, и менее- на краях). Орбитали р-электронов представляют собой гантели, направленные вдоль осей координат. Облака d –электронов имеют более сложную форму. Метод гибридизации орбиталей исходит из предположения, что при образовании молекул вместо исходных s-, р-, d-,f- орбиталей (облаков) образуются такие равноценные «смешанные» или гибридные электронные облака, которые вытянуты по направлению к соседним атомам, благодаря чему достигается более полное их перекрывание с электронными облаками других атомов. На гибридизацию затрачивается энергия, за то она окупается более полным перекрыванием. Получается более прочная молекула. Затраченная на гибридизацию энергия окупается энергией, выделяющейся при образовании связи. Пример –молекула метана.В результате перекрывания четырех гибридных sр3 орбиталей атома углерода и 4 s орбиталей 4-х атомов водорода, образуется тетраэдрическая модель молекулы метана с четырьмя σ связями, под углом 1090. Если в молекуле гибридизуется 3-р орбитали, то sр2 гибридизация – молекула этилена, если 2 орбитали sр – гибридизция (ацетилен). У элементов 3 и последующих периодов в образовании гибридных облаков участвуют и d-электроны. В этом случае образуются 6 равноценных гибридных облака, вытянутых к вершинам октаэдра sр3 d2-гибридизация. Такую гибридизацию имеет центральный атом комплексного иона. Этим объясняется их октаэдрическая структура. Ковалентная связь обладает направленностью. Область перекрывания располагается в определенном направлении по отношению к взаимодействующим атомам. Характер распределения электронов по молекулярным орбиталям позволяет объяснить магнитные свойства частиц. Молекулы, суммарный спин которых равен нулю, проявляют диамагнитные свойства, т.е. во внешнем магнитном поле их собственные магнитные моменты ориентируются против направления поля. Молекулы, суммарный спин которых отличен от нуля, проявляют парамагнитные свойства, т.е. во внешнем магнитном поле их собственные магнитные моменты ориентируются в направлении поля. Таким образом молекула Н2 диамагнитна. Геометрическая форма молекул зависит от направленности химической связи. Ядра атомов молекул имеющих sр-гибридизацию атомных орбиталей расположены в одной плоскости, sр2 –направлены к вершинам треугольника, sр3 – к верщинам тетраэдра (стр 119).
7) Ковалентная связь. Метод молекулярных орбиталей (МО). Связывающие и разрыхляющие МО. Энергетические диаграммы гомоядерных двухатомных молекул. Порядок связи. Магнитные свойства молекул и молекулярных ионов.
Метод
МО:
молек. рассматривается как единое целое,
кажд. е принадлежит молекуле в целом и
движется в поле всех ее ядер и электронов;
состояние i-го электрона описывается
одноэл-й волновой функцией ![]() ,
характеризуемой определенным набором
квантовых чисел; квадрат модуля волновой
функции определяет плотность е облака;
полное описание состояния е хар-ет
молекулярная спин-орбиталь, выражаемая
произведением МО и спиновой функции
ψS; каждой МО соотв. опред. энергия, кот.
слагается из кин. Е е, пот Е притяжения
е ко всем ядрам и усредненной пот Е
отталкивания данного е от всех остальных
е; совокупность МО, называемая Эл.
конфигурацией молекулы, строится на
основе фундаментальных положений
квантовой механики: принципа
наим. Е, принципа Паули и правила Хунда;
движение е взаимно независимое, и общая
волновая функция основного состояния
молекулы задается как произведение
одноэлектронных волновых функций
,
характеризуемой определенным набором
квантовых чисел; квадрат модуля волновой
функции определяет плотность е облака;
полное описание состояния е хар-ет
молекулярная спин-орбиталь, выражаемая
произведением МО и спиновой функции
ψS; каждой МО соотв. опред. энергия, кот.
слагается из кин. Е е, пот Е притяжения
е ко всем ядрам и усредненной пот Е
отталкивания данного е от всех остальных
е; совокупность МО, называемая Эл.
конфигурацией молекулы, строится на
основе фундаментальных положений
квантовой механики: принципа
наим. Е, принципа Паули и правила Хунда;
движение е взаимно независимое, и общая
волновая функция основного состояния
молекулы задается как произведение
одноэлектронных волновых функций ![]() ;
образование МО упрощенно рассматривается
как лин.комб. АО.
При сложении волновых
функций 2х валентных АО разных атомов,
имеющих близкие значения Е, е плотность
между ядрами ат. увеличивается, это
ведет к обр. связывающей
МО,
имеющей более низкое значение Е, чем
исходные АО.(
;
образование МО упрощенно рассматривается
как лин.комб. АО.
При сложении волновых
функций 2х валентных АО разных атомов,
имеющих близкие значения Е, е плотность
между ядрами ат. увеличивается, это
ведет к обр. связывающей
МО,
имеющей более низкое значение Е, чем
исходные АО.( ![]() ).
При
вычитании волновых функций 2х валентных
АО разных атомов, имеющих близкие
значения Е, е плотность между ядрами
уменьшается, что ведет к образованию разрыхляющей
МО,
имеющей более высокое значение Е, чем
исходные АО.
ЛК невалентных АО, как
показывают квантовомех. расчеты, приводит
к образованию МО, имеющих вид и Е, близкую
к виду и Е исходн. АО, поэтому такие
орбитали условно называют несвязывающими.
Для возникновения МО требуются опред.
условия: Е АО должны быть соизмеримы; е
облака взаимодейств. атомов должны
макс. перекрываться, чем значительнее
перекрывание, тем прочнее связь;
макс. перекрывание возможно для АО,
обладающих одинаковыми свойствами
симметрии относительно оси
молекулы.
Образование МО возможно
при ЛК: 2 s-АО (ss СМО, s*s РМО), 2 pz-АО (spz,s*pz),
2 px АО(ppx, p*px), аналогично с py. Разрешенные
комбинации: s – s,pz,dz*2; pz-ан-но; px-px,dxz(py
ан-но); dxz – px,dxz (dyx, dxy – ан-но); dz*2 – s, pz,
dz*2, dx2-y2 – dx2-y2.
От H2 до N2 s2pz после
p2px=2py, а от О2 до Ne2 наоборот…
Порядок
связи:
).
При
вычитании волновых функций 2х валентных
АО разных атомов, имеющих близкие
значения Е, е плотность между ядрами
уменьшается, что ведет к образованию разрыхляющей
МО,
имеющей более высокое значение Е, чем
исходные АО.
ЛК невалентных АО, как
показывают квантовомех. расчеты, приводит
к образованию МО, имеющих вид и Е, близкую
к виду и Е исходн. АО, поэтому такие
орбитали условно называют несвязывающими.
Для возникновения МО требуются опред.
условия: Е АО должны быть соизмеримы; е
облака взаимодейств. атомов должны
макс. перекрываться, чем значительнее
перекрывание, тем прочнее связь;
макс. перекрывание возможно для АО,
обладающих одинаковыми свойствами
симметрии относительно оси
молекулы.
Образование МО возможно
при ЛК: 2 s-АО (ss СМО, s*s РМО), 2 pz-АО (spz,s*pz),
2 px АО(ppx, p*px), аналогично с py. Разрешенные
комбинации: s – s,pz,dz*2; pz-ан-но; px-px,dxz(py
ан-но); dxz – px,dxz (dyx, dxy – ан-но); dz*2 – s, pz,
dz*2, dx2-y2 – dx2-y2.
От H2 до N2 s2pz после
p2px=2py, а от О2 до Ne2 наоборот…
Порядок
связи: ![]() -
показатель прочности молекулы. Если ПС
= 0, значит энергия связи в молекуле равна
нулю.
Магнитные
свойства молекул:
характер распределения е по МО может
объяснить магн. св-ва частиц. Молекулы,
суммарный спин которых = 0 проявляют
диамагнитные свойства (во внешнем
магнитном поле их собственные магнитные
моменты ориентируются против магнитного
поля), суммарный спин которых не равен
0 проявляют парамагнитные св-ва (наоборот).
-
показатель прочности молекулы. Если ПС
= 0, значит энергия связи в молекуле равна
нулю.
Магнитные
свойства молекул:
характер распределения е по МО может
объяснить магн. св-ва частиц. Молекулы,
суммарный спин которых = 0 проявляют
диамагнитные свойства (во внешнем
магнитном поле их собственные магнитные
моменты ориентируются против магнитного
поля), суммарный спин которых не равен
0 проявляют парамагнитные св-ва (наоборот).
олее универсальным квантово-механическим методом описания химической связи является метод молекулярных орбиталей (МО). В этом методе состояние электронов в многоатомной системе описывается молекулярными орбиталями подобно тому, как состояние электронов в атоме характеризуется атомными орбиталями. Метод основан на следующих принципах: -молекула рассматривается как единое целое, каждый электрон принадлежит молекуле в целом и движется в поле всех ее ядер и электронов; -состояние i-го электрона описывается одноэлектронной волновой функцией Ψi, характеризуемой определенным набором квантовых чисел; -функция Ψi. называемая молекулярной орбиталью, в отличие от атомной орбитали является многоцентровой и делокализованной; -квадрат модуля волновой функции |Ψ|i 2 , как и для электрона в атоме, определяет плотность электронного облака; -полное описание состояния электрона характеризует молекулярная спин-орбиталь, выражаемая произведением МО и спиновой функции ΨS; -каждой МО соответствует определенная энергия, которая слагается из кинетической энергии электрона, потенциальной энергии притяжения электрона ко всем ядрам и усредненной потенциальной энергии отталкивания данного электрона от всех остальных электронов; -совокупность МО, называемая электронной конфигурацией молекулы, строится на основе фундаментальных положений квантовой механики: принципа наименьше энергии, принципа Паули, правила Хунда; -движение электронов взаимно независимое, и общая волновая функция основного состояния молекулы задается как произведение одноэлектронных волновых функций Ψ=П Ψ; -образование МО упрощенно рассматривается как линейная комбинация атомных орбиталей. Способ построения МО путем линейной комбинации атомной орбитали, наиболее часто используют для приближенных расчетов электронного строения и реакционной способности молекул. При сложении волновых функций двух валентных электронов атомные орбитали разных атомов (А и В), имеющих близкие значения энергии, электронная плотность между ядрами атомов увеличивается (под действием сил притяжения положительно заряженных атомных ядер). Это ведет к образованию связывающей молекулярной орбитали, имеющей более низкое значение энергии, чем исходные атомные орбитали: Ψ = а Ψ А + b Ψ В, где Ψ –симметричная волновая функция связывающей молекулярной орбитали; Ψ А и Ψ В –волновые функции атомных орбиталей атомов А и В соответственно; а и b – коэффициенты, учитывающие вклад соответствующих атомных орбиталей в образование МО, при этом а= b, если А и В – атомы одного элемента, и b>а, если электроотрицательность атома В выше. При вычитании волновых функций (имеют разные знаки) двух валентных атомных орбиталей разных атомов, имеющих близкие значения энергии, электронная плотность между ядрами атомов уменьшается , что ведет к образованию разрыхляющей молекулярной орбитали(химическая связь не образуется), имеющей более высокое значение энергии, чем исходных атомных орбиталей: Ψ * = а Ψ А - b Ψ В десь Ψ* - антисимметричная волновая функция разрыхляющей молекулярной орбитали. (стр 130). Показателем прочности молекулы может служить порядок связи , который определяется разностью чисел электронов, находящихся на связывающей молекулярной орбитали и электронов, находящихся на разрыхляющей молекулярной орбитали: ПС =(nе (СМО) – nе(РМО))/2, где nе (СМО) – число электронов на связывающих МО; nе(РМО) - число электронов на разрыхляющих МО. По существу. Порядок связи – другая форма понятия валентности. Энергетическую диаграмму напиши на обратной стороне листа стр 132 рис 4.15. Характер распределения электронов по молекулярным орбиталям позволяет объяснить магнитные свойства частиц. Молекулы, суммарный спин которых равен нулю, проявляют диамагнитные свойства, т.е. во внешнем магнитном поле их собственные магнитные моменты ориентируются против направления поля. Молекулы, суммарный спин которых отличен от нуля, проявляют парамагнитные свойства, т.е. во внешнем магнитном поле их собственные магнитные моменты ориентируются в направлении поля. Таким образом молекула Н2 диамагнитна. Заряд иона зависит от количества свободных, не вступивших в химическую связь электронов, имеющих определенный спин (суммарный спин не равен нулю), следовательно ионы диамагнитны.
8)Кристаллы. Элементарные кубические ячейки и их характеристика (наименьшее число частиц, необходимых, для построения ячейки). Координационное число, кратчайшее расстояние между частицами. Расчет эффективных радиусов атомов для расчета кубических решеток. Виды связи в кристаллах. Атомные, молекулярные, ионное и металлические кристаллы.
Для кристаллического состояния характерно строго определенное расположение частиц во всем объеме – дальний порядок. (свойства – постоянная Т плавления, анизотропность) Это обуславливает анизотропию, или векторность свойств, кристаллов – различие физических свойств, таких как теплопроводность, сжимаемость, прочность на разрыв, коэффициент преломления света – в разных направлениях. Располагаясь в кристалле определенным образом, частицы образуют крист. решетку – трехмерное упорядоченное геометрическое распределение в пр-ве точек, называемых узлами. Элементарная ячейка – это мысленно выделенная часть кристаллической решетки, включающая все элементы симметрии данного кристалла, праллельная трансляция которой по всем направлениям дает тело кристалла. (n-число частиц, требуемое для построения куба, K –координационное число (число одинаковых частиц, расположенных на кратчайшем расстоянии от данной частицы (число ближайших соседей)))Кубическая система: элементарный куб (тип NaCl), n=1, K=6, но т.к. в узлах элем. ячейки находятся ионы Na и Cl n=1/2NaCl, т.е. требуется половина молекулы для построения ячейки. Объемоцентрированный куб – тип CsCl. K=8, n=1/8*8+1=2. Так же нужно учесть, что в узлах находятся ионы Cs и Cl. В металлической связи: Гранецентрированная КР, К=8, кратчайшее расстояние между центрами атомов = половине диагонали куба, n=2(число ат, необх. для постр.), плотность упаковки ~68% K, Na, W… Гранецентрированная кубическая решетка: K=12, расстяние=половине диагонали грани, n=4, плотность упаковки ~74%, Cu, Ni g-Fe, Pb….. В соответствии с природой составляющих частиц крист. решетки м.б. ионными, атомными(ковалентными или металлическими) и молекулярными. Ионные КР построены из катионов и анионов, между кот. действуют электростатические силы притяжения. Ионы м.б. простыми, как, например в кристалле NaCl, или сложными, как в кристалле (NH4)2SO4. Строение таких кристаллов определяется соотношением радиусов ионов и принципом электронейтральности кристалла. Всл-е ненаправленного и ненасыщаемого хар-ра ионной связи, для ионных крист. хар-ны высокие Т плавления и большая твердость. Однако ИК отличаются повышенной хрупкостью, т.к. механическое воздействие приводит к нарушению правильного расположения разноименно заряженных ионов, что уменьшает Е их взаимодействия. В узлах атомно-ковалентной КР находятся атомы одинак. или разл. Э, осуществляющие направленные или ковалентные связи, кол-во которых определяется валентными возможностями атомов. В металлических КР атомы удерживаются металлической связью, которая определяет построение решетки по принципу плотной упаковки и такие хар-е св-ва металлов, как высокая тепло- и электропроводность. В узлах молекулярной КР расположены молекулы, связанные между собой слабыми межмолекулярными силами. Если основной вклад в межмол. взаим. внос. силы Ван-дер-Ваальса, крист. хар-ся высокими значениями корд. чисел (для I2, напр. К=12). Если между мол. действ. направленные водородные связи, К меньше и зависит от е строения отриц-но поляризованного атома. У льда каждая молекула тетраэдрически связана с соседними, и К=4, соответственно. Т.к. Е межмол. взаим. невелика, для в-в с молекулярной КР хар-ны низкие ТП, летучесть, невысокая твердость.
Одно и тоже вещество, в зависимости от условий (температуры, давления) может находиться в газообразном, жидком или твердом состоянии. При затвердевании веществ поступательное движение их частиц сменяется колебательным около точки, в которой была застигнута частица в момент затвердевания вещества. За исключением немногих природных веществ и произведений техники (стекло) твердые вещества имеют кристаллическое строение. Для кристаллического состояния характерно строго определенное расположение частиц во всем объеме – дальний порядок. Это определяет анизотропию или векторность свойств кристаллов – различие физических свойств: теплопроводность, сжимаемость, прочность на разрыв, коэффициент преломления света – в разных направлениях. Располагаясь в кристалле определенным образом, частицы образуют кристаллическую решетку – трехмерное упорядоченное геометрическое распределение в пространстве точек, называемых узлами. Это определяет внешнюю форму вещества в виде кристалла, ограниченного плоскими гранями, сходящимися в точечных вершинах, и прямолинейными ребрами. В соответствии с природой составляющих частиц кристаллические решетки могут быть атомными ( в узлах находятся атомы, связанные друг с другом ковалентными связями); молекулярными (в узлах находятся молекулы, связанные между собой межмолекулярными силами); ионными (в узлах располагаются, чередуясь друг с другом положительно и отрицательно заряженные ионы, они связаны друг с другом силами электростатического притяжения); металлическими ( в узлах находятся атомы металлов, между которыми свободно движутся общие для этих атомов электроны). Веществ с атомной решеткой сравнительно мало (алмаз, кремний ..), веществ с молекулярной решеткой много (к ним относятся неорганические вещества с ионной связью. Решетки различных веществ отличаются между собой не только по природе образующих частиц, но и по взаимному расположению частиц в пространстве – по своему строению. Каждую решетку можно охарактеризовать ее элементарной ячейкой - наименьшей частью кристалла, имеющей все особенности структуры данной решетки. Существует 14 типов элементарных ячеек, которые систематизированы на основе симметрии. Важнейшей характеристикой элементарной ячейки, помимо типа кристаллической решетки, являются: кратчайшее расстояние между частицами, образующими данный тип элементарной ячейки; координационное число К – число одинаковых частиц, расположенных на кратчайшем расстоянии от данной частицы (число ближайших соседей); число частиц n, необходимых для построения элементарной ячейки.
В кристалле NаСl – каждый ион окружен шестью ближайшими ионами противоположного знака, а в кристалле СsСl – восемью.
9. Термодинамические системы и их классификация. Параметры системы. Первый закон термодинамики. Понятие о термодинамической функции состояния. Внутренняя энергия и энтальпия. Применение первого закона термодинамики к изохорному, изобарному, изотермическому и адиабатному процессам. стандартные условия. Стандартная энтальпия образования вещества. Закон Гесса и следствия из него. Теплой эффект химической реакции. Термохимические расчеты. Закон Кирхгоффа
Термодинамическая
система - часть пространства, выделенная
для рассмотрения и отделенная от
окружающей среды реальной или условной
границей. Обмен с окружающей средой -
открытая система; нет обмена - закрытая.
Равновесное состояние - при к-ом ее св-ва
неизменны во времени и в ней отсутствуют
потоки в-ва или энергии.
Количество
теплоты Q, сообщенное системе, расходуется
на увеличение ее внутренней энергии DU
и на совершение работы А системой, т.е.
Q=DU+A. Для элем. проц. с б.м. изменением
параметров: δQ=dU+δA=dU+pdV+δA’, где δA’ –
сумма др. видов работ (электр., сил пов-го
натяж.). В термомеханических системах
δA’=0.
Для идеального газа: T=const: 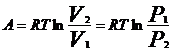 .
V=const Q=DU. P=const:
.
V=const Q=DU. P=const: ![]() ,
, ![]() ,
H=U+pV – жнтальпия, по физ. смыслу энтальпия –
энергия расширения системы.
Тепловым
эффектом хим. реакции называют
кол-во теплоты, выделяемое или поглощаемое
в результате осуществления хим. процесса
в термомеханической системе (δA’=0) при
постоянном давлении или объеме и
равенстве температур исходных веществ
и продуктов. Раздел химической
термодинамики, изучающий тепловые
эффекты реакций и фазовых превращений,
называют термохимией.
Различают ТЭ
при const V и P.
,
H=U+pV – жнтальпия, по физ. смыслу энтальпия –
энергия расширения системы.
Тепловым
эффектом хим. реакции называют
кол-во теплоты, выделяемое или поглощаемое
в результате осуществления хим. процесса
в термомеханической системе (δA’=0) при
постоянном давлении или объеме и
равенстве температур исходных веществ
и продуктов. Раздел химической
термодинамики, изучающий тепловые
эффекты реакций и фазовых превращений,
называют термохимией.
Различают ТЭ
при const V и P. ![]() ,
, ![]() .
Тепловые эффекты
.
Тепловые эффекты ![]() и
и ![]() ,
приведенные к стандартным
термод. условиям (p=101325Па=1атм=760мм
рт. ст., Т=298,15 К), называют стандартной
внутренней энергией и стандартной
энтальпией соответственно.
Стандартные
тепловые эффекты при постоянном давлении
и постоянном объеме связаны уравнением:
,
приведенные к стандартным
термод. условиям (p=101325Па=1атм=760мм
рт. ст., Т=298,15 К), называют стандартной
внутренней энергией и стандартной
энтальпией соответственно.
Стандартные
тепловые эффекты при постоянном давлении
и постоянном объеме связаны уравнением: ![]() ,
T=298 K.
Стандартной энтальпией образования
вещества (
,
T=298 K.
Стандартной энтальпией образования
вещества (![]() )
называют тепловой эффект реакции
образования 1 моль данного вещества из
соответствующего кол-ва простых веществ,
находящихся в стандартных условиях.
Стандартная энтальпия м.б. отрицательной
(получаемое в-во более термод. стабильно),
=0 (для простых в-в в стандартных
состояниях), больше нуля. При горении
в-в всегда выделяется тепло, поэтому
станд. энт. сгорания всегда отрицательна
для в-в, способных окисляться в кислороде,
или равна 0 для негорючих соед.
Закон
Гесса:
Тепловой эффект хим. реакции определяется
только видом и состоянием исходных
веществ и продуктов, но не зависит от
пути процесса.
1-е
следствие:
тепловой эффект реакции = сумме энтальпий
образования продуктов за вычетом суммы
энтальпий образования исходных веществ
с учетом стехиометрических коэффициентов.
2-е следствие:
Тепловой эффект реакции = сумме энтальпий
сгорания исходных веществ за вычетом
суммы энтальпий сгорания продуктов с
учетом стехиом. коэф.
)
называют тепловой эффект реакции
образования 1 моль данного вещества из
соответствующего кол-ва простых веществ,
находящихся в стандартных условиях.
Стандартная энтальпия м.б. отрицательной
(получаемое в-во более термод. стабильно),
=0 (для простых в-в в стандартных
состояниях), больше нуля. При горении
в-в всегда выделяется тепло, поэтому
станд. энт. сгорания всегда отрицательна
для в-в, способных окисляться в кислороде,
или равна 0 для негорючих соед.
Закон
Гесса:
Тепловой эффект хим. реакции определяется
только видом и состоянием исходных
веществ и продуктов, но не зависит от
пути процесса.
1-е
следствие:
тепловой эффект реакции = сумме энтальпий
образования продуктов за вычетом суммы
энтальпий образования исходных веществ
с учетом стехиометрических коэффициентов.
2-е следствие:
Тепловой эффект реакции = сумме энтальпий
сгорания исходных веществ за вычетом
суммы энтальпий сгорания продуктов с
учетом стехиом. коэф.
Термодинамическая система – часть пространства, выделенная для рассмотрения и отделенная от окружающей среды реальной (межфазовой) или условной границей. Системы могут быть изолированными, закрытыми (замкнутыми) и открытыми. Изолированная система характеризуется постоянством массы m, объема V, энергии U (m=соnst, V= соnst, U= соnst) она не обменивается с окружающей средой ни веществом, ни энергией. Закрытая система обменивается с окружающей средой только энергией и не обменивается веществом (m= соnst, V не соnst, Uне соnst). В открытой системе осуществляются оба указанных вида обмена с окружающей средой (m не const, V не соnst, U не соnst). Состояние системы определяется ее физическими и химическими свойствами (объем, давление, температура, химический состав, внутренняя энергия, энтальпия, энтропия и др.), которые подразделяются на параметры состояния и функции состояния. Параметры состояния– свойства системы, выбранные в качестве независимых переменных. Функция состояния – величина, определяемая этими параметрами, однозначно характеризует систему и не зависит от пути ее перехода из одного состояния в другое. (если для 1 моля идеального газа параметрами состояния выбрать давление и температуру, то функцию состояния объем можно рассчитать по ура нению состояния Менделеева-Клапейрона РV=RТ). Первый закон термодинамики вытекает из более общего закона сохранения энергии. Для термодинамической системы он формулируется следующим образом: количество теплоты Q, сообщенное системе, расходуется на увеличение ее внутренней энергии dU и на совершение работы W системой, т.е. Q = dU + W Для элементарных процессов с бесконечно малыми изменениями параметров принимает вид dQ=dU+dW= dU+рdV+dW! Где рdV –работа расширения системы; dW! – сумма других видов работ (электрической, сил поверхностного натяжения и др). Внутренняя энергия соответствует функции состояния системы, поэтому перед символом U поставлен знак полного дифференциала. Теплота и работа не являются функциями состояния, и их бесконечно малые количества обозначены буквой d. Вышеприведенные уравнения описывают первый закон термодинамики в интегральной и дифференциальной формах соответственно. В термомеханических системах при протекании процессов совершается только работа расширения или сжатия, т.е. dW!=0. и dQ= dU+рdV Согласно этому закону внутренняя энергия является однозначной функцией состояния вещества (или совокупности вещества) и зависит только от параметров состояния, тогда как по отдельности каждая из величин, определяющих внутреннюю энергию (теплота Q, работа W) зависит от пути процесса, переводящего реагенты в продукты. Другой функцией состояния системы является энтальпия – тепловой эффект реакции при постоянном давлении (dН). Теплота Q, выделившаяся или поглощенная в химической реакции, называется тепловым эффектом реакции. Его можно измерить в специальных приборах – калориметрах. Изотермический процесс (Т=соnst) в идеальном газе силы межмолекулярного взаимодействия равны нулю. Внутренняя энергия идеального газа зависит от температуры, количества вещества и не зависит от давления и объема, поэтому для данных условий U=соnst; dU=0 dQТ= dW = рdV ; QТ= W теплота, сообщенная системе, в изотермическом процессе полностью расходуется на совершение работы расширения. используя р=RТ/V и проинтегрировав получим W= RТln(V/V1 = RТln(р/р1), т.к. по закону Бойля-Мариотта (р V)Т = соnst Изохорный процесс (V= соnst) при постоянном объеме dV=0, значит работа расширения газа dW= рdV=0 и dQ V = dU или Q = U2- U1 =dd U В изохорном процессе теплота, сообщенная системе, полностью расходуется на увеличение ее внутренней энергии и характеризует изменение состояния системы. Изобарный процесс (р= соnst) постоянную величину р можно внести под знак дифференциала, поэтому работа расширения dW = рdV =dW = d(рV) и dQр=dU+ d(рV) = d(U +рV)= ?Н Величины рV и U характеризуют состояние системы. Их сума Н=U+рV также соответствует функции состояния, которую называют энтальпией. По физическому смыслу энтальпия есть энергия расширения системы. В изобарном процессе теплота, сообщенная системе расходуется на увеличение внутренней энергии и н совершение работы расширения против сил внешнего давления и характеризует изменение состояния системы. Сложные вещества можно условно или реально синтезировать из соответствующего количества простых веществ в стандартных термодинамических условиях. За стандартное состояние твердого вещества при Т=2980К принимают его чистый кристалл под давлением р=101,3кПа. За стандартное состояние жидкого вещества при при Т=2980К принимают чистую жидкость под давлением р=101,3кПа. Для газообразного при этой же температуре вещеста стандартным является состояние условного идеального газа, имеющего летучесть f=101,3 кПа и свойства бесконечно разреженного газа. Стандартной энтальпией образования вещества (df Н0298) называют тепловой эффект реакции образования 1 моль данного вещества из соответствующего количества простых веществ, находящихся в стандартных условиях. Русский ученый Г.И.Гесс в 1840г экспериментально установил основной закон термохимии: тепловой эффект химической реакции определяется только видом и состоянием исходных веществ и продуктов. Но не зависит от пути процесса. Первое следствие: тепловой эффект реакции равен сумме энтальпий образования продуктов за вычетом суммы энтальпий образования исходных веществ с учетом стехиометрических коэффициентов. drН298О =E v'i dfН' 298i О -Evi dfН 298 i О i i Второе следствие: тепловой эффект реакции равен сумме энтальпий сгорания исходных веществ за вычетом суммы энтальпий сгорания продуктов с учетом стехиометрических коэффициентов. drН298О = Ev'i dсН' 298i О -Evi dсН 298 i О i i
10. Второй закон термодинамики. Функция состояния -энтропия. Расчет изменения энтропии при изобарном и изохорном процессах, при изотермическом расширении идеального газа, при смешении идеальных газов.
Постулат
Клаузиуса:
теплота не может самопроизвольно
переходить от менее нагретого тела к
более нагретому. В применении к
элементарному обратимому процессу : ![]() ,
где числитель – алгебраическая сумма
теплоты, полученной и отданной рабочим
телом (идеальным газом) при совершении
процесса, Дж, Т – абсолютная т-ра.
,
где числитель – алгебраическая сумма
теплоты, полученной и отданной рабочим
телом (идеальным газом) при совершении
процесса, Дж, Т – абсолютная т-ра. ![]() -
приведенная теплота. Энтропия есть
функция состояния, изменение которой
= алгебраической сумме приведенной
теплоты всех элементов обратимого
процесса. Т.к. А, совершаемая сист. в
необр проц. < раб., сов. в обр проц:
-
приведенная теплота. Энтропия есть
функция состояния, изменение которой
= алгебраической сумме приведенной
теплоты всех элементов обратимого
процесса. Т.к. А, совершаемая сист. в
необр проц. < раб., сов. в обр проц: ![]() .
В изолированной системе знак изменения
энтропии является критерием направленности
самопроизвольного процесса: если DS=0,
т.е. S достигла своего макс. значения, то
система находится в состоянии термод.
равновесия. Если DS>0, то процесс
самопроизвольно протекает в прямом
направлении, если DS<0, то самопр.
протекать может лишь обратный
процесс.
Термодинамической
вероятностью w называют
число микросостояний, через которое
можно реализовать данное макросостояние
системы.
.
В изолированной системе знак изменения
энтропии является критерием направленности
самопроизвольного процесса: если DS=0,
т.е. S достигла своего макс. значения, то
система находится в состоянии термод.
равновесия. Если DS>0, то процесс
самопроизвольно протекает в прямом
направлении, если DS<0, то самопр.
протекать может лишь обратный
процесс.
Термодинамической
вероятностью w называют
число микросостояний, через которое
можно реализовать данное макросостояние
системы.
![]() ,
где kб- константа Больцмана. По
Больцману:
энтропия – функция термодинамической
вероятности того или иного состояния
индивидуального вещества или
системы. Согласно
этому Ур-ю, второе начало термод. можно
сформулировать так:
всякая
изолированная система самопроизвольно
стремится принять состояние,
характеризующееся максимальной
термодинамической вероятностью.
,
где kб- константа Больцмана. По
Больцману:
энтропия – функция термодинамической
вероятности того или иного состояния
индивидуального вещества или
системы. Согласно
этому Ур-ю, второе начало термод. можно
сформулировать так:
всякая
изолированная система самопроизвольно
стремится принять состояние,
характеризующееся максимальной
термодинамической вероятностью.
![]() ,
, ![]() .
Эти Ур-ия можно рассматривать в процессах
изохорного и изобарного нагрева и охл.
конденсированных веществ.
.
Эти Ур-ия можно рассматривать в процессах
изохорного и изобарного нагрева и охл.
конденсированных веществ. ![]() .
.


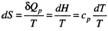
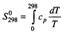
Вопрос о направлении и условиях самопроизвольного протекания процесса решается в рамках второго закона термодинамики. Одна из первых формулировок второго начала термодинамики – постулат Клаузиуса гласит: теплота не может самопроизвольно переходить от менее нагретого тела к более нагретому. Рассмотрев работу тепловой машины, Клаузиус ввел новую, не известную ранее функцию состояния системы, названную энтропией, для полного дифференциала которой в применении к элементарному обратимому процессу получил выражение dS = dQобр Т Где dQобр – алгебраическая сумма теплоты, полученной и отданной рабочим телом (идеальным газом) при совершении процесса, Дж; Т- абсолютная температура, К. По Клаузиусу энтропия есть функция состояния, изменение которой равно алгебраической сумме приведенной теплоты всех элементов обратимого процесса. В изолированной системе знак изменения энтропии является критерием направленности самопроизвольного процесса: - если dS =0 (S= Smax, энтропия достигла своего максимального значения), то система находится в состоянии термодинамического равновесия; -если dS> 0 (Sa Smax, энтропия возрастает), то процесс самопроизвольно протекает в прямом направлении, т.е. термодинамически возможен; - если dS< 0 (Sa Smin, энтропия убывает), то самопроизвольно протекать может лишь обратный процесс, связанный с увеличением энтропии, прямой процесс термодинамически невозможен. Расчет изменения энтропии при изобарном и изохорном процессах, при изотермическом расширении идеального газа, при смешении идеальных газов. Стр 196-197 А.А. Гуров. Термодинамической вероятностью w называют число микросостояний, через которые можно реализовать данное макросостояние системы. В отличие от математической вероятности, равной или меньшей единице, термодинамическая вероятность представляет собой очень большую величину6 значение w имеет порядок 104, для системы, состоящей из десятка частиц, в то время, как й моль вещества содержит 6,02 ×1023 структурных единиц. Связь энтропии с термодинамической вероятностью установи Больцман: S=kБ ln w уравнение названо его именем., где kБ – константа Больцмана (универсальная газовая постоянная, рассчитанная на одну молекулу) = 1,38×10-23 Дж\К. Таким образом по Больцману энтропия есть функция термодинамической вероятности того или иного состояния индивидуального вещества или системы. Согласно уравнению Больцмана второе начало термодинамики можно сформулировать: всякая изолированная система самопроизвольно стремится принять состояние, характеризующееся максимальной термодинамической вероятностью. Третий закон термодинамики. Абсолютная энтропия веществ. Расчет изменения энтропии реакции. Зависимость энтропии от температуры. Абсолютные значения внутренней энергии и энтальпии невозможно вычислить даже для простых систем. Абсолютные значения энтропии рассчитываются как для простых, так и для сложных веществ, поскольку у них имеется начальная точка в шкале отсчета, устанавливаемаятретьим законом термодинамики. Третье начало термодинамики формулируется следующим образом: при температуре абсолютного нуля (Т = 0К) энтропия идеального кристалла любого простого или сложного вещества равна нулю Действительно, в бездефектном кристалле вещества существует абсолютный порядок в расположении частиц. При абсолютном нуле возможно единственное состояние системы, при котором частицы «застывают» в узлах кристаллической решетки. У неидеальных кристаллов, смесей, твердых растворов, стеклообразных структур всегда имеется нулевая энтропия (Sо>0), связанная с дефектами кристаллической решетки, возможностью различной ориентации частиц в пространстве и другими причинами. В 1911 г немецкий физик Планк определил, что значения энтропии вещества являются абсолютными, поскольку статистически при термодинамической температуре, стремящейся к нулю, энтропия всех веществ, становящихся идеальными кристаллами, обращается в ноль. Стандартную энтропию вещества обозначают символом SТО (р=101,3 кПа), ее можно определить при любом значении температуры, однако для удобства сравнения величин SТО для различных веществ их определяют обычно при стандартных термодинамических условиях (р=101,1кПа, Т=198 К) и обозначают S298О (Дж/моль К). Стандартная энтропия любого вещества всегда положительная величина (S298О>0). Стандартные энтропии образования ионов в водных растворах (df S298О(иона)) определяются для процессов образования и гидратации ионов, они могут быть как положительными, так и отрицательными. Изменения энтропии (dS) в процессах могут положительными, отрицательными и равными нулю. Энтропия представляет собой функцию состояния системы, поэтому стандартное изменение энтропии в результате осуществления химической реакции (стандартную энтропию реакции drS298О) можно вычислить как разность между суммой стандартных энтропий продуктов реакции и суммой стандартных энтропий исходных веществ с учетом стехиометрии процесса drS298О = Sn' i S' 298 О -Sni S298 О i i Энтропия вещества тем выше, чем меньше упорядоченность частиц этого вещества. Вблизи абсолютного нуля все вещества находятся в твердом состоянии. Если при стандартных термодинамических условиях вещество находится в кристаллическом состоянии, то в интервале температур от 0 до 298К энтропия изменяется только за счет повышения температуры, значит при постоянном давлении dS = dQр\Т = dН\Т = Ср dТ\Т.
11. Термодинамические функции состояния: энергия Гиббса, энергия Гельмгольца. Критерий самопроизвольного протекания химических процессов в закрытых системах. Методы расчета изменения энергии Гиббса. Реакции при стандартных условиях.
Реальные
процессы проводятся, как правило, в
закрытых системах в изобарно-изотермических
или изохорно-изотермических условиях.
Критерием направленности самопроизвольного
процесса в этих случаях является знак
изменения энергии
Гиббса DG или
энергии Гельмгольца DА в системе.
G=H-TS=U+pV-TS,
A=U-TS. TS хар-ет связанную с частицами
системы энергию, т.е. ту часть полн. Е
сист., кот. рассеив. в окр. среде в виде
теплоты (потерянная работа). \энергия
Гиббса (Гельмг.) – та часть полн. Е
системы, кот. м.б. превр. в работу в
изоб-изот Илии изох-изот проц. Е G и A –
функции состояния системы, их абс.
значения не поддаются вычислению. Для
выяснения критерия направленности
самопр. проц. в закр. сист: dG=dU+dpV+dVp-TdS-dTS,
в силу TdS>=dU+pdV => dG<=-SdT+VdP. Для
изобарно-изотермического: dG<=0, ![]() .
В закрытой системе знак изменения
энергии Гиббса является критерием
направленности самопроизвольного
процесса при проведении его в изоб.-изот.
условиях. При DG=0 система нах. в сост.
термод. равн.
Изохорно-изот. проц. в
закр. сист. терм-ки возможен при DА<0,
невозм, при DА>0, система нах. в термод.
равн. при =0.
При const T, p=101,3 кПа:
.
В закрытой системе знак изменения
энергии Гиббса является критерием
направленности самопроизвольного
процесса при проведении его в изоб.-изот.
условиях. При DG=0 система нах. в сост.
термод. равн.
Изохорно-изот. проц. в
закр. сист. терм-ки возможен при DА<0,
невозм, при DА>0, система нах. в термод.
равн. при =0.
При const T, p=101,3 кПа: ![]() -
Ур-ие Гиббса-Гельмгольца. При равенстве
стандартной Е Гиббса нулю в системе
устанавливается подвижное ТР, положение
которого может смещаться в любую сторону
при изменении внешних факторов. DG при
298 К можно рассчитать по стандартным Е
Гиббса обр-я исходных веществ и продуктов
реакции
-
Ур-ие Гиббса-Гельмгольца. При равенстве
стандартной Е Гиббса нулю в системе
устанавливается подвижное ТР, положение
которого может смещаться в любую сторону
при изменении внешних факторов. DG при
298 К можно рассчитать по стандартным Е
Гиббса обр-я исходных веществ и продуктов
реакции ![]() .
Стандартной энергией Гиббса образования
в-ва называют ст. Е Гиббса р-ции обр-я 1
моль данного соединения из простых в-в,
находящихся в термод-ки устойчивых
модификациях,кто провед. в станд. термод
. услов.
Химический
потенциал:
для открытой системы G=f(p, T, ni), ni- кол-во
в-ва i-го компонента, моль. Масса системы
при этом const, но ее состав изменяется.
Полный дифференциал :
.
Стандартной энергией Гиббса образования
в-ва называют ст. Е Гиббса р-ции обр-я 1
моль данного соединения из простых в-в,
находящихся в термод-ки устойчивых
модификациях,кто провед. в станд. термод
. услов.
Химический
потенциал:
для открытой системы G=f(p, T, ni), ni- кол-во
в-ва i-го компонента, моль. Масса системы
при этом const, но ее состав изменяется.
Полный дифференциал : 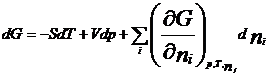 .
Хим. потенциал i-го компонента:
.
Хим. потенциал i-го компонента: 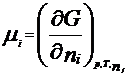 .
По физ. смыслу : ХП означает изм. G сист.
в PT процессе при доб. 1 моль i-го комп. к
беск. большому кол-ву смеси, чтобы ее
состав был const (nj=const). Для ид.
р-ров:
.
По физ. смыслу : ХП означает изм. G сист.
в PT процессе при доб. 1 моль i-го комп. к
беск. большому кол-ву смеси, чтобы ее
состав был const (nj=const). Для ид.
р-ров: ![]() ,
, ![]() .
. ![]() -стандартный
хим. потенциал i-го комп. (при отн-ном
давлении
-стандартный
хим. потенциал i-го комп. (при отн-ном
давлении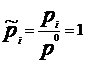 ),
ХЭ и-ое – мольная доля I компонента.
Второе уравнение справедливо для любых
ид-х р-ров, первое – для газовых. Для
индивид в-ва станд. хим. пот = станд. Е
Гиббса обр-я в-ва. При проведении реакции
в PT условиях:
),
ХЭ и-ое – мольная доля I компонента.
Второе уравнение справедливо для любых
ид-х р-ров, первое – для газовых. Для
индивид в-ва станд. хим. пот = станд. Е
Гиббса обр-я в-ва. При проведении реакции
в PT условиях: ![]() .
Хим. переменная:
.
Хим. переменная: 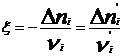 .
(глубина, степень протекания реакции,
число пробегов).
.
(глубина, степень протекания реакции,
число пробегов). ![]() .
Получим:
.
Получим: ![]() .
– общие условия хим. равновесия и самопр.
протек. реакции, идущей в закр. системе,
при p, T=const. Реальные процессы проводятся,
как правило, в закрытых системах в
изобарно-изотермических (р,Т=соnst)
или
изохорно-изотермических (V, Т= соnst)
условиях. Критерием направленности
самопроизвольного процесса в этих
случаях является знак изменения энергии
Гиббса dG или энергии Гельмгольца dА в
системе.
Энергия
Гиббса G
= Н –ТS = U + рV –ТS.
Энергия
Гельмгольца А
= U – ТS,
при этом G
= f(р,Т); А = f(V,Т).
Уравнения также можно
представить в виде: Н
= G +ТS; U = А
+ ТS.
Где величина ТS характеризует связанную
с частицами системы энергию, т.е. ту
часть полной энергии системы, которая
рассеивается в окружающей среде в виде
теплоты (так называемая потерянная
работа).
Энергия
Гиббса (или энергия Гельмгольца)
характеризуют ту часть полной энергии
системы, которая может быть превращена
в работу в изобарно-изотермическом (или
в изохорно-изотермическом) процессе
(так называемая полезная работа,
совершаемая системой). Энергия Гиббса
и энергия Гельмгольца являются функциями
состояния системы, их абсолютные значения
не поддаются вычислению.
В
закрытой системе знак изменения энергии
Гиббса является критерием направленности
самопроизвольного процесса при
проведении его в изобарно-изотермических
условиях:
-
при dG = 0 (G=Gmin , энергия Гиббса имеет
минимальное значение) система находится
в состоянии термодинамического
равновесия;
- при dG < 0 (Ga Gmin , энергия
Гиббса убывает) процесс самопроизвольно
протекает в прямом направлении, т.е.
термодинамически возможен;
-
при dG > 0 (Ga Gmin, энергия Гиббса возрастает)
самопроизвольно протекает только
обратный процесс, прямой процесс
термодинамически невозможен.
Стандартную
энергию Гиббса химической реакции при
Т = 298К можно рассчитать двумя способами:
используя уравнение Гиббса-Гельмгольца:
drGТ0 = dr НТ0 - ТdrSТ0, в которое нужно подставить
указанное значение Т, и по стандартным
энергиям Гиббса образования исходных
веществ и продуктов реакции
dfG2980.
Стандартную энтальпию реакции
dr Н2980 находят по первому
drН298О = Sn'i
dfН' 298i О -Sni dfН 298 i О
i
i
или
второму следствию из закона Гесса
drН298О
= Sn'i dсН' 298i О -Sni dсН 298 i О
i
i
а стандартную энтропию реакции
drS298О
= Sn' i S' 298 О -Sni S298 О.
i
i
При
расчете необходимо учитывать, что
единицей измерения энтальпии является
килоджоуль, а энтропии Дж\К.
Энергия
Гиббса является функцией состояния
системы, поэтому ее можно рассчитать с
использованием табличных значений
стандартных энергий Гиббса образования
исходных веществ и продуктов
реакции
drG298О
= Sn'i dfG' 298i О -Sni dfG 298 i О
i
i
Стандартной
энергией Гиббса образования
вещества dfG298 О называют
стандартную энергию Гиббса реакции
образования 1 моль данного соединения
из простых веществ, находящихся в
термодинамически устойчивых модификациях,
которая проведена в стандартных
термодинамических условиях.
В
отсутствие справочных данных по dfG298 О
какого-либо сложного вещества эту
величину можно вычислить по уравнению
dfG298О = dfН298 О - 298 df S 298 О, где dfН298 О и
df S 298 О – стандартные энтальпия и энтропия
соответственно реакции образования 1
моль этого соединения из простых веществ,
находящихся в термодинамически устойчивых
состояниях, проведенной при стандартных
термодинамических условиях.
.
– общие условия хим. равновесия и самопр.
протек. реакции, идущей в закр. системе,
при p, T=const. Реальные процессы проводятся,
как правило, в закрытых системах в
изобарно-изотермических (р,Т=соnst)
или
изохорно-изотермических (V, Т= соnst)
условиях. Критерием направленности
самопроизвольного процесса в этих
случаях является знак изменения энергии
Гиббса dG или энергии Гельмгольца dА в
системе.
Энергия
Гиббса G
= Н –ТS = U + рV –ТS.
Энергия
Гельмгольца А
= U – ТS,
при этом G
= f(р,Т); А = f(V,Т).
Уравнения также можно
представить в виде: Н
= G +ТS; U = А
+ ТS.
Где величина ТS характеризует связанную
с частицами системы энергию, т.е. ту
часть полной энергии системы, которая
рассеивается в окружающей среде в виде
теплоты (так называемая потерянная
работа).
Энергия
Гиббса (или энергия Гельмгольца)
характеризуют ту часть полной энергии
системы, которая может быть превращена
в работу в изобарно-изотермическом (или
в изохорно-изотермическом) процессе
(так называемая полезная работа,
совершаемая системой). Энергия Гиббса
и энергия Гельмгольца являются функциями
состояния системы, их абсолютные значения
не поддаются вычислению.
В
закрытой системе знак изменения энергии
Гиббса является критерием направленности
самопроизвольного процесса при
проведении его в изобарно-изотермических
условиях:
-
при dG = 0 (G=Gmin , энергия Гиббса имеет
минимальное значение) система находится
в состоянии термодинамического
равновесия;
- при dG < 0 (Ga Gmin , энергия
Гиббса убывает) процесс самопроизвольно
протекает в прямом направлении, т.е.
термодинамически возможен;
-
при dG > 0 (Ga Gmin, энергия Гиббса возрастает)
самопроизвольно протекает только
обратный процесс, прямой процесс
термодинамически невозможен.
Стандартную
энергию Гиббса химической реакции при
Т = 298К можно рассчитать двумя способами:
используя уравнение Гиббса-Гельмгольца:
drGТ0 = dr НТ0 - ТdrSТ0, в которое нужно подставить
указанное значение Т, и по стандартным
энергиям Гиббса образования исходных
веществ и продуктов реакции
dfG2980.
Стандартную энтальпию реакции
dr Н2980 находят по первому
drН298О = Sn'i
dfН' 298i О -Sni dfН 298 i О
i
i
или
второму следствию из закона Гесса
drН298О
= Sn'i dсН' 298i О -Sni dсН 298 i О
i
i
а стандартную энтропию реакции
drS298О
= Sn' i S' 298 О -Sni S298 О.
i
i
При
расчете необходимо учитывать, что
единицей измерения энтальпии является
килоджоуль, а энтропии Дж\К.
Энергия
Гиббса является функцией состояния
системы, поэтому ее можно рассчитать с
использованием табличных значений
стандартных энергий Гиббса образования
исходных веществ и продуктов
реакции
drG298О
= Sn'i dfG' 298i О -Sni dfG 298 i О
i
i
Стандартной
энергией Гиббса образования
вещества dfG298 О называют
стандартную энергию Гиббса реакции
образования 1 моль данного соединения
из простых веществ, находящихся в
термодинамически устойчивых модификациях,
которая проведена в стандартных
термодинамических условиях.
В
отсутствие справочных данных по dfG298 О
какого-либо сложного вещества эту
величину можно вычислить по уравнению
dfG298О = dfН298 О - 298 df S 298 О, где dfН298 О и
df S 298 О – стандартные энтальпия и энтропия
соответственно реакции образования 1
моль этого соединения из простых веществ,
находящихся в термодинамически устойчивых
состояниях, проведенной при стандартных
термодинамических условиях.
12. Химическое равновесие в гомогенных закрытых системах. Константы равновесия, связь между ними и способы расчета. Параметры, влияющие на состояние равновесия. Принцип Ле-Шателье. Применение уравнения изобары, изохоры и изотермы химической реакции для анализа смещения равновесия.
Под ХИМИЧЕСКИМ
РАВНОВЕСИЕМ понимают
достигаемое с 2х противоположных сторон
и неизменное во времени при постоянных
давлении, объеме и температуре состояние
системы, сод-ей в-ва, способные к хим.
взаимодействию. Различают истинное
(достигается только в закрытых системах,
хар-ся термод. устойчивостью, подвижностью,
обе реакции, прямая и обр-я, в зависимости
от условий могут протекать самопроизвольно,
динамический характер – непр-е протекание
с одинак. скор как прямой, так и обр.
р-ции) и замороженное ХР(лишь по 1-му
признаку тождественно истинному).
Гомогенные
– р-ции, протекающие в одной фазе –
газовой или жидкой. Хар-ся отсутствием
пов-ти раздела между реаг. и прод. Все
уч. нах. в 1 агрег. сост. и сотавл. 1
фазу.
Количественно ХР хар-ют:
равновесным составом р-ой смеси;
равновесной степенью превр(конверсии)
реаг; равновесным выходом продуктов;
константой хим. равновесия.
![]() .
Активностью (фугитивностью) называют
величину, при посдтановке кот. вместо
конц.(парц. давлю) в выр-ия, вывед-е для
ид. сист., можно примен. их к идеальным
системам. Равновесный выход продукта:
например, для в-ва D (1-го из прод.)
.
Активностью (фугитивностью) называют
величину, при посдтановке кот. вместо
конц.(парц. давлю) в выр-ия, вывед-е для
ид. сист., можно примен. их к идеальным
системам. Равновесный выход продукта:
например, для в-ва D (1-го из прод.) ![]() .
Для реагентов:
.
Для реагентов: ![]() .
Константа
равновесия –
позволяет судить о полноте протекания
реакции при тех или иных условиях. Кравн
есть величина постоянная для данной
хим. р-ции при данных Т, давлении и в
данном растворителе = отношению
произведения равновесных активностей(или
фугитивностей для газов) прод-ов реакции,
взятых в степенях = их стехиом. коэф., к
анал. произвед. для исх. в-в в состоянии
хим. равн. – одна из формулировок закона
действующих масс Гульдберга-Вааге для
хим. равновесия.
Для гомог. хим.
равновесий, устанавливающихся в ид.
жидких и газообразных растворах, Кравн
можно выразить и через равновесные
молярные концентрации и равновесные
молярные доли, а для равновесий в газовых
смесях – равновесные парциальные
давления:
.
Константа
равновесия –
позволяет судить о полноте протекания
реакции при тех или иных условиях. Кравн
есть величина постоянная для данной
хим. р-ции при данных Т, давлении и в
данном растворителе = отношению
произведения равновесных активностей(или
фугитивностей для газов) прод-ов реакции,
взятых в степенях = их стехиом. коэф., к
анал. произвед. для исх. в-в в состоянии
хим. равн. – одна из формулировок закона
действующих масс Гульдберга-Вааге для
хим. равновесия.
Для гомог. хим.
равновесий, устанавливающихся в ид.
жидких и газообразных растворах, Кравн
можно выразить и через равновесные
молярные концентрации и равновесные
молярные доли, а для равновесий в газовых
смесях – равновесные парциальные
давления:
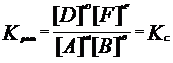 ,
, 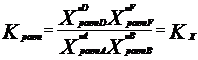 ,
,  Взаимосвязь
между эмпирическими константами:
Взаимосвязь
между эмпирическими константами: ![]() .
Vм-мольный объем смеси. Выводы: если
газоф. р. протек. без измен. числа молей
газообр. в-в участников, Dn=0, то все
константы не зависят от общего (суммарного)
давления в системе, безразмерны и равны.
Размерность констант зависит от формы
записис стехиом. Ур-ия. (и числ. значения
так же разнятся). Для газа активность =
фугитивности.
Если Кравн>1, то полож.
равновесия смещено вправо (преобл.
продукты) и наоборот.
.
Vм-мольный объем смеси. Выводы: если
газоф. р. протек. без измен. числа молей
газообр. в-в участников, Dn=0, то все
константы не зависят от общего (суммарного)
давления в системе, безразмерны и равны.
Размерность констант зависит от формы
записис стехиом. Ур-ия. (и числ. значения
так же разнятся). Для газа активность =
фугитивности.
Если Кравн>1, то полож.
равновесия смещено вправо (преобл.
продукты) и наоборот.
![]() .
В
любой изолированной системе в состоянии
равновесия ее энтропия максимальна и
постоянна. Стандартная константа
равновесия:
.
В
любой изолированной системе в состоянии
равновесия ее энтропия максимальна и
постоянна. Стандартная константа
равновесия: ![]() .
Связь
между константами равновесия и термод.
хар-ми сист. устанавливается уравнениями
изотермы, изобары и изохоры Х
р-ции.
.
Связь
между константами равновесия и термод.
хар-ми сист. устанавливается уравнениями
изотермы, изобары и изохоры Х
р-ции.
уравнение
изотермы в
общем виде: 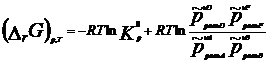 .
Оно имеет неск. разновидностей, для
р-ций, протек. в ид. р-рах, в него вход.
константа равновесия K0C (или Kx) и
относительные неравновесные молярные
концентрации C(~)i.(или неравн. молярн.
доли Xi) компонентов.
Стандартное
уравнение изотермы хим. р-ции или
уравнение стандартного сродства.
Уравнение
изобары хим. р. определяет зависимость
константы равновесия Kr0 от Т и в диффер.
форме выводится на основании стандартного
уравнения изотермы и дифф-го уравнения
Гиббса-Гельмгольца для станд. условий
.
Оно имеет неск. разновидностей, для
р-ций, протек. в ид. р-рах, в него вход.
константа равновесия K0C (или Kx) и
относительные неравновесные молярные
концентрации C(~)i.(или неравн. молярн.
доли Xi) компонентов.
Стандартное
уравнение изотермы хим. р-ции или
уравнение стандартного сродства.
Уравнение
изобары хим. р. определяет зависимость
константы равновесия Kr0 от Т и в диффер.
форме выводится на основании стандартного
уравнения изотермы и дифф-го уравнения
Гиббса-Гельмгольца для станд. условий ![]() ,
и получаем искомое Ур-ие
изобары
,
и получаем искомое Ур-ие
изобары ![]() ,
или – внести Т под знак диф-ла (для
удобства графического построения).
Уравнение изобары в инт. форме –
проинтегрировать исходное в узком инт.
т-р (где можно считать энтальпию
постоянной).
Если газофазная р-ция
осущ-ся в закр. сист при пост. объеме,
то: ан-но выводу для
изобары (только
тут энергия Гельмгольца и внутр. Е и
Кс):
,
или – внести Т под знак диф-ла (для
удобства графического построения).
Уравнение изобары в инт. форме –
проинтегрировать исходное в узком инт.
т-р (где можно считать энтальпию
постоянной).
Если газофазная р-ция
осущ-ся в закр. сист при пост. объеме,
то: ан-но выводу для
изобары (только
тут энергия Гельмгольца и внутр. Е и
Кс): ![]() ,
в интегральной форме (проинтегрировать).
Факторы,
влияющие на хим. равновесие:
1.
Введение катализатора и изменение
концентрации компонентов. Катализатор
не влияет на Кравн, поэтому не вызывает
его смещения. Ускоряет в равн. степ. как
прямую, так и обратную реакцию. Изменение
сост-я равн. в рез. изм. внешн. условий
наз. смещением или сдвигом положения
равновесия. Чаще всего прих. сталкиваться
с смещ. пол.равн. в следствие а)изменение
конц. или парц. давлений 1 или сразу неск.
в-в, участников р-ции, б)изменения Т
системы, в) изменения общего давления
в системе, г) за счет введения в газообр.
равновесн. сист. инертного газа, т.е.
разбавления ее.
Принцип
Ле Шателье-Брауна:
если на систему, находящуюся в состоянии
истинного хим. равновесия, оказывать
внешнее воздействие путем изменения
какого-либо из условий (Ci, T, pi, pобщ),
определяющих положение равновесия, то
в сист. происх. изменение равновесного
состава и смещение положения равновесия
в направлении того процесса, протекание
которого ослабляет эффект (влияние)
этого воздействия.
,
в интегральной форме (проинтегрировать).
Факторы,
влияющие на хим. равновесие:
1.
Введение катализатора и изменение
концентрации компонентов. Катализатор
не влияет на Кравн, поэтому не вызывает
его смещения. Ускоряет в равн. степ. как
прямую, так и обратную реакцию. Изменение
сост-я равн. в рез. изм. внешн. условий
наз. смещением или сдвигом положения
равновесия. Чаще всего прих. сталкиваться
с смещ. пол.равн. в следствие а)изменение
конц. или парц. давлений 1 или сразу неск.
в-в, участников р-ции, б)изменения Т
системы, в) изменения общего давления
в системе, г) за счет введения в газообр.
равновесн. сист. инертного газа, т.е.
разбавления ее.
Принцип
Ле Шателье-Брауна:
если на систему, находящуюся в состоянии
истинного хим. равновесия, оказывать
внешнее воздействие путем изменения
какого-либо из условий (Ci, T, pi, pобщ),
определяющих положение равновесия, то
в сист. происх. изменение равновесного
состава и смещение положения равновесия
в направлении того процесса, протекание
которого ослабляет эффект (влияние)
этого воздействия.
Химические реакции заключаются во взаимодействии реагентов с образованием продуктов. Не следует, однако полагать, что направление химической реакции только одно. В действительности, химические реакции протекают и в прямом, и в обратном направлениях. Реагенты n Продукты. Такие реакции называются обратимыми. Обратимые (и необратимые) химические реакции бывают как гомогенными, так и гетерогенными. Гомогенными называют реакции, протекающие в одной фазе – газовой или жидкой. Они характеризуются отсутствием поверхности раздела между реагентами и продуктами, взаимодействие которых осуществляется во всем объеме реакционной смеси. У гомогенных обратимых реакций все участники находятся в одном агрегатном состоянии и составляют одну фазу, например: Реакция между газообразными веществами СН4(г) + СО2(г) n 2СО(г) + 2Н2(г) Реакция этирификации, протекающая в водной среде. Равновесие, имеющее место у таких реакций, называется гомогенным. Обратимые гомогенные и гетерогенные реакции в отличие от необратимых идут не конца, т.е. не до полного исчезновения реагентов: они «прекращаются» прежде, чем будут полностью израсходованы их исходные вещества (если они были взяты в стехиометрических соотношениях), поэтому в реакционной смеси у таких реакций всегда присутствуют и исходные вещества и продукты их взаимодействия. максимальный выход продуктов у них менее 100% и соответствует равновесному составу. Такие реакции протекают до установления в них определенного концентрационного предела, общего для их прямого и обратного направлений, называемого состоянием химического равновесия. Именно с наступлением последнего и связывают прекращение протекания реакции в целом. Равновесие в системе наступает в результате стремления ее к минимальному значению энергии и максимальному хаотическому распределению в пространстве, характеризующемуся энтропией. Достигнутое и неизменное при состоянии химического равновесия соотношение достаточно больших, отчетливо определяемых аналитически концентраций всех веществ называют положением химического равновесия. Одна из важнейших количественных характеристик химического равновесия – константа равновесия, позволяющая судить о полноте протекания реакции при тех или иных условиях. В общем случае применительно к любым химическим системам, как идеальным, так и реальным, константа равновесия Кравн обратимой химической реакции есть величина постоянная при данных температуре, давлении и в данном растворителе. Для гомогенных химических равновесий, устанавливающихся в идеальных жидких и газообразных (газовых смесях) растворах, константу равновесия можно выразить через равновесные молярные концентрации и равновесные молярные доли, а для равновесий в газовых смесях – через равновесные парциальные давления: (для реакции nАА(Г) + nВВ(Г) n nD D(Г) + n F F(Г) Кравн = [D] nD [F] n F = КС - моль/м3 [А] nА [В] n В Кравн =Хравн DnD Хравн F n F = КХ – безразмерная величина ХравнА nА Хравн В n В Кравн = Рравн DnD Рравн F n F = КР – Па (атм) РравнА nА Рравн В n В Коэффициенты активности и фугитивности компонентов таких систем равны единице, поэтому активности становятся равными концентрациям, а фугитивности – давлениям. Активностью (фугитивностью) называют величину, при подстановке которой вместо концентрации (парциального давления) в выражения, выведенные для идеальных систем, можно применить их к реальным системам. Константы КС, КХ, КР иногда называют концентрационными или эмпирическими константами равновесия, поскольку для их расчета используются экспериментально определяемые значения равновесной молярной концентрации (в квадратных скобках), парциальные давления компонентов рравн i , равновесные молярные доли Хравн i . Кр = Кс (RТ)rnг ; Кр = Кх робщrnг ; Кс = Кх Vм-rnг где Vм мольный объем смеси (раствора); rnг = (nD + n F ) – (nА + n В) – изменение числа молей газообразных веществ, рассчитываемое по разности между суммами стехиометрических коэффициентов продуктов и реагентов. (стр 331 -333 А.А.Гуров) Связь между константами равновесия и термодинамическими характеристиками системы устанавливается уравнениями изотермы, изобары и изохоры химической реакции. Эти уравнения впервые были выведены Вант-Гоффом (1886г), поэтому в литературе их часто называют его именем, а именно: изотерма, изобара и изохора Вант-Гоффа. Каждое из них имеет несколько разновидностей и используется либо в дифференциальной, либо в интегральной формах. Уравнение изотермы в общем виде связывает энергию Гиббса, константу равновесия и начальные (или текущие), т.е. неравновесные концентрации (активности в случае реальных растворов) или неравновесные парциальные давления (фугитивности в случае реальных газовых смесей) всех компонентов химической реакции. (rr G)р,Т = - RТ ln Кр0 + R Т ln рDn D р F n F рАn А р В n В Которое и называют уравнением изотермы химической реакции в общем виде. По уравнению изотермы вычисляют энергию Гиббса реакции, а также определяют направление и глубину ее самопроизвольного протекания при данных условиях, если известны стандартная константа равновесия для данной температуры и исходный состав произвольно приготовленной реакционной смеси, т.е. неравновесные относительные, например, парциальные давления рi всех компонентов в начальный момент времени. Очевидно, что чем больше абсолютное значение (rr G)р,Т , тем менее равновесна система. -если в реакционной смеси присутствуют или только продукты реакции или только исходные вещества, то (rr G)р,Т ®±¥ для любой реакции; - (rr G)р,Тñ 0 реакция протекает в обратном направлении справа налево до установления равновесия; -(rr G)р,Тá 0 реакция протекает самопроизвольно в прямом направлении; -(rr G)р,Т= 0 отвечает состоянию химического равновесия; -Если величина, стоящая под знаком логарифма во втором слагаемом равна единице (это возможно, например при начальных относительных парциальных давлениях всех компонентов, равных единице, т.е. компоненты находятся в стандартных условиях), то само слагаемое равно нулю, а уравнение изотермы трансформируется:
(rr G)р,Т = - RТ ln Кр0 или rr GТ0 = - RТ ln Кр0 |
и его называют стандартным уравнением изотермы химической реакции или уравнением стандартного сродства. С его помощью можно вычислить Кр0 без экспериментального изучения равновесия. Уравнение изобары химической реакции определяет зависимость константы равновесия Кр0 от температуры и в дифференциальной форме выводится на основании стандартного уравнения изотермы
rr GТ0 = - R ln Кр0 Т |
и дифференциального уравнения энергии Гиббса – Гельмгольца для стандартных условий
rr GТ0 = rr НТ0 + Т d (rr GТ0) d Т |
После преобразований получают уравнение изобары:
d ln Кр0 = rr Н0Т d Т RТ2 |
Анализируя его можно установить, что, чем больше абсолютная величина теплового эффекта и чем ниже температура, тем больше значение производной, следовательно, сильнее сказывается изменение температуры на константе равновесия и в большей степени смещается положение равновесия. Уравнение полезно при вычислении графическим путем тепловых эффектов реакции. Если температурная зависимость константы равновесия выражена в координатах ln Кр0 - 1/ Т. Газофазная реакция осуществляется в закрытой системе, при постоянном объеме (V = соnst). Уравнение изохоры химической реакции в дифференциальной форме:
d ln К с0 = rr U0Т d Т RТ2 |
В интегральной форме:
ln К 0с2 = rr U0Т (Т2-Т1) К 0с1 RТ1 Т2 |
Где rr U0Т - изменение стандартной внутренней энергии реакции; К с0 - безразмерная стандартная константа равновесия, выраженная через относительное равновесные молярные концентрации компонентов. К внешним факторам, влияющим на состояние равновесия любой системы (гомогенной или гетерогенной), относятся температура, давление, внешние поля (электрическое, магнитное, гравитационное и др.). Если значение последнего фактора существенно не отличается от нормального им пренебрегают. Обычно рассматривают воздействие только двух важных факторов:температуры, давления. Влияние изменения внешних условий на состояние равновесия устанавливается термодинамическим положением, впервые сформулированным Брауном, которое называется правилом смещения положения подвижного равновесия, или принципом Ле Шателье: если на систему, находящуюся в состоянии истинного химического равновесия, оказывать внешнее воздействие путем изменения какого-либо из условий (Сi, Т, рi. Робщ), определяющих положение равновесия, то в системе происходит изменение равновесного состава и смещение положения равновесия в направлении того процесса, протекание которого ослабляет эффект (влияние) этого воздействия.
13. Химическое равновесие в гетерогенных процессах. Правило фаз Гиббса-Коновалова. Константа равновесия для гетерогенной реакции. Термодинамические понятия: фаза, независимый компонент, число степеней свободы (для гомогенных и гетерогенных равновесных химических реакций.
 Если
все компоненты находятся в разных фазах,
то равновесии называется гетерогенным.
Если
все компоненты находятся в разных фазах,
то равновесии называется гетерогенным.

![]()
![]()



 Для
гетерогенных систем константы равновесия
такие же, как и для гомогенных, только
указываются параметры веществ, находящихся
в жидком или газовом сост.
Для
гетерогенных систем константы равновесия
такие же, как и для гомогенных, только
указываются параметры веществ, находящихся
в жидком или газовом сост.
![]()
![]()
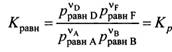 Константа
равновесия –
позволяет судить о полноте протекания
реакции при тех или иных условиях. Кравн
есть величина постоянная для данной
хим. р-ции при данных Т, давлении и в
данном растворителе = отношению
произведения равновесных активностей(или
фугитивностей для газов) прод-ов реакции,
взятых в степенях = их стехиом. коэф., к
анал. произвед. для исх. в-в в состоянии
хим. равн. – одна из формулировок закона
действующих масс Гульдберга-Вааге для
хим. равновесия.
Константа
равновесия –
позволяет судить о полноте протекания
реакции при тех или иных условиях. Кравн
есть величина постоянная для данной
хим. р-ции при данных Т, давлении и в
данном растворителе = отношению
произведения равновесных активностей(или
фугитивностей для газов) прод-ов реакции,
взятых в степенях = их стехиом. коэф., к
анал. произвед. для исх. в-в в состоянии
хим. равн. – одна из формулировок закона
действующих масс Гульдберга-Вааге для
хим. равновесия.
В гетерогенных системах вещества находятся различных фазах. Если вещества химически не взаимодействуют, то такую систему называют химически нереагирующей системой. В гетерогенных химически реагирующих системах вещества чаще всего взаимодействуют на границе раздела фаз. Равновесие, устанавливающееся в таких системах, называют гетерогенным химическим равновесием. Для гетерогенных процессов: - конденсированные фазы (жидкие и твердые), не улетучивающиеся в ходе реакции и не переходящие полностью в раствор и их количества не оказывают влияния на равновесие; -в выражение константы равновесия обратимых гетерогенных реакций входят только парциальные равновесные давления газообразных веществ, взятых в степенях стехиометрических коэффициентов. Для уравнения: Ст + СО2 г «2СОг Кр = р2равнСО/ рравнСО2 -константы равновесия Кр и Кр0 и Кс и Кс0 зависят от свойств всех веществ, реагирующих в гетерогенной реакции, а не только газов. -для обратимых гетерогенных реакций введение дополнительного количества жидких или твердых компонентов (реагентов, продуктов) в реакционную смесь или их выведение из нее не влечет за собой изменения состава и, следовательно, не нарушает состояния равновесия, т.е. не смещает его положения; -смещение положения равновесия в обратимых гетерогенных реакциях при изменении температуры вне зависимости от агрегатного состояния реагентов и продуктов подчиняется общим закономерностям. Правило фаз (уравнение) Гиббса, известное также как закон фазового равновесия, было сформулировано в 1876 г. Оно относится к одному из общих законов физики и химии и является выражением второго закона термодинамики применительно к фазовым равновесиям. С его помощью можно описать и гомогенные и гетерогенные химические равновесия, а именно: -термодинамически установить (предсказать) возможность или невозможность существования данной гомогенной или гетерогенной системы; -вычислить максимальное число фаз, которые одновременно могут находиться в равновесии при данных условиях; -априори характеризовать фазовый состав сколь угодно сложной многокомпонентной системы при заданном числе независимых переменных, определяющих равновесие; -предсказать по числу степеней свободы поведение исследуемой системы, т.е. установить варианты возможных фазовых превращений при изменении одного, двух и более внешних условий (при сохранении остальных параметров состояния системы постоянными). В общем виде правило фаз Гиббса формулируется следующим образом: число термодинамических степеней свободы, или вариантность равновесной термодинамической системы С, определяется как разность числа независимых компонентов системы К и числа сосуществующих фаз Ф плюс число внешних факторов n, влияющих на равновесие. Его математическое выражение записывается в виде простого соотношения: С = К – Ф + n. Анализируя это выражение можно сделать следующие выводы: -число степеней свободы равновесной системы возрастает с увеличением числа независимых компонентов и понижается с увеличением числа сосуществующих фаз; - число степеней свободы может быть равно либо нулю, либо целому положительному числу, т.е. С³0; - так как число степеней свободы не может быть отрицательным, то число сосуществующих фаз в равновесной термодинамической системе в общем случае не может превышать суммы К + n. Числом термодинамических степеней свободы, или вариантностью равновесной термодинамической системы С, называют максимальное число независимых интенсивных термодинамических параметров состояния фаз равновесной системы, произвольное и независимое изменение которых в определенных пределах не влияет на качественный и количественный состав системы, а именно не вызывает исчезновения одних и образования других фаз. Независимыми компонентами называют составляющие систему вещества, наименьшее число которых необходимо и достаточно для образования и однозначного выражения состава каждой фазы данной системы при любых условиях ее существования. Примеры на стр 358 Гуров Для вышеприведенной реакции: число существующих фаз Ф = 2 (т, г); К = 3 -1 =2; n. = 2 (р, Т); С = 2 Фаза (газообразная, жидкая, твердая) – гомогенная область. Гомогенными называются реакции, протекающие в одной фазе (в смеси газов, жидком растворе или твердой фазе). Скорость – важнейшая количественная кинетическая характеристика любой химической реакции. Наиболее общим является определение скорости реакции r как скорости возрастания степени завершенности реакции ξ, которую, называют химической переменной. Пусть в закрытой системе протекает необратимая химическая реакция: ν А А + ν В В → ν D D + ν F F где А, В, D, F – участники реакции; ν А ν В ν D ν F соответствующие стехиометрические коэффициенты. Тогда химическая переменная ξi , характеризующая глубину протекания реакции по i –му компоненту, определяется как ξi = n i - n 0 i ν i Где n i - число молей i – компонента в момент времени t; n 0 i - число молей того же компонента в начальный момент времени (t = 0); ν i - стехиометрический коэффициент при i компоненте в уравнении реакции. Стехиометрические коэффициенты у исходных веществ берутся со знаком «-«, а у продуктов реакции – со знаком «+». Это связано с тем, что количество продуктов реакции увеличивается. При таком выборе знаков ξi всегда положительна независимо от выбора участников реакции.
14. Скорость гомогенной химической реакции. Кинетические кривые. Факторы, влияющие на скорость реакции. Основной постулат химической кинетики (зависимость скорости реакции от концентраций реагирующих веществ). Константа скорости, порядок реакции и молекулярность.
Гомогенные
реакции –
реакции, протекающие в одной фазе. Скорость
реакции –
скорость возрастания степени завершенности
реакции x, которую называют хим.
переменной. ![]() ,
ni- число молей, в мом. времени t, ni0-в
начале., ni-стехиом. коэф. (для исх. в-в
берется с -, прод с +). Скорость гомогенной
хим. реакции – скорость изменения
глубины протекания реакции в единицу
времени в единице объема:
,
ni- число молей, в мом. времени t, ni0-в
начале., ni-стехиом. коэф. (для исх. в-в
берется с -, прод с +). Скорость гомогенной
хим. реакции – скорость изменения
глубины протекания реакции в единицу
времени в единице объема: ![]() .
Скорость реакции по i-ому компоненту:
.
Скорость реакции по i-ому компоненту: ![]() .,
., ![]() .
Для системы с постоянным объемом:
.
Для системы с постоянным объемом: ![]() .
Используют +, если скорость определяется
по образующемуся компоненту и
наоборот.
Под кинетической
кривой понимают
график зависимости концентрации реагента
или продукта от времени.
Уравнение, описывающее
зависимость скорости хим. процесса от
С называется кинетическим
уравнением процесса.
Основной постулат химической кинетики(закон
действующих масс – Гульдберг и Вааге):
скорость реакции в каждый момент времени
при постоянной температуре ~ произведению
концентраций реаг-х в-в, возведенных в
некоторые степени.
.
Используют +, если скорость определяется
по образующемуся компоненту и
наоборот.
Под кинетической
кривой понимают
график зависимости концентрации реагента
или продукта от времени.
Уравнение, описывающее
зависимость скорости хим. процесса от
С называется кинетическим
уравнением процесса.
Основной постулат химической кинетики(закон
действующих масс – Гульдберг и Вааге):
скорость реакции в каждый момент времени
при постоянной температуре ~ произведению
концентраций реаг-х в-в, возведенных в
некоторые степени. ![]() ,
, ![]() .
Коэффициент пропорциональности k –
константа скорости реакции.
Константы
сильно зависят от Т. Показатель степени
– порядок
реакции по
дан. реаг. Общий пор. реак. q=n+m. Для элем.
реакции порядок по дан. реаг. = числу
частиц, участвующих в превращении.
Лимитирующая стадия – самая медленная
стадия сложной ХР.
.
Коэффициент пропорциональности k –
константа скорости реакции.
Константы
сильно зависят от Т. Показатель степени
– порядок
реакции по
дан. реаг. Общий пор. реак. q=n+m. Для элем.
реакции порядок по дан. реаг. = числу
частиц, участвующих в превращении.
Лимитирующая стадия – самая медленная
стадия сложной ХР.
Константа
скорости р-ции:
![]() ,
c- трансмиссионный коэффициент (учитывает
вероятность перехода АК в продукты
р-ции)
,
c- трансмиссионный коэффициент (учитывает
вероятность перехода АК в продукты
р-ции)
Скорость гомогенной реакции в целом определяется как скорость изменения глубины протекания реакции в единицу времени в единице объема:
r = 1 d ξi V dt |
Где V- объем системы; t – время. Определенная таким образом скорость реакции не зависит от выбора компонентов и будет практически одинаковой для разных веществ, участвующих в реакции. Скорость реакции по i – компоненту
r i = ± 1 d n i V dt |
Она характеризует изменение количества i вещества n i (в молях) в единицу времени в единице объема. Таким образом, скорость реакции (для системы с переменным объемом) в целом и скорости реакции по отдельным компонентам связаны следующим соотношением
r = 1 r i ν i |
Скорость реакции при постоянном объеме представляет собой изменение концентрации данного компонента в единицу времени. Скорость реакции есть положительная величина.
r i = ± d С i уравнение также характеризует мгновенную скорость реакции dt |
Средняя скорость реакции в интервале времени от t1 до t2 определяется соотношением
r ср = ± С2 - С1 = ± ΔС где С1 и С2 молярные концентрации любого участника реакции в момент t2 - t1 Δt времени t1 и t2 соответственно. |
На скорость гомогенной реакции могут оказывать влияние следующие факторы: природа реагирующих веществ; концентрации реагирующих веществ; давление (если в реакции участвуют газы); температура; наличие специфических веществ, ускоряющих реакцию (катализаторы) или замедляющих ее (ингибиторы); световое или радиационное излучение. Основной постулат химической кинетики базируется на большом числе экспериментальных данных и выражает зависимость скорости реакции в каждый момент времени от концентрации реагирующих веществ: скорость реакции в каждый момент времени при постоянной температуре пропорциональна произведению концентраций реагирующих веществ, возведенных в некоторые степени. r = k СА n С В n Коэффициент пропорциональности k называют константой скорости химической реакции. Константы скоростей значительно отличаются для разных реакций и сильно зависят от температуры.
15. Дифференциальные и интегральные уравнения скорости для необратимых реакций нулевого и первого и второго порядков. Константа скорости. Кинетические кривые. Период полупревращения. Методы определения порядков реакции.
Время
(период) полупревращения –
время, необходимое для хим. превращения
половины начального кол-ва компонента.
Методы
определения порядка реакции:
Метод подстановки – подстановка экс.
данных в какое-либо из известных
уравнений. Прав-ть выбора опред. по тому,
как хорошо сохраняется константа
скорости.
Метод Вант-Гоффа (опр-е по
нач. скоростям): Если проходит реакция
А+В->Прод-ты, то ![]() .
Для опр. пор. по в-ву А проводят серию
эксп. при пост. нач. конц. в-ва В и различных
нач. конц. А.
.
Для опр. пор. по в-ву А проводят серию
эксп. при пост. нач. конц. в-ва В и различных
нач. конц. А. ![]() ,
где lgk’=lgk+lgCob*m. При различн. конц. А
измеряют нач. скорости, смотря график
зависимости lgr0 от lgCoA. Тангенс угла
наклона равен порядку р-ции по А (tga=n).
Аналогично для В. Можно оценить и
расчетным путем, если известны нач.
скорости при 2х нач. конц. в-ва:
,
где lgk’=lgk+lgCob*m. При различн. конц. А
измеряют нач. скорости, смотря график
зависимости lgr0 от lgCoA. Тангенс угла
наклона равен порядку р-ции по А (tga=n).
Аналогично для В. Можно оценить и
расчетным путем, если известны нач.
скорости при 2х нач. конц. в-ва: ![]() .
Берем отношение скоростей, логарифмируем
и выделяем n.
Метод определения по
периоду полупревращения. Основан на
зависимости ППП от начальной концентрации
в-ва.
Влияние Т на скорость хим.
реакций.
Основное влияние Т оказывает
на константу скорости.
.
Берем отношение скоростей, логарифмируем
и выделяем n.
Метод определения по
периоду полупревращения. Основан на
зависимости ППП от начальной концентрации
в-ва.
Влияние Т на скорость хим.
реакций.
Основное влияние Т оказывает
на константу скорости.
Кинетические
ур-я односторонних реакций: реакции
0-го порядка.![]() ,
интегрируя от начальной концентрации
до конц. в мом. врем. t:
,
интегрируя от начальной концентрации
до конц. в мом. врем. t: ![]() ,
С=С0-kt. ,
,
С=С0-kt. , ![]() (св-ва
сама!). 0-й порядок имеет место в тех сл.,
когда убыль в-ва в рез. протек. хим. реак.
восполняется доставкой его из др.
фазы. Реакции
1-го порядка:
(св-ва
сама!). 0-й порядок имеет место в тех сл.,
когда убыль в-ва в рез. протек. хим. реак.
восполняется доставкой его из др.
фазы. Реакции
1-го порядка: ![]() ,
, ![]() =>
после преобразований:
=>
после преобразований: ![]() ,
, ![]() .
Среднее время жизни молекулы t=1/k1,
соотв-ет уменьшению исх. конц. в е
раз.
Реакции
2-го порядка:
1. В р-ции участвуют 2 реагента, конц. кот.
=.
.
Среднее время жизни молекулы t=1/k1,
соотв-ет уменьшению исх. конц. в е
раз.
Реакции
2-го порядка:
1. В р-ции участвуют 2 реагента, конц. кот.
=. ![]() ,
, ![]() .
2. Начальные концентрации реагентов не
равны:
.
2. Начальные концентрации реагентов не
равны: ![]() .
. ![]() .
3. Р-ция 2А->P.
.
3. Р-ция 2А->P. ![]() ,
множитель 2 означает, что в каждой элем.
реакции превращаются 2 молекулы вещества
А.
,
множитель 2 означает, что в каждой элем.
реакции превращаются 2 молекулы вещества
А.
Показатель степени, в которой концентрация реагента входит в кинетическое уравнение, называют порядком реакции по данному реагенту. Общий порядок q реакции есть алгебраическая сумма показателей степеней при концентрациях всех реагентов, которые входят в кинетическое уравнение: q=n+m Для элементарной реакции порядок по данному реагенту равен числу частиц данного реагента, участвующих в элементарном химическом акте. Например для элементарной реакции : 2N О + Сl2 ® 2NО Сl молекулярность равна 3 (количество молекул, участвующих в реакции). Порядок реакции по NО равен 2, а по Сl – 1. общий порядок реакции равен 3, всегда положителен и целочислен. Показатель степени, в которой концентрация реагента входит в кинетическое уравнение, называют порядком реакции по данному реагенту (для элементарных реакций - это коэффициент, стоящий перед реагентом в уравнении реакции). Общий порядок реакции равен сумме порядков по каждому компоненту. В качестве критерия скорости реакции нередко используется период полупревращения t1/2 , равный времени, в течение которого концентрация реагента уменьшается вдвое по сравнению с начальной концентрацией. Под кинетической кривой понимают график зависимости концентрации реагента или продуктов реакции от времени Наиболее часто встречаются реакции нулевого, первого, второго, иногда третьего порядков. Нулевой порядок имеет место в тех случаях, когда убыль вещества в результате протекания химической реакции восполняется доставкой его из другой фазы. Нулевой порядок наблюдается если скорость процесса лимитируется подачей энергии, необходимой для активации реагирующих молекул. Например, при фотохимических реакциях определяющим фактором может служить количество поглощенного излучения, а не концентрация вещества. Часто в каталитических реакциях скорость зависит от концентрации катализатора, а не от концентрации реагирующих веществ. Кинетическое уравнение для реакция нулевого порядка имеет вид: - dС/ dt = k. Разделяя переменные (- dС= k dt ) и интегрируя в пределах от начальной концентрации Со (t =0) до текущей концентрации С в момент времени t: - ∫СС0 dС = ∫0t k dt получают С = С0 - k t реакции нулевого порядка имеют следующие особенности: -в соответствии с последним выражением в реакциях нулевого порядка концентрация исходного вещества линейно уменьшается о времени (кинетическая кривая - линейная зависимость стр 247); -константа скорости k = (С0 – С ) /t. Ее размерность такая же как и размерность скорости реакции; -время полупревращения t1/2, при С = С0/2 t1/2 = С0 / 2 k, т.е. для реакции нулевого порядка t1/2 пропорционально начальной концентрации исходного вещества. Первый порядок имеют реакции диссоциации или разложения молекул Н2 → 2Н; радиоактивного распада 22286Rn →4 2α +21882Rn (радон). Для реакций первого порядка кинетическое уравнение имеет вид- dС/ dt = k1 С. Разделяя переменные - dС/С = k1 dt и интегрируя полученное уравнение, находят ∫СС0 -dС/С = (integ)0t k1 dt, находят -(lnС – lnС0) = k1t lnС = lnС0 – k1 t ; С = С0exp(-k1 t) реакции первого порядка имеют особенности: - график зависимости lnС от t выражается прямой линией. Это значит, что в полулогарифмических координатах, при разных начальных концентрациях прямые будут параллельны между собой; -концентрация исходного вещества стремится к нулю при t-> oo. Полностью вещество реагирует за бесконечно большой промежуток времени; -скорость реакции также отличается экспоненциальной зависимостью r = k1С0e- k1 t -размерность константы скорости реакции первого порядка соответствует обратному времени и ее можно выразить в обратных секундах, минутах, часах и т.д; -время полупревращения при С = С0/2 t1/2 = ln2 /k, т.е. не зависит от начальной концентрации реагирующего вещества; -кинетическая кривая реакции первого порядка инвариантна при линейном преобразовании концентрации (график имеет вид верхней части равносторонней гиперболы стр 249). Примеры реакций второго порядка : Н2 + I2 ->2НI ; или 2NО 2->2NО + О2 если в реакции участвуют два компонента, концентрации которых равны, то дифференциальное уравнение имеет вид - dС/dt = k2 С × С. (или С2) Разделение переменных и интегрирование в пределах от Со до С приводит к следующему результату: 1/С - 1/С0 = k2 t из уравнения следует, что концентрация исходных веществ зависит от времени следующим образом: С = С0 / (1+ k2 С0 t) Полученные уравнения позволяют отметить следующие особенности реакций второго порядка: -при равенстве начальных концентраций реагирующих веществ обратная концентрация линейно зависит от времени (стр 251) 1/С = 1/С0 + k2 t -константа скорости k2 = (1/ t) ( 1 /С - 1 /С0) = (1/t) (С0 – С) /СС0 где k – л моль-1 с-1 -время полупревращения при С=С0 /2 (1 /С0 /2) – (1 /С0) = k2 t1 /2 отсюда t1/2 = 1 / (k2 С0) в отличие от реакций первого порядка время полупревращения реакций второго порядка обратно пропорционально начальной концентрации.