

Кислоты
Кислотами называются сложные вещества, диссоциирующие в водных растворах с образованием гидратированных катионов водорода:
НСl + Н2О = Н3О+ + Сl-
НNО3 + Н2О = Н3О+ + NО-3
По своему составу кислоты делятся на бескислородные (НСl, Н2S, НСN, HI и др.) и кислородные (H2SО4, НNО3, H3PO4, H2SiO3 и др.)
Для названия кислот обычно используются русская номенклатура согласно которой названия бескислородных кислот производится от названия неметалла с прибавлением суффикса «исто» (реже «о») и слова водородная:
Н2S – сероводородная кислота;
НСl – хлористоводородная кислота;
НСN – цианистоводородная кислота.
В случае кислородосодержащих кислот их названия зависят от степени окисления элемента – кислотообразователя. Если элемент проявляет свою высшую степень окисления, то к его названию прибавляют окончание «ная» или «овая», например:
H2S6+О4 – серная кислота;
Н2W6+О4 – вольфрамовая кислота;
HMn7+O4 – марганцовая кислота;
HN5+O3 – азотная кислота.
Для кислот с более низкой степенью окисления к названию кислотообразователя добавляется окончание «истая».
H2S4+О3 – сернистая кислота;
HN3+O2 – азотистая кислота;
H2P3+O3 – фосфористая кислота.
При проявлении элементом еще более низкой степени окисления к его названию прибавляется окончание «оватистая»:
HBr1+O – бромноватистая кислота;
H3P1+O2 – фосфорноватистая кислота.
Наконец, для кислот, в которых кислотообразующий элемент проявляет промежуточную степень окисления по сравнению с соответствующими кислотами, имеющими названия с окончанием «ная» и «истая», к его названию добавляется окончание «оватая». Так четыре кислоты, которые могут образовать хлор в зависимости от проявляемой им степени окисления можно назвать так:
НСl1+О – хлорноватистая кислота;
НСl3+О2 – хлористая;
НСl5+О3 – хлорноватая кислота;
НСl7+О4 – хлорная кислота.
Название кислоты определяется и числом молекул воды, присоединенных к молекуле ангидрида кислоты. В этом случае перед названием наименее гидратированной формы кислоты ставится приставка – «мета», а перед названием более гидратированной кислоты приставка «орто». Так, при взаимодействии фосфорного ангидрида с водой могут быть получены мета - и ортофосфорные кислоты:
HPO3 – метафосфорная кислота;
H3PO4 – ортофосфорная кислота.
Кислоты, молекулы которых содержат более одного кислотного остатка, называются поликислотами. Их можно рассматривать как продукты присоединения к кислородосодержащей кислоте ангидрида той же или другой кислоты. Так, при растворении хромового ангидрида в воде получается хромовая кислота:
CrO3 + H2O = H2CrO4
Чем концентрированнее водный раствор CrO3, тем более конденсированные кислоты он содержит:
H2CrO4 * CrO3 – дихромовая кислота H2Cr2O7
трихромовая кислота H2Cr3O10
тетрахромовая кислота H2Cr4O13.
Существуют и другие разновидности кислот. Так, кислоты, в или все атомы кислорода замещены серой, называются (H2S2O3), а кислоты, содержащие в своем составе «перекисную цепочку» - надкислотами (H2S2O8). первая кислота называется тиосерной, а вторая – надсерной кислотой.
Названия наиболее распространенных в практической деятельности человека кислот и соответствующих им солей предоставлены в таблице 1.
При составлении графических формул всех перечисленных выше видов кислот следует учитывать, что в молекулах кислородосодержащих кислот атомы водорода, способные замещаться атомами металлов, связаны с элементом – кислотообразователем через атом кислорода. В молекулах бескислородных кислот атом водорода связан непосредственно с атомами неметалла:
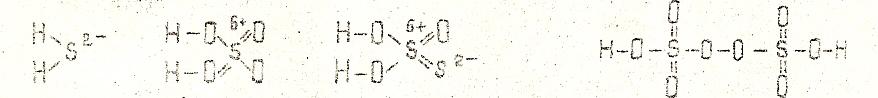
(H2S) (H2SO4) (H2S2O3) (H2S2O8)
Число атомов водорода, которые способны замещаться атомами металла, определяют основность кислот. Так,
НNО3, НСl, HI , НСlО4 – одноосновные
H2SiO3, H2CrO4, H2SO4 – двухосновные
Н3ВО3, H3PO4 – трехосновные кислоты.
У некоторых кислот их основность не совпадает с числом содержащихся в их молекулах атомов водорода. например, в фосфористой (H3PO4) кислоте основность равна двум, а в фосфорноватистой (H3PO2) – единице:
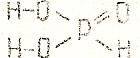
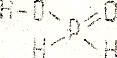
В первой кислоте два атома водорода (соединенные с атомами кислорода) способны замещаться на атомы металла, во второй только один.
Двух- и многоосновные кислоты диссоциируют ступенчато:
H3PO4
![]() Н+
+ H2PO-4
Н+
+ H2PO-4
H2PO-4
![]() Н+
+ HPO2-4
Н+
+ HPO2-4
HPO2-4
![]() Н+
+ PO3-4
Н+
+ PO3-4
В связи с этим валентность кислотного остатка (H2PO-4, HPO2-4, PO3-4) определяется количеством атомов водорода, посылаемых в раствор молекулой кислоты.
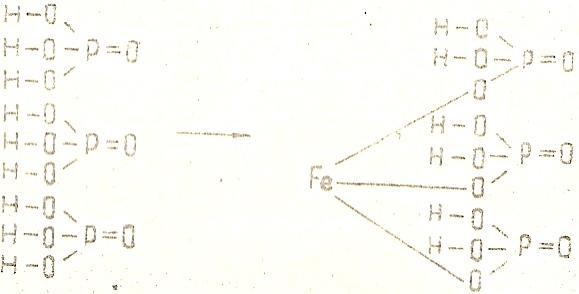
К важнейшим свойствам кислот относится их способность взаимодействовать с гидроксидами (реакция нейтрализации), основными и амфотерными оксидами с образованием солей:
3НСl + Fe (OH)3 = FeCl3 + 3H2O
H2SO4 + CuO = CuSO4 + H2O
6HCl + Al2O3 = 2AlCl3 + 3H2O
Помимо этого кислоты взаимодействуют с металлами. В зависимости от активности металла (его положения в ряду напряжений металлов) и природы кислоты в результате таких реакций выделяется либо водород, либо продукты восстановления окисляющей кислоты:
Zn
+ H2SO4
(разб)
= ZnSO4
+ H2
Mn
+ 2HCl = MnCl2
+ H2
Cu0 + H2SO4 (конц) = Cu2+SO4 + S4+O2 +2H2O
3CuO
+ 8HN5+O3
= Cu (NO3)2
+ 2N2+O
 +
4H2O
+
4H2O
Водные растворы кислот имеют кислый вкус и окрашивают индикаторы: лакмус в красный, метилоранж – в розовый цвет. Фенолфталеин в кислой среде не меняет окраски.
Номенклатура кислот и солей
Таблица 1.
|
Формула |
Название кислоты |
Номенклатура солей |
|
|
русская |
международная |
||
|
1 |
2 |
3 |
4 |
|
H2S |
Сероводородная |
Сернистые |
Сульфиды |
|
H2SO4 |
Серная |
Сернокислые |
Сульфаты |
|
H2SO3 |
Сернистая |
Сернистокислые |
Сульфиты |
|
H2S2O3 |
Тиосерная (серноватистая) |
Тиосернокислые (серноватистокислые) |
Тиосульфаты |
|
H3PO4 |
Ортофосфорная |
Ортофосфорнокислые |
Ортофосфаты |
|
HPO3 |
Метафосфорная |
Метафосфорнокислые |
Метафосфаты |
|
H3PO3 |
Фосфористая |
Фосфористокислые |
Фосфиты |
|
HNO3 |
Азотная |
Азотнокислые |
Нитраты |
|
HNO2 |
Азотистая |
Азотистокислые |
Нитриты |
|
H2CO3 |
Угольная |
Углекислые |
Карбонаты |
|
H2SiO3 |
Кремниевая |
Кремниевокислые |
Силикаты |
|
H3BO3 |
Ортоборная |
Ортоборнокислые |
Ортобораты |
|
HBO2 |
Метаборная |
Метаборнокислые |
Метабораты |
|
H2B4O7 |
Тетраборная |
Тетраборнокислые |
Тетрабораты |
|
HCl |
Хлористоводородная (соляная) |
Хлористые |
Хлориды |
|
HBr |
Бромистоводородная |
Бромистые |
Бромиды |
|
HI |
Йодистоводородная |
Йодистые |
Иодиды |
|
HCN |
Цианистоводородная |
Цианистые |
Цианиды |
|
H2CrO4 |
Хромовая |
Хромовокислые |
Хроматы |
|
H2Cr2O7 |
Дихромовая |
Дихромовокислые |
Дихроматы |
|
H3CrO3 |
Ортохромистая |
Ортохромистокислые |
Ортохромиты |
|
HCrO2 |
Метахромистая |
Метахромистокислые |
Метахромиты |
|
HMnO4 |
Марганцовая |
Марганцовокислые |
Перманганаты |
|
1 |
2 |
3 |
4 |
|
H2MnO4 |
Марганцовистая |
Марганцовистокислые |
Манганаты |
|
HClO4 |
Хлорная |
Хлорнокислые |
Перхлораты |
|
HF |
Фтористоводородная (плавиковая) |
Фтористые |
Фториды |
|
H3AsO4 |
Ортомышьяковая |
Ортомышьяковокислые |
Ортоарсенаты |
|
H3AsO3 |
Ортомышьяковистая |
Ортомышьяковистокислые |
Ортоарсениты |
|
CH3COOH |
Уксусная |
Уксуснокислые |
Ацетаты |
|
HCOOH |
Муравьиная |
Муравьинокислые |
Формиаты |
|
H2C2O4 |
Щавелевая |
Щавелевокислые |
Оксалаты |
|
HCN5 |
Роданистоводородная |
Роданистые |
Роданиды |
Основными методами получения кислот являются:
-
Взаимодействие неметалла с водородом с последующим растворением полученного продукта в воде:
Н2 + S = Н2S
-
Взаимодействие ангидридов кислот с водой:
SO3 + Н2O = Н2SO4
-
Взаимодействие галогенов с водой:
Cl2 + Н2O = НCl + НClО
-
Вытеснение слабых кислот из их солей более сильными кислотами:
Na2SiO3
+
Н2SO4
=
Na2SO4
+
Н2SiO3
-
окисление неметалла раствором сильной кислоты – окислителя:
3Р + 5 НNО3 + 2Н2О = 3Н3РО4 + 5NО
СОЛИ
Солями называются продукты полного или неполного замещения атомов водорода в молекуле кислоты атомами металла или гидроксильных групп в молекуле основания кислотными остатками. В связи с этим различают средние (нормальные), кислые и основные соли.
Средние соли – продукты полного замещения атомов водорода в кислоте или гидроксильных групп в основании.
Н2SO4
 Na2SO4
Na2SO4


Наименования средних солей бескислородных кислот по международной номенклатуре производится от латинского корня названия элемента – кислотообразователя с добавлением окончания «ид». Затем дается название металла с указанием его степени окисления (если металл имеет переменные степени окисления):
ВаS – сульфид бария
CuCl2 – хлорид меди (II)
NaCN – цианид натрия
CrN – нитрид хрома (III)
Mn3P2 – фосфид марганца (II)
NH4SCN – роданид аммония
Русские названия таких солей составляются из названия неметалла с окончанием «истый» («истое») и названия металла. Например:
ВаS – сернистый барий
NaCN – цианистый барий
NH4SCN – роданистый аммоний
В случае, если металл, образующий среднюю бескислородную соль, проявляет переменную степень окисления, то окончание «истый» характерно для солей с низшей степенью окисления, а для солей с более высокой степенью окисления вводится окончание «ный» («ная», «ное»). Например:
Fe2+Cl2 – хлористое железо
Fe3+Cl3 – хлорное железо
Названия средних солей кислородосодержащих кислот (HNO3, H2SO4, H3PO4, H2CO3 и др.) по международной номенклатуре складывается из латинского названия кислотообразователя с добавлением окончания «ит» (если элемент – кислотообразователь проявляет низшую степень окисления) и окончание «ат» (если элемент – кислотообразователь проявляет высшую степень окисления). Затем дается название металла в родительном падеже с указанием степени окисления (если она переменная). Так, соли H5+NO3, H2S6+O4, H3P5+O4, H2C5+O3, Н2Si4+O3 называются соответственно: нитратами, сульфатами, фосфатами, карбонатами, силикатами, а соли с азотистой H3+NO2 и сернистой H2S4+O3 – нитритами, сульфитами:
Na2SO4 – сульфат натрия
Na2SO3 – сульфит натрия
Cu SO4 – сульфат меди (II)
Cu (NO3)2 – нитрат меди (II)
Если кислотные остатки образованы элементом, обладающим различными степенями окисления, то названия образованных ими солей отличаются еще и приставками. Например, средние соли четырех кислот, образованных хлором, называются:
NaCl1+О – гипохлорит натрия;
NaCl3+О2 – хлорит натрия;
NaCl5+О3 – хлорат натрия;
NaCl7+О4 – перхлорат натрия.
Название последней соли, как и солей марганцовой кислоты (НMnO4), содержит приставку «пер». Это исторически сложившееся исключение, так как с такой приставки начинаются названия солей надкислот:
Na2S2O3 – персульфат натрия;
K2Cr2O12 – пероксохромат калия.
Если элемент образует соединения, в которых он присутствует в форме определенной функциональной группы или сложного иона (например: сульфурил, тионил и т.д.), то название соответствующих солей закачивается названием этой группы в родительном падеже:
SOCl2 – хлорид тионила;
SOF2 – фторид сульфурила.
Основными способами получения средних солей являются:
-
Взаимодействие металла с неметаллом:
Са + Cl2 = СаCl2
-
Взаимодействие металла с кислотой:
Fе + Н2SO4 = FеSO4 + Н2
-
Взаимодействие металлов с растворами солей менее активных металлов:
CuSO4 + Fе = Cu + FеSO4
-
Взаимодействие основного оксида с кислотным:
MgO + SO3 = MgSO4
-
Взаимодействие основного оксида с кислотой:
MgO + Н2SO4 = MgSO4 + Н2О
-
Взаимодействие кислотного оксида с гидроксидом:
2NaОН + SO3 = Na2SO3 + Н2О
-
Взаимодействие гидроксида с кислотой:
NaОН + НCl = NaCl + Н2О
-
Взаимодействие гидроксида (щелочи) с солью:
2NaОН
+ CuCl2
= Cu(ОН)2
 +
2 NaCl
+
2 NaCl
-
Взаимодействие кислоты с солью:
НCl
+ АgNО3
=
АgCl + НNО3
+ НNО3
-
Взаимодействие соли с солью:
ВаCl2
+
К2SO4
= ВаSO4 + 2КCl
+ 2КCl
-
Взаимодействие аммиака с кислотой:
2NН3 + Н2SO4 = (NН4)2SO4
Многие соли хорошо растворимы в растворителях, обладающих большими величинами диэлектрической проницаемости, например, в воде: это соли уксусной, азотной кислот, соли серной кислоты (кроме солей Са2+, Pb2+, Sz2+ и Ва2+), соли галогеноводородных кислот НCl, НВr, НI (кроме солей Аg+, Pb2+) и другие. Из солей Н2SiO3, H3PO4, Н2СO3 растворимы лишь соли щелочных металлов и аммония. (Смотри табл. 2).
За исключением солей, образованных сильными основаниями и сильными кислотами, все соли при растворении подвергаются гидролизу:
ZnCl2
+ HOH![]() ZnOHCl
+ HCl
ZnOHCl
+ HCl
В
ионном виде: Zn2+
+ HOH![]() ZnOH+
+
H+
ZnOH+
+
H+
или: Li2СО3
+ HOH![]() LiHCО3
+ LiОH
LiHCО3
+ LiОH
в
ионном виде: Li2-3
+ HOH![]() HCО-3
+ ОH-
HCО-3
+ ОH-
Для нормальных (средних) солей характерны следующие реакции, протекающие в направлении образования малорастворимых или малодиссоциированных веществ:
Растворимость некоторых веществ и оснований в воде. Таблица 2.

-
С солями: ВаCl2 + Na2SO4 = ВаSO4
 +
NaCl
+
NaCl -
С кислотами: Na2SO3 + 2HCl = 2 NaCl + Н2О + СO2

-
Со щелочами: CuCl2 + 2КОН = Cu(ОН) 2
 +
2КCl
+
2КCl -
С металлами (более активными, чем тот, который входит в состав соли): Нg (NО3)2 + Cu(NО3)2 + Нg
-
Термическая диссоциация: MgСO3
 MgO
+ СO2
MgO
+ СO2
Кислые соли образуют многоосновные (двух- и более) кислоты при неполном замещении их атомов водорода на металл.
Соли этого типа можно получить:
-
При взаимодействии кислоты с недостаточным количеством гидроксида: КОН + H3PO4 = КН2РО4 + Н2О
недостаток
2 КОН + H3PO4 = К2НРО4 + 2Н2О
-
При взаимодействии кислоты с недостаточным количеством основного оксида: 2H3PO4 + СаО = Са(H2PO4)2 + Н2О
-
При взаимодействии кислоты со средней солью, имеющей тот же кислотный остаток: H2SO4 + Na2SO4 = 2 Na НSO4
-
При взаимодействии щелочи или соли с ангидридом кислоты, взятом в избытке: КОН + СO2 = КН СO3
СаСO3 + СO2 + Н2О = Са (НСO3)2
Названия кислых солей составляется по международной номенклатуре аналогично названиям нормальных солей, только присутствие в молекуле соли атомов водорода отмечается приставкой «гидро» с соответствующим греческим числительным («моно»-, ди -, три - и т.д.). Числительное «моно» обычно опускается:
Са(НSO4)2 – гидросульфат кальция;
КН2РО4 – дигидрофосфат калия;
К2НРО4 – гидрофосфат калия;
Na НСO3 – гидрокарбонат натрия.
Иногда в названиях солей двухосновных кислот приставка «гидро» - заменяется приставкой «би»:
Na НСO3 – бикарбонат натрия
КНSO4 – бисульфат калия
Na НSO3 – бисульфит натрия.
Названия кислых солей в русской номенклатуре подобны названиям нормальных солей с добавлением слова кислый. Количество замещенных в кислоте атомов водорода указывается прилагательным «однозамещенный», «двузамещенный» (в случае трехосновной и выше кислоты).
Ва (Н2РО4)2 – кислый фосфорнокислый барий однозамещенный
К2НРО4 – кислый фосфорнокислый калий двузамещенный
Na НSO4 – кислый сернокислый натрий
валентность кислотного остатка в кислой среде определяется количеством атомов водорода кислоты, замещенных на атомы металла:
Mg (Н2РО4)2 – (один атом водорода в молекуле замещен на атом металла)
К2НРО4 – (два атома водорода замещены на атомы металла).
В водных растворах соли такого типа диссоциируют на положительно заряженные ионы металла (катионы) и отрицательно заряженные ионы кислого остатка (анионы):
К2НРО4
![]() 2К+
+ НРО2-4
2К+
+ НРО2-4
Са
НСО3)2
![]() Са2+
+
2НСО-3
Са2+
+
2НСО-3
В таких растворах частично присутствуют и ионы Н+, РО3-4, СО2-3.
Кислые
соли можно перевести в нормальные
действием на них избытка гидроксида:
Са (НСО3)2
+ Са (НО)2
= 2 СаСО3 + 2Н2О
+ 2Н2О
Основные соли («гидроксо» - соли) являются продуктами неполного замещения гидроксильных групп в гидроксиде кислотными остатками. Они могут быть получены:
-
из многокислотных гидроксидов при их неполной нейтрализации:
Cu (ОН) 2 + Н2SO4 = (CuОН) 2 SO4 + 2Н2О
недостаток
Са (ОН) 2 + HCl = СаОН Cl + Н2О
-
при взаимодействии средней соли с гидроксидом:
Вi(NО3)2 + 2КОН = Вi(ОН) 2NО3 + 2КNО3
недостаток
В средние соли основные соли переводятся действием на них избытка кислоты: ZnОН NО3 + НNО3 = Zn (NО3)2 + Н2О
Количество образованных данных гидроксидом основных солей зависит от его кислотности. Однокислотные гидроксиды (NаОН, КОН и т.д.) основных солей не образуют. Двухкислотным гидроксидам соответствуют основные соли одного типа:
Zn(ОН)2
 Zn
ОН Cl
Zn
ОН Cl
Ва
(ОН)2
 ВаОН)2
SO4
ВаОН)2
SO4
Трехкислотным гидроксидам – двух типов:
Fе(ОН)3
 Fе(ОН)2
Cl
Fе(ОН)2
Cl
Fе(ОН)3
 Fе
ОН Cl2
Fе
ОН Cl2
Валентность основного остатка определяется количеством гидроксильных групп в гидроксиде, замещенных кислотными остатками:
Аl(ОН)3
 [Аl(ОН)2]+2
SO2-4
[Аl(ОН)2]+2
SO2-4
Аl(ОН)3
 Аl
ОН2
+
SO2-4
Аl
ОН2
+
SO2-4
Диссоциация таких солей в водных растворах протекает по схеме:
[Аl(ОН)2]2
SO4
![]() 2
Аl(ОН)+2
+ SO2-4
2
Аl(ОН)+2
+ SO2-4
Аl
ОН
SO4![]() Аl(ОН)2+
+ SO2-4
Аl(ОН)2+
+ SO2-4
Русские названия основных солей составляются аналогично названиям средних солей, но при этом добавляется слово «основной» («основная», «основное») и указывается количество замещенных в гидроксиде гидроксильных групп (для многокислотных гидроксидов). Например,
(SrOH)2 SO4 – основной сернокислый стронций;
Cr(OH)2 Cl – основной хлористый хром однозамещенный;
Fe OH CO3 – основное углекислое двузамещенное железо (III)
По международной номенклатуре названия основных солей повторяют названия соответствующих средних солей с добавлением приставки «гидроксо» и с указанием с помощью греческого числительного количества гидроксильных групп в этих солях:
(SrOH)2 SO4 – гидросульфат стронция;
Cr(OH)2 Cl – дигидроксохлорид хрома (III);
Fe OH CO3 – гидроксокарбонат железа (III).
иногда при образовании основной соли происходит отщепление воды из основного остатка с выделением «оксосоли». Такие соли уже не содержат гидроксильных групп:
Вi(ОН)2 = SiOCl + Н2О
Sb(ОН)2Cl + SbОCl + Н2О
Помимо этого «оксосоли» могут образоваться в результате замещения гидроксильных групп в гидратных соединениях на кислотный остаток.
UO2(ОН)2
 UO2Cl
UO2Cl
или Н2 UO4
CrO2
(OH)2 CrO2
(NО3)2
CrO2
(NО3)2
или H2 CrO4
Названия «оксосолей» включают в себя название металла, образующего «оксо» - группу, с окончанием «ил» и латинское название соответствующей кислоты: ВiОCl – хлорид висмутила;
SbО NО3 – нитрат стибила;
UO2Cl2 – хлорид уранила;
CrO2(NО3)2 – нитрат хромила.
Особые группы солей образуют двойные, смешанные, комплексные (о них вы узнаете в соответствующем разделе практикума) соли, галогенангидриды, карбиды, нитриды, силициды, бориды.
Двойные соли образованы одной и той же кислотой, но двумя различными металлами. Формулы и названия таких солей обычно начинаются с катиона, имеющего более низкую степень окисления. Кислотный остаток называется аналогично средним солям:
NаАlF4 – фторид натрия-алюминия;
КАl(SO4)2*24 Н2О – сульфат калия-алюминия;
N H4 MgРО4 – фосфат аммония (магния).
Смешанные соли в своем составе содержат анионы различных кислот и катионы одного и того же металла. Например, соль СаCl(ClО) – соль соляной (НCl) и хлорноватистой (НClО) кислот, ВаCl(NО3) – соль соляной и азотной кислот. В соответствии с этим соль СаCl(ClО) называется хлоридгипохлорит кальция, а соль ВаCl(NО3) – хлорид-нитрат бария.
Галогенангидриды – продукты полного или неполного замещения гидроксильных групп в молекулах кислородосодержащих кислот атомами галогенов. Так, фосфористой кислоте Н3РО3 (или Р(ОН)3) соответствуют галогенангидриды РCl3, РОВr3; азотистой кислоте НNО2, (или NООН) – NОCl и т.д.
В соответствии с этим их можно назвать следующим образом:
РCl3 – трихлорангидрид фосфористой кислоты;
РОCl3 – трихлорангидрид ортофосфорной кислоты;
NОCl –хлорангидрид азотистой кислоты.
Характерным свойством для многих галогенагидридов, является их способность при взаимодействии с водой или парами воды образовывать необратимо две кислоты – галогеноводородную и соответствующую кислородосодержащую:
NОCl + Н2О = НNО2 + НCl
РВr3 + 3Н2О = Н3РО3 + 3НВr
Карбиды – соединения металлов и некоторых неметаллов с углеродом. В зависимости от характера межатомных связей карбиды делятся на четыре группы:
-
Солеобразные карбиды образуют главным образом элементы I, II, III и частично IV, V и VI групп. Их можно рассматривать как продукты замещения металлом атомов водорода в:
-
метане CH4 (Be2C, Al4C3) Be
 C
C
 Be
Be -
ацетилене CH
 CH (CaC2,
Na2C2,
ZnC2,
Ag2C2)
CH (CaC2,
Na2C2,
ZnC2,
Ag2C2)
 Na
– C
C
– Na
Na
– C
C
– Na
-
Карбиды с ковалентной связь. (SiC, B4C3), отличающиеся тугоплавкостью, жаростойкостью, высокой твердостью и химической инертностью.
-
Карбиды – фазы внедрения атомов углерода в кристаллическую решетку из атомов металла (TiC, ZnC, VC, Mo2C, W2C).
-
Карбиды типа Fe3C (Mn3C, CO3C, Ni3C). Эти карбиды растворяются в разбавленных кислотах с выделением смеси углеродов с водородом и обладают более низкими температурами плавления и твердостью.
Основным методом получения карбидов является прокаливание при высоких температурах соответствующих металлов или их оксидов с углем в инертной Ar, He или восстановительной Н2, СО атмосфере.
Нитриды – соединения металлов и некоторых неметаллов с азотом. Подобно карбидам, они делятся по типу химической связи на несколько групп:
-
Ионные или Солеобразные нитриды. Их образуют s-элементы (щелочные и щелочноземельные металлы) и металлы подгрупп меди и цинка (Li3N, Zn3N2, Cu3N2, Вe3N2 и др.)
Li
N
 Li Zn
= N – Zn – N = Zn
Li Zn
= N – Zn – N = Zn
Li
Все представители этой группы являются полупроводниками, при высоких температурах неустойчивы, легко взаимодействуют с Cl2, F2, P с разбавленными кислотами, водородом.
-
Ковалентные нитриды (AlN, BN, Si3N4) Al N, В N образуются Р - элементами и обладают свойствами либо полупроводника, либо диэлектрика. Устойчивость их значительно выше, чем у ионных нитридов. В машиностроении в последнее время широко используется нитрид бора (боразон), обладающий твердостью, близкой к алмазу, стойкостью к нагреванию до 2000С (алмаз сгорает при 900С).
-
Металлоподобные нитриды образуются за счет ионной и металлической связи d и f – элементами (TiN, VN, CrN, Cr2N и др.). Такие связи очень прочны, так как образованы при внедрении атомов азота в кристаллическую решетку металла за счет не только внешних s-электронов, но и глубоко расположенных d –электронов. Многие металлоподобные нитриды являются хорошими сверхпроводниками, обладают высокой химической стойкостью по отношению к кислороду воздуха, кислотам, расплавленным металлам.
Получаются
нитриды либо при непосредственном
взаимодействии металлов и азота: 2Al
+ N2
 2AlN,
либо действием на оксиды металлов
азотом или аммиаком:
2AlN,
либо действием на оксиды металлов
азотом или аммиаком:
TiО2
+ 2С + ½ N2
 TiN
+ 2СО
TiN
+ 2СО
3Cu2O
+ 2NH3
 2Cu3N
+ 3H2O
2Cu3N
+ 3H2O
Силициды – соединения кремния с металлами и некоторыми неметаллами (KSi, CaSi, SiC, Si3N4, NiSi4). Силициды очень похожи на карбиды, однако они еще больше, чем карбиды приближаются к интерметаллическим соединениям. Для получения силицидов используют следующие методы:
-
непосредственное соединение металлов с порошкообразным кремнием в защитной среде инертных газов
Nа
+ Si
 NаSi
NаSi
-
взаимодействием оксидов металлов с кремнием в вакууме
ВаО
+ 2 Si
 ВаSi
+ SiО
ВаSi
+ SiО
-
восстановлением оксидов карбидом кремния
МеО + SiС = МеS + СО
Силициды Cr, Mo, W, Ti, Zr и др. играют значительную роль при создании жаропрочных сплавов, стабилизируя их при высоких температурах.
Подобными же свойствами обладают бориды этих же металлов. Бориды – соединения металлов и некоторых неметаллов (С, Si) с бором солеобразного (Mg3B2 Mg=B–Mg–B=Mg) и несолеобразного (B4C, CaB6, BaB6, AlB2 и др.) типа.
Многие бориды имеют высокую твердость, химическую стойкость, тугоплавкость (барид циркония ZsB2 плавится при 3040С).
Графические формулы солей
составляются исходя из графических формул соответствующих кислот, заменой атомов водорода в них на атомы металла (с учетом его степени окисления). Например, составление графической формулы средней соли карбоната натрия Nа2СО3 начинают с написания графической формулы угольной кислоты Н2СО3:
Н+2С4+О2-3 
затем заменяют одновалентные атомы водорода, выделенные пунктиром, на одновалентные атомы натрия:
Nа2С4+О2-3 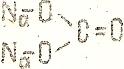
Из формулы следует, что кислород в данном соединении двухвалентен, углерод – четырех. Графическая формула соли двухвалентного металла карбоната кальция СаСО3 будет иметь вид:
Са2+С4+О2-3 
Если в составе молекулы соли несколько кислотных остатков, то ее графическая формула составляется исходя из соответствующего количества графических формул кислот:
Fe3+2 (SO4)2-3 – соль серной кислоты Н2SO4

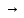

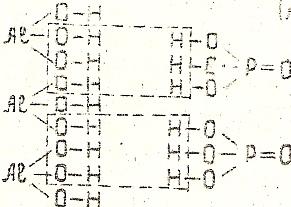
Графические формулы кислых солей, у которых в составе молекул имеются атомы водорода, незамещенные на металл, составляются аналогично формулам средних солей:
КНSO4 – гидросульфат калия
 -
один атом водорода в кислоте
-
один атом водорода в кислоте
замещен на атом калия
Fe(Н2РО4)3 – дигидроортофосфат железа (III):
В каждой из трех молекул фосфорной кислоты два атома водорода остаются в составе молекулы соли, и лишь один замещается атомом железа. В соответствии с валентностью этого металла он замещает три атома водорода (по одному в каждой из трех молекул кислоты).
Графические формулы основных солей, производных гидроксидов, составляются следующим образом: вначале пишется графическая формула гидроксида, затем соответствующее число гидроксильных групп в нем замещается на кислотный остаток. Например: СаОНNО3 – гидроксонитрат кальция
Са(ОН)2
НNО3
[Cr(OH)2]2SO4 - дигидроксосульфат хрома (III).
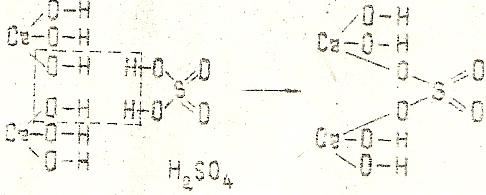
Кислотный остаток серной кислоты замещает по одной гидроксильной группе в каждой молекуле гидроксида железа (III).
(AlОН)3(РО4)2 – гидроксоортофосфат алюминия
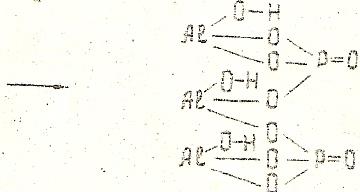
В трех молекулах гидроксида алюминия остается по одной гидроксильной группе, остальные группы ОН замещаются, соответственно, двумя кислотными остатками ортофосфорной кислоты.
Распространенные тривиальные названия некоторых неорганических соединений.
Таблица 3
|
Название |
Формула |
|
1 |
2 |
|
Алебастр |
CaSO4 0,5 H2O |
|
Аммиачная вода |
NH3 (водный раствор) |
|
Ангидрит |
CaSO4 |
|
Антихлор |
Na2S2O3 |
|
Барит |
BaSO4 |
|
Баритовая вода |
Ba(OH)2 (водный раствор) |
|
Белая сажа |
SIO2 |
|
Белый мышьяк |
As2O3 |
|
Бура |
Na2B4O7 10H2O |
|
Гипс |
CaSO4 2H2O |
|
1 |
2 |
|
Глауберова соль |
Na2SO4 10 H2O |
|
Глинозем |
Al2O3 |
|
Горькая соль |
MgSO4 7H2O |
|
Известь гашеная |
Ca(OH)2 |
|
Известь негашеная |
CaO |
|
Известь хлорная |
Ca(ClO)2 2H2O |
|
Каломель |
Hg2Cl2 |
|
Карборунд |
SiC |
|
Кварц |
SiO2 |
|
Киноварь |
HgS |
|
Коагулянт железный |
Fe2(SO4)3 |
|
Корунд |
Al2O3 |
|
Крон свинцовый |
PbCrO4 |
|
Купорос медный |
CuSO4 5H2O |
|
Ляпис |
AgNO3 |
|
Магнезит |
MgCO3 |
|
Магнезия жженая |
MgO |
|
Малахит |
CuCО3 Cu(OH)2 |
|
Нашатырный спирт |
NH3 (водный раствор) |
|
Нашатырь |
NH4Cl |
|
Пергидроль |
H2O 2 (30% раствор) |
|
Поташ |
K2CO3 |
|
Известь пушонка |
Ca(OH)2 |
|
Сода каустическая |
NaOH |
|
Соль кальцинированная |
Na2CO3 |
|
Сода питьевая |
NaHCO3 |
|
Сурик железный |
Fe2O3 |
|
Сухой лед |
CO2 (твердый) |
|
Хромпик |
K2Cr2O7 или Na2Cr2O7 2H2O |
|
Цементит |
Fe3C |
Экспериментальная часть
-
Взаимодействие основного оксида с кислотой.
В пробирку поместить небольшое количество (0,5 г) оксида свинца и 3- 4 мл разбавленной соляной кислоты. Смесь нагревать 2-3 минуты и дать отстояться. Слить жидкость в другую пробирку и охладить ее. наблюдать выпадение в осадок белых кристаллов хлорида свинца. Написать уравнение реакции образования этой соли.
-
Взаимодействие металла с кислотой.
В две пробирки налить по 1мл НСl и бросить в первую из них кусочек железа, во вторую – цинка. Какой газ выделяется? Написать уравнение реакции.
-
Взаимодействие кислоты с основанием. (Реакция нейтрализации).
В фарфоровую чашечку налить 5 мл 2Н раствора соляной кислоты, а затем по каплям приливать 4 мл 2н раствора едкого натра (NаОН) до получения при перемешивании нейтрального раствора (проверить лакмусовой бумажкой). Раствор выпарить над пламенем горелки. Написать уравнение реакции получения хлорида натрия.
-
Взаимодействие кислоты с солью:
-
к 1-2 мл раствора силиката натрия (Nа2 Si О3) прилить по каплям 12%-й раствор соляной кислоты, смесь встряхнуть. Что наблюдается? Написать уравнение реакции;
-
в пробирку с 1-2мл разбавленной соляной кислоты прибавить несколько капель раствора азотнокислого серебра (АgNО3). Что выпадет в осадок? Написать уравнение реакции;
-
в две пробирки прилить 1-2 мл раствора серной кислоты (Н2SO4), затем в первую пробирку добавить раствор хлорида бария ВаСl2, во вторую – раствор нитрата свинца Pb(NO3)2. Каков цвет образовавшихся осадков? Написать уравнение реакции.
-
Взаимодействие кислотного оксида с гидроксидом.
Через
раствор гидроксида кальция Са(ОН)2
(известковую воду) пропустить из аппарата
Киппа углекислый газ (СО2),
полученный в процессе реакции СаСО3
+ 2НСl
= СаСl2
+ СО2
 +
Н2О.
Наблюдать выпадение осадка карбоната
кальция, который при дальнейшем
пропускании СО2
растворяется. Почему? Написать уравнения
соответствующих реакций.
+
Н2О.
Наблюдать выпадение осадка карбоната
кальция, который при дальнейшем
пропускании СО2
растворяется. Почему? Написать уравнения
соответствующих реакций.
-
В две пробирки прилить 2-3 мл раствора нитрата свинца, затем в первую из них добавить раствор хромата калия, во вторую – иодида калия. Каков цвет образовавшихся осадков? Составить уравнения реакций.
-
Взаимодействие соли с гидроксидом:
-
к раствору сульфата меди CuSO4 прилить раствор едкого натра. Отметить цвет полученного осадка, написать уравнение реакции;
-
в пробирку с раствором хлорида кобальта (II) CoCl2 прилить по каплям раствор едкого натра. Обратить внимание на изменение цвета осадка. Составить уравнения реакций, учитывая при этом, что вначале образуется основная соль кобальта, а затем уже – гидроксид кобальта (II).
-
Получение двойной соли.
Смешать в небольшом стаканчике равные объемы насыщенных растворов сульфата алюминия и сульфата калия. Полученный раствор выпарить на слабом огне на одну третью часть и дать остыть. Через некоторое время наблюдать выделение мелких кристаллов алюминиевых квасцов. Написать уравнение реакции.
Замечания и меры предосторожности
-
Кислоты и щелочи наливать следует осторожно. Если кислота или щелочь случайно попала на кожу, необходимо быстро смыть ее большим количеством воды из водопроводного крана, а затем нейтрализовать, соответственно, либо 3-5% раствором гидрокарбоната натрия (NaHCO3), либо 3-5% раствором уксусной кислоты.
-
Отработанные кислоты и щелочи слить в специальные склянки.
-
В специально отведенную склянку следует вылить и растворы солей серебра.
-
При работе следует учитывать также токсичность таких веществ, как соли свинца, хрома (VI), меди.
СТРОЕНИЕ АТОМА
Цель работы состоит в том, чтобы на основе современной теории строения атома научить студентов записывать электронные формулы химических элементов и объяснять их химические свойства.
ПРОГРАММА КОЛЛОКВИУМА
Планетарная модель атома. Строение атома по Бору. Основные представления квантово-механической теории о строении атома. Электронные облака. Волновая функция. Квантовые числа. Главное, орбитальное, магнитное и спиновое квантовые числа. Принцип Паули. Следствия из принципа Паули. Правило Хунда. Электронные структуры атомов. Взаимосвязь строения электронной оболочки атома элемента с его расположением в системе Д.И.Менделеева. Электронные формулы атомов элементов.
В конце XIX века был сделан ряд великих научных открытий, которые привели к огромному прогрессу в понимании строения материи. Для химии особое значение имели открытия рентгеновских лучей (К.Рентген, 1895 г.), явления радиоактивности (А. Беккерель, 1895 г.), электрона (Дж.Дж. Томсона, 1897 г.).
Изучение катодных лучей позволило установить, что электрон характеризуется массой, является носителем наименьшего отрицательного заряда и содержится в атомах всех элементов. Результаты опытов с радиоактивными материалами могли быть объяснены в предположении, что атомы радиоактивных элементов распадаются и превращаются в атомы других элементов. Все это свидетельствовало о сложной природе атома и стимулировало интенсивные исследования его внутренней структуры.
ПЛАНЕТАРНАЯ МОДЕЛЬ АТОМА
Электронейтральность атома при наличии в нем электронов приводила к выводу о существовании в атоме области, несущей положительный заряд.
Изучая
движение
 – частиц, проходящих сквозь тонкие
металлические пластинки в вакууме, Э.
Резерфорд в 1911 году выяснил, что весь
положительный заряд и основная массса
атома сосредоточены в центральной,
очень малой по объему, части атома – в
его ядре. Исходя из этого Резерфорд
предположил, что электроны вращаются
вокруг ядра, причем центробежная сила
электрона равна кулоновской силе
притяжения его к ядру:
– частиц, проходящих сквозь тонкие
металлические пластинки в вакууме, Э.
Резерфорд в 1911 году выяснил, что весь
положительный заряд и основная массса
атома сосредоточены в центральной,
очень малой по объему, части атома – в
его ядре. Исходя из этого Резерфорд
предположил, что электроны вращаются
вокруг ядра, причем центробежная сила
электрона равна кулоновской силе
притяжения его к ядру:
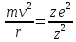 ,
где m
– масса
,
где m
– масса
v – скорость движения электрона
ze – заряд ядра
Теория строения атома по Резерфорду была крупным шагом вперед. Однако она имела следующие недостатки:
Она не могла объяснять устойчивость атома;
С точки зрения теории Резерфорда нельзя было также объяснить природу атомных спектров (по Резерфорду спектры должны быть сплошными, в действительности они являются линейчатыми).
СТРОЕНИЕ АТОМА ВОДОРОДА ПО БОРУ
Исходным положением теории Бора был очень смелый для того времени, противоречащий классическим представлениям постулат о том, что в атоме, который не подвержен достаточно сильным воздействиям, электрон движется, не излучая энергии. Этот постулат объяснял устойчивость атомов (действительно, если энергия электрона при его движении не убывает и радиус вращения, а значит, электрон не упадет на ядро).
Н. Бор предположил также, что электрон движется, не излучая энергии, по определенным орбитам, радиус которых связан со скоростью вращения электрона соотношением

m – масса электрона
h
– постоянная Планка, равна 6,626* Дж*сек
Дж*сек
n – переменная величина, которая может принимать только целочисленные значения (1, 2, 3, 4….).
Если подставить в формулу числовые значения постоянных величин и произвести соответствующие вычисления, то получим:
 (A)
(A)
Зная радиусы «дозволенных» орбит, можно найти энергию электрона из них:

В этой формуле n – единственная переменная величина. Так как n принимает определенные значения (1, 2, 3…), энергия электрона может принимать только определенные значения, т.е. энергия электрона изменяется определенными порциями – квантуется. Расчеты, произведенные Бором для атома водорода, оказались в замечательном согласии с экспериментальными данными.
При
объяснении природы атомных спектров
Н. Бор использовал квантовую теорию
излучения, разработанную М. Планком в
1900 году, согласно которой лучистая
энергия испускается и поглощается
телами не непрерывно, а дискретно, то
есть отдельными порциями – квантами.
При этом энергия Е каждой такой порции
связана с частотой излучения
 соотношением, получившим название
уравнение Планка:
соотношением, получившим название
уравнение Планка:

h – постоянная Планка
Картина
возникновения атомного спектра по Бору
такова. В атомах, не подвергающихся
достаточно сильному внешнему воздействию,
электрон движется по ближайшей к ядру
«дозволенной» орбите, т.е. обладает
минимальной энергией
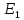 .
Это соответствует самому устойчивому,
нормальному или основному состоянию
атома. Если подвергнуть атомы воздействию
внешнего источника энергии, то электрон
может поглотить квант энергии. При этом
энергия электрона
.
Это соответствует самому устойчивому,
нормальному или основному состоянию
атома. Если подвергнуть атомы воздействию
внешнего источника энергии, то электрон
может поглотить квант энергии. При этом
энергия электрона
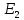 станет большей, чем его энергия в
нормальном состоянии.
станет большей, чем его энергия в
нормальном состоянии.

Атом,
энергия которого больше, чем энергия в
нормальном состоянии, называется
возбужденным атомом. Электрон в
возбужденном атоме движется уже не по
первой «дозволенной» орбите, а по одной
из более удаленных, при чем по тем более
удаленной, чем больший квант энергии
 поглотил электрон. Таким образом,
поглощая квант энергии, электрон с
ближайшей к ядру орбиты переходит на
более или менее удаленную.
поглотил электрон. Таким образом,
поглощая квант энергии, электрон с
ближайшей к ядру орбиты переходит на
более или менее удаленную.
Возбужденное
состояние атома является неустойчивым.
В результате атом вновь переходит в
основное состояние с энергией
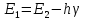 ,
при этом электрон возвращается на
ближайшую орбиту, выделяя квант энергии
,
при этом электрон возвращается на
ближайшую орбиту, выделяя квант энергии
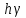 .
Величина
.
Величина
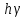 определяется разностью энергий электрона
в возбужденном атоме
определяется разностью энергий электрона
в возбужденном атоме
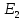 и атоме в основном состоянии
и атоме в основном состоянии
 :
:
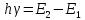 ,
откуда:
,
откуда:
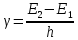
Частота электромагнитного излучения атомов каждого элемента принимает ограниченный ряд значений, поскольку энергетические состояния электрона в атоме могут быть не любыми, а строго определенными. По этой причине атомный спектр не сплошной, а представляет собой совокупность разделенных интервалами спектральных линий. Таким образом, все представления о строении атома по Бору можно кратко сформулировать тремя постулатами:
Электрон может вращаться вокруг ядра не по любым, а только по некоторым определенным круговым орбитам. Эти орбиты получили название стационарных
Двигаясь по стационарной орбите, электрон не излучает электромагнитной энергии.
Излучение происходит при скачкообразном переходе электрона с одной стационарной орбиты на другую. При этом испускается или поглощается квант электромагнитного излучения, энергия которого равна разности энергии атома в конечном и исходном состоянии.
Однако модель Бора, блестяще подтвердившаяся для атома водорода, привела к совершенно разочаровывающим результатам для атомов с двумя и более электронами (и тем более молекул). Непригодность этой модели для описания многоэлектронных систем означала необходимость введения каких-то радикально новых представлений, из которых дискретность состояний связанных электронов вытекала бы как естественное, внутренне присущее им свойство, а не являлось бы, как у Бора, результатом необъяснимых постулированных ограничений, наложенных на законы классического движения частиц.
Основные положения квантовой механики. Современная теория строения атома основана на законах, описывающих движение микрочастиц (микрообъектов). Поскольку масса и размеры микрочастиц чрезвычайно малы по сравнению с массами и размерами макроскопических тел, свойства и закономерности движения отдельной микрочастицы качественно отличаются от свойств и закономерностей движения отдельной микрочастицы, качественно отличаются от свойств и закономерностей движения макроскопического тела, которые были изучены классической физикой. В 20 годы XX века возник новый раздел физики, описывающий движения и взаимодействия микрочастиц – квантовая (или волновая) механика. Создание квантовой механики связано с разработкой концепции о корпускулярно-волновой природе элементарных частиц, образующих атом. Эта теория основана на представлении о квантовой энергии, волновом характере движения микрочастиц и вероятном методе описания микрообъектов.
В 1924 г. французский физик Луи де Бройль выдвинул предположение о том, что все виды материи обладают волновыми свойствами. Он пришел к выводу, что если масса взаимосвязана с энергией в соответствии с уравнением Эйнштейна

то эта энергия должна характеризоваться частотой, описываемой соотношением Планка

Отсюда следует, что

или
после установки вместо
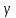 эквивалентного выражения
эквивалентного выражения


Согласно этому уравнению масса фотона, то есть его корпускулярная характеристика, обратно пропорциональна его длине волны, то есть его корпускулярная характеристика, обратно пропорциональна его длине волны, то есть волновой характеристике. В более общем случае де Бройль предложил заменить скорость света на скорость материальной частицы υ
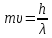
Таким образом, любая движущаяся частица должна обладать волновыми свойствами и характеризоваться длиной волны и частотой, связанной с её движением. Предположение де Бройля получило экспериментальное подтверждение для малых частиц, таких, как электроны и нейтроны, когда удалось обнаружить дифракцию этих частиц на решетке кристаллов.
Кажущуюся двойственную природу микрочастиц объясняет установленный Вернером Гейзенбергом в 1927 году принцип неопределенности: невозможно одновременно определить и скорость (или импульс p= mυ) и положение микрочастицы (её координаты). Математическое выражение принципа неопределенности имеет вид

Произведение
неопределенности положения ( )
и скорости (
)
и скорости (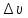 )
никогда не может быть меньше
)
никогда не может быть меньше
 .
.
Из
этого соотношения следует: чем точнее
определены координаты частицы (чем
меньше неопределенность
 ),
тем менее определенной становится
величина её скорости (больше
),
тем менее определенной становится
величина её скорости (больше
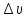 )
и наоборот.
)
и наоборот.
Для
макрочастиц (тяжелых частиц) величина
отношения
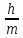 очень мала, поэтому для них справедливы
законы классической механики, в рамках
которых скорость и положение частицы
могут быть точно определенны одновременно.
очень мала, поэтому для них справедливы
законы классической механики, в рамках
которых скорость и положение частицы
могут быть точно определенны одновременно.
Квантование энергии, волновой характер движения микрочастиц – все это показывает, что классическая механика совершенно непригодна для описания поведения микрочастиц. Так, состояние электрона в атоме нельзя представить как движение материальной частицы по какой-либо орбите. Квантовая механика отказывается от уточнения положения электрона в пространстве; она заменяет классическое понятие точного нахождения частицы понятием статистической вероятности нахождения электрона в данной точке пространства или элементе объема dυ вокруг ядра.
ЭЛЕКТРОННОЕ ОБЛАКО
Поскольку
движение электрона имеет волновой
характер, квантовая механика описывает
его движение в атоме при помощи так
называемой волновой функции ψ. В разных
точках атомного пространства эта функция
принимает разные значения. Математически
это записывается равенством
 ,
где
,
где
 - координаты точки. Физический смысл
волновой функции объяснить пока трудно.
Имеет определенный физический смысл
ее квадрат
- координаты точки. Физический смысл
волновой функции объяснить пока трудно.
Имеет определенный физический смысл
ее квадрат
 .
Он характеризует вероятность нахождения
электрона в данной точке атомного
пространства. Величина
.
Он характеризует вероятность нахождения
электрона в данной точке атомного
пространства. Величина
 представляет собой вероятность
обнаружения рассматриваемой частицы
в элементе объема dυ.
представляет собой вероятность
обнаружения рассматриваемой частицы
в элементе объема dυ.
Рис.
1
Очевидно, чем прочнее связь электрона с ядром, тем электронное облако меньше по размерам и плотнее по распределению заряда. Электронное облако часто изображают в виде граничной поверхности (охватывающей примерно 90% электронного облака). Пространство вокруг ядра, в котором наиболее вероятно пребывание электрона, называется орбиталью.
Вычисления вероятности нахождения электрона в данном месте атома (молекулы) и его энергии – сложная математическая проблема. Она решается с помощью волнового уравнения Шредингера.
ВОЛНОВОЕ УРАВНЕНИЕ ШРЕДИНГЕРА
В 1926 году Эрвин Шредингер предположил уравнение, которое в волновой механике играет такую же роль, какую закон Ньютона играет в классической механике.
Уравнение Шредингера связывает волновую функцию ψ с потенциальной энергией электрона и его полной энергией Е:

Где
 – сумма вторых производных волновой
функции по координатам x,
y,
z,
m
– масса электрона, h
– постоянная Планка.
– сумма вторых производных волновой
функции по координатам x,
y,
z,
m
– масса электрона, h
– постоянная Планка.
Различным
функциям
 которые являются решением волнового
уравнения, соответствуют свои значения
энергии
которые являются решением волнового
уравнения, соответствуют свои значения
энергии
 .
Таким образом, квантование энергии
микросистемы непосредственно вытекает
из решения волнового уравнения. Волновая
функция, являющаяся решением Шредингера,
называется орбиталью.
.
Таким образом, квантование энергии
микросистемы непосредственно вытекает
из решения волнового уравнения. Волновая
функция, являющаяся решением Шредингера,
называется орбиталью.
Энергия электрона, момент его количества движения и z-компонента момента составляют вместе полную систему физических величин, определяющих движение электрона в кулоновском поле ядра. Дискретные значения величин выражаются определенными числами, которые называются квантовыми и обозначаются n, l и m. Переменная величина n – это главное квантовое число. Оно же определяет энергию электрона, принимает любое значение натурального ряда чисел: 1, 2, 3, 4… Остальные две переменные величины l и m могут принимать лишь строго определенные значения в зависимости от значения n и поэтому, также как и n, называются квантовыми числами.
Орбиталь можно однозначно описать с помощью этих квантовых чисел. Итак, энергия электрона в атоме величина квантовая, возможные энергетические состояния электрона в атоме определяются величиной главного квантового числа в соответствии с уравнением:
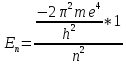
Где m – масса электрона, e – заряд электрона.
Наименьшей энергией электрон обладает при n = 1, с увеличением n энергия электрона возрастает.
Состояние электрона, характеризующееся определенным значением главного квантового числа, принято называть энергетическим уровнем электрона в атоме: при n=1 электрон находится на первом энергетическом уровне, при n = 2 на втором и т.д.
Главное квантовое число определяет размеры электронного облака. Для того, чтобы увеличить размеры электронного облака нужно часть его удалить на большее расстояние от ядра. Этому препятствуют силы электростатистического притяжения электрона к ядру, преодоление которых требует затраты энергии. Поэтому большим размерам электронного облака соответствует более высокая энергия электрона в атоме, и следовательно, большее значение главного квантового числа n. Электроны, характеризующиеся одним и тем же значением главного квантового числа, образуют в атоме электронные облака приблизительно одинаковых размеров.
Для энергетических уровней электрона в атоме, соответствующих различным значениям n, приняты следующие обозначения:
|
гларное квантовое число n |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
обозначение энергетического уровня |
K |
Z |
M |
N |
O |
P |
Q |
Орбитальное квантовое число. Изучение атомных спектров показало, что спектральные линии мультиплетны, т.е. они состоят из нескольких тонких близко расположенных линий. Мультиплётность (расщепление) линий свидетельствует о том, что электроны, находящиеся на одном и том же уровне, несколько отличаются по запасу энергии. Следовательно, энергетические уровни представляют совокупность энергетических подуровней. Наличие подуровней обуславливается различием форм электронных облаков. Форму электронного облака характеризует орбитальное (азимутальное) квантовое число l. Оно определяет значение (М) орбитального момента количества движения электрона

и принимает значения от 0 до (n – 1).
Орбитальное квантовое число обычно обозначают буквами в соответствии со схемой:
|
значение орбитального квантового числа |
0 |
1 |
2 |
3 |
4 |
5 |
|
обозначение |
s |
p |
d |
f |
g |
h |
Соотношения между значениями n и l приведены в таблице 2.
|
n |
l |
Обозначение орбитали электронного облака |
Число значений l при данном n |
|
1 |
0 |
1s |
1 |
|
2 |
0,1 |
2s , 2p |
2 |
|
3 |
0,1,2 |
3s , 3p , 3d |
3 |
|
4 |
0, 1, 2, 3 |
4s , 4p , 4d , 4f |
4 |
|
5 |
0, 1, 2, 3, 4 |
5s , 5p , 5d , 5f , 5g |
5 |
Таким образом, для электрона первого энергетического уровня (n=1) возможна только одна форма электронного облака, для второго энергетического уровня (n=2) возможны две формы, для третьего уровня (n=3) – три и т.д. Согласно квантомеханическим расчетам s – орбитали имеют форму шара, p – орбитали – форму гантели, d – и f – орбитали более сложные формы. Формы граничных поверхностей s-, p-, d- орбиталей показанных на рисунке 2. На изображении граничной поверхности часто указывают также знак волновой функции.
Для обозначения состояния электрона главное квантовое число ставят перед символом орбитального квантового числа. Например, 4s означает электрон, у которого n=4 и l=0 (облако имеет форму шара); 2p означает электрон, у которого n=2 и l=1(облако имеет форму гантели) и т.д.
МАГНИТНОЕ КВАНТОВОЕ ЧИСЛО. ПРОСТРАНСТВЕННАЯ ОРИЕНТАЦИЯ ЭЛЕКТРОННЫХ ОБЛАКОВ
Магнитное
квантовое число
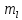 определяет значение
определяет значение
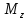 проекции орбитального момента количества
движения на выделенное направление,
например, на ось z
проекции орбитального момента количества
движения на выделенное направление,
например, на ось z
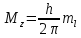
 характеризует
пространственное расположение электронных
облаков и может иметь следующие значения:
0, ±1, ±2,…. ±l.
характеризует
пространственное расположение электронных
облаков и может иметь следующие значения:
0, ±1, ±2,…. ±l.
Число значений магнитного квантового числа зависит от орбитального квантового числа и определяется соотношением 2l+1.
|
Орбитальное квантовое число (l) |
Магнитное
квантовое
число ( |
Число
орбиталей (облаков) с данным значением
|
|
0 |
0 |
1 |
|
1 |
1, 0, -1 |
3 |
|
2 |
2, 1, 0, -1, -2 |
5 |
|
3 |
3, 2, 1, 0, -1, -2, -3 |
7 |
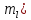
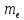
s
– состоянию отвечает одна орбиталь, p
– состоянию – три, d
– состоянию – пять, f
– состоянию – 7 и т.д. Орбитали с одинаковой
энергией называются вырожденными. Общее
число орбиталей данного энергетического
уровня равно
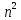 .
.
По
характеру ориентации в пространстве p
– орбитали обозначаются
 .
d
– Орбитали, ориентированные своими
лопастями по осям координат, обозначают
.
d
– Орбитали, ориентированные своими
лопастями по осям координат, обозначают
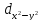 и
и
 ,
d
–орбитали, ориентированные лопастями
между осями координат, обозначают
,
d
–орбитали, ориентированные лопастями
между осями координат, обозначают
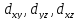 (рис.2).
(рис.2).

Рис. 2 Формы s -, p-, d –орбиталей
Спиновое квантовое число
Изучение
тонкой структуры атомных аспектов
показало, что кроме различия размеров
облаков, их формы и характера расположения
друг относительно друга электроны
различаются спином. Упрощенно спин
можно представить как собственное
вращение электрона вокруг своей оси.
Для характеристики спина электрона
вводится четвертое квантовое число
 , называемое спиновым. Оно имеет значения
, называемое спиновым. Оно имеет значения
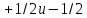 .
.
Подведем
некоторые итоги сказанному. Состояние
электрона в атоме может быть полностью
описано с помощью четырех квантовых
чисел: n,
l,
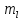 ,
,
 .
Они характеризуют энергию, спин
электрона, размер, форму и положение в
пространстве электронного облака.
.
Они характеризуют энергию, спин
электрона, размер, форму и положение в
пространстве электронного облака.
Принцип Паули
В
1925 году Паули высказал принцип: в атоме
не может быть двух электронов, имеющих
одинаковый набор всех четырех квантовых
чисел. Иными словами, данными значениями
квантовых чисел n,
l,
 ,
,
 может характеризоваться только один
электрон. Для любого другого электрона
в атоме должно быть иным значение хотя
бы одного из квантовых чисел.
может характеризоваться только один
электрон. Для любого другого электрона
в атоме должно быть иным значение хотя
бы одного из квантовых чисел.
Из
принципа Паули непосредственно вытекает,
что на одной орбитали может находиться
лишь два электрона с
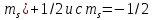 .
Следовательно, в s
– состоянии /одна орбиталь / может быть
только два электрона, в p
– состоянии / три орбитали / - шесть, в d
– состоянии - / пять орбиталей / десять,
в f
– состоянии /семь орбиталей / - четырнадцать
электронов и т.д.
.
Следовательно, в s
– состоянии /одна орбиталь / может быть
только два электрона, в p
– состоянии / три орбитали / - шесть, в d
– состоянии - / пять орбиталей / десять,
в f
– состоянии /семь орбиталей / - четырнадцать
электронов и т.д.
Из
значений квантовых чисел вытекает, что
общее квантовое число возможных орбиталей
на каждом уровне равно
 , где n
- главное квантовое число. Если учесть,
что на каждой орбитали может быть только
два электрона с противоположными
спинами, то максимальное число электронов,
соответствующее данному энергетическому
уровню, определяется формулой
, где n
- главное квантовое число. Если учесть,
что на каждой орбитали может быть только
два электрона с противоположными
спинами, то максимальное число электронов,
соответствующее данному энергетическому
уровню, определяется формулой
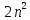 .
Отсюда следует, что на первом энергетическом
уровне должно находиться не более 2
электронов, на втором – не более 8 и
т.д., как указано в таблице 3.
.
Отсюда следует, что на первом энергетическом
уровне должно находиться не более 2
электронов, на втором – не более 8 и
т.д., как указано в таблице 3.
|
n |
l |
|
|
Емкость подуровня |
Емкость уровня |
|
1 |
0/s/ |
0 |
±1/2 |
2 |
2 |
|
2 |
0/s/ 1/p/ |
0 +1, 0, -1 |
±1/2 |
2 6 |
8 |
|
3 |
0/s/ 1/p/ 2/d/ |
0 +1, 0, -1 +2, +1, 0, -1, -2 |
±1/2 |
2 6 10 |
18 |
|
4 |
0/s/ 1/p/ 2/d/ 3/f/ |
0 +1, 0, -1 +2, +1, 0, -1, -2 +3, +2, +1, 0, -1, -2, -3 |
±1/2 |
2 6 10 14 |
32 |
Принципы построения электронных оболочек атомов
Наиболее
устойчиво состояние атома, в котором
электроны имеют наиболее низкую энергию,
т.е. находятся в наиболее близких ядру
слоях. Последовательность энергетических
состояний в порядке возрастания энергии
орбитали многоэлектронных атомов можно
представить следующим образом: 1s
< 2s
< 2p
< 3s
< 3p
< 4s
< 3d
< 4p
< 5s
< 4d
< 5 p
< 6s
<5d
 4f
< 6p
< 7s
<6d
4f
< 6p
< 7s
<6d
 5f
и т.д.
5f
и т.д.
Порядок заполнения орбиталей данного подуровня подчиняется правилу Хунда: устойчивому состоянию атома соответствует такое распределение электронов в пределах энергетического уровня, при котором абсолютное значение суммарного спина атома максимально.
Иными словами, орбитали данного подслоя заполняются сначала по одному, затем по второму электрону. Электроны с противоположными спинами на одной и той же орбитали образуют двухэлектронное облако (спариваются) и их суммарный спин равен нулю.
Электронные структуры атомов
-
Отметим, что правило Хунда не запрещает другого распределения электронов в пределах подуровня. Оно лишь утверждает, что максимальное значение суммарного спина атома соответствует устойчивому, т.е. невозбужденному состоянию, в котором атом обладает наименьшей возможной энергией, при любом другом распределении электронов энергия атома находиться в возбужденном, неустойчивом состоянии.
-
В каждом энергетическом уровне заполнение электронами начинается с s – подуровня. Элементы, в атомах которых заполняется s – подуровень, называются s – элементами – это элементы и 2 главных подгрупп, p – элементы (заполняется p – подуровень) – это элементы 3, 4, 5, 6, 7, 8 групп главных подгрупп (кроме гелия): d- элементы (заполняется d – подуровень с отстаиванием на один период: в 4-ом периоде заполняется 3d, в пятом – 4d, в шестом – 5d, в седьмом – 6d ) – это элементы всех побочных подгрупп. f – элементы (заполняется f – подуровень с отстаиванием на 2 периода, в шестом периоде заполняется 4f, в седьмом - 5f) – это лантаноиды и актиноиды.
|
|
1 |
2 |
3 |
4 |
5 |
6 |
|
|
|
|
|
f |
d |
s |
|
|
|
|
|
d |
p |
|
|
|
|
|
|
p |
s |
|
|
|
|
|
d |
s |
|
|
|
|
|
|
p |
|
|
|
|
|
|
|
s |
|
|
|
|
|
|
p |
|
|
|
|
|
|
|
s |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
s |
|
|
|
|
|
 n=
n=
При составлении схем распределения электронов в атоме используется следующими обозначениями: клетка – орбиталь, стрелка – электрон, направление его стрелки – ориентация его спина. Первый период системы Менделеева состоит из двух элементов. В атоме водорода 1 электрон и он должен находиться на первом энергетическом уровне, т.е. электронная формула «невозбужденного» атома водорода 1S.
ыы .
В атоме лития три электрона. Два из них,
как в атоме гелия, займут 1s
– ячейку, а третий, в соответствии с
принципом Паули, займет другую ячейку.
Так как самая выгодная после 1s
– ячейки – это 2s
– ячейка, ее и займет третий электрон:
.
В атоме лития три электрона. Два из них,
как в атоме гелия, займут 1s
– ячейку, а третий, в соответствии с
принципом Паули, займет другую ячейку.
Так как самая выгодная после 1s
– ячейки – это 2s
– ячейка, ее и займет третий электрон:



 S
P
n=2
S
P
n=2
 n=1
n=1
Аналогично введены электронные структуры атомов бериллия и бора. Четвертый электрон бериллия делит ячейку 2s с другим s – электроном. Пятый электрон бора «вынужден» занять менее энергетически выгодную 2p – ячейку:


 S
P S P
S
P S P
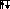 n=2
n=2
n=2
n=2
Be
1 n=1 B 1
n=1 B 1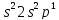 n=1
n=1
Ниже приведены электронные формулы некоторых атомов элементов 2 периода.
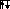



 S
P
S
P
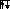 n=2
N
n=2
N

N n=1
Начиная с кислорода, 2 p – орбитали заполняются по второму электрону:
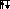




 n=2
O
n=2
O
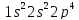
 N
n=1
N
n=1
У неона восемь внешних электронов образуют высокосимметричную структуру из четырех двухэлектронных облаков:


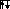





 n=2
Ne
n=2
Ne
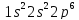
Ne n=1
В атоме неона достигается максимально возможное число электронов второго энергетического уровня.
У элементов 3 периода заполняется 3 энергетический уровень /n=3/, состоящий из 3s -, 3p - и 3d – орбиталей.



3s 3p 3d
У двух первых элементов /Na и Mg/ заполняются s – орбитали, у последующих 6 элементов три p – орбитали, например:

 Na
Na
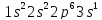





 P
P
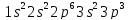




 Ar
Ar

У атомов Na, P и Ar в двух первых слоях повторяется структура атома неона, поэтому здесь показан характер заполнения только внешнего слоя. У Ar внешний уровень заполнен не полностью. Остаются не заполненными 3d – орбитали.
У элементов 4 периода заполняется четвертый энергетический уровень /n=4/. У двух первых элементов, калия и кальция, заполняется орбиталь s – подуровня. Появление электрона в 4 s –подуровне при наличии 3 d – орбиталей обуславливается экранированием ядра плотным и симметричным электронным слоем. У элементов следующих за Ca энергетически более выгодным является 3d – состояние:
|
Sc |
|
|
V |
|
|
Mn |
|
|
Zn |
|
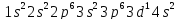

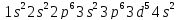

После заполнения у последующих шести элементов /Ga – Kr/ заполняется p – орбитали внешнего слоя, т. е. 4-го.
|
Ga |
|
|
Kr |
|
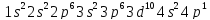

Таким образом, 4-й период начинается двумя s – элементами и заканчивается шестью p – элементами, но в отличии от 2-го и 3-го периодов между s – и p – элементами распологаются десять d – элементов.
В 5-м периоде заполнение электронных орбиталей происходит как в 4-м периоде, а именно: у двух первых /s –элементов Rb и Sr/ заполняется s –орбиталь, а у последующих 10 элементов заполняются d – орбитали предвнешнего энергетического уровня и у последних 6 элементов – 5p – орбитали внешнего энергетического уровня.
|
Rb |
|
|
Tc |
|
|
Xe |
|
|
|
|



Шестой
период, как и предыдущие, начинается с
двух s
– элементов /Cr,
Ba/.
У Za
/ z=57,
расположенного непосредственно после
Ba,
появляется 5d
– электрон и его электронная структура
соответствует:
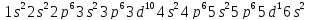 . Однако уже у следующего за лантаном
элемента Ce/z=58/
начинается застройка подуровня 4f
и его электронная структура:
. Однако уже у следующего за лантаном
элемента Ce/z=58/
начинается застройка подуровня 4f
и его электронная структура:
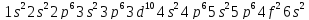
Таким образом, начиная с церия, происходит последовательное заполнение всех орбиталей 4f – подуровня. Расположенные в этой части шестого периода 14 лантаноидов относятся к f – элементам и близки по свойствам к лантану. Характерной особенностью построения электронных оболочек их атомов является то, что при переходе к последующему f – элементу новый электрон занимает место в глубоко расположенном, третьем снаружи слое. Благодаря отсутствию у атомов лантаноидов существенных различий в структуре внешнего и предвнешнего электронных слоев, все лантаноиды проявляют большое сходство в химических свойствах.
Заполнение 5d – подуровня, начатое у лантана, возобновляется у гафния /z=72/ и заканчивается у ртути /z=80/. После этого, как и в предыдущих периодах, располагается шесть p – элементов. Здесь происходит построение 6p – подуровня: оно начинается у галлия /z=81/ и заканчивается у благородного газа радона /z=86/, которым и завершается шестой период.
Седьмой пока незавершенный период системы элементов, построен аналогично шестому. После двух s – элементов /Fr и Ra/ и одного d – элемента /Ac/ здесь расположено 14f – элементов, свойства которых проявляют известную близость к свойствам актиния. Эти элементы, начиная с Th и кончая Zr, обычно объединяют под общим названием актиноидов. Непосредственно за актиноидами расположен Ku и элемент с z=105. Оба эти элементы искусственно получены группой ученных во главе с академиком Флеровым, они принадлежат к d – элементам и завершают известную часть периодической системы элементов.
ХИМИЧЕСКАЯ СВЯЗЬ
Наиболее важными характеристиками химической связи является длина связи (межъядерные расстояния), направленность связи, энергия связи.
Энергия химической связи
Мерой прочности химической связи является энергия связи. Ее величина определяется работой, необходимой для разрушения связи, или количеством энергии, выделившейся при образовании вещества из отдельных атомов. Для двухатомных молекул энергия связи равна по величине энергии диссоциации. Например, энергия связи Н-Н в молекуле водорода равна 435 кДж/ моль ( 104 ккал/ моль). Это значит, что при образовании1 моля газообразного водорода из изолированных атомов по уравнению
Н
+ Н =
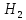 + 435 кДж/моль
+ 435 кДж/моль
Выделяется
435 кДж/моль. Такое же количество энергии
должно быть затрачено на распад 1 моля
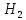 до атомарного состояния (энергия
диссоциации).
до атомарного состояния (энергия
диссоциации).
При
образовании многоатомных молекул,
содержащих одинаковые связи (например,
молекул
 или воды), средняя энергия связи в
пересчете на 1 моль вещества определяется
делением энергии образования этого
вещества из изолированных атомов на
число связей. Например:
или воды), средняя энергия связи в
пересчете на 1 моль вещества определяется
делением энергии образования этого
вещества из изолированных атомов на
число связей. Например:
С
+ 4Н =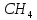 + 1647 кДж/моль
+ 1647 кДж/моль
2Н
+ О =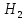 О
+ 924 кДж/моль
О
+ 924 кДж/моль
Отсюда средние энергии связей С-Н и О-Н равны, соответственно, 1647 : 4 = 412 кДж/моль и 924 : 2 = 462 кДж/моль. Для определения энергии единичной связи необходимо средние энергии разделить на число Авогадро.
 Все
связи в каждой из рассмотренных молекул
энергетически равноценны. Однако, при
последовательном отрыве атомов истинная
энергия разрыва связи (энергия диссоциации)
может существенно отличаться от средней.
Так, отрыв первого атома водорода от
молекулы воды по уравнению
Все
связи в каждой из рассмотренных молекул
энергетически равноценны. Однако, при
последовательном отрыве атомов истинная
энергия разрыва связи (энергия диссоциации)
может существенно отличаться от средней.
Так, отрыв первого атома водорода от
молекулы воды по уравнению
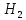 О
Н + ОН – 493 кДж/моль с образованием
атомарного водорода и свободного
радикала ОН, требует затраты энергии
493 кДж/моль. Для отрыва второго атома
водорода необходимо затратить лишь 430
кДж/моль. В процессе последовательной
диссоциации могут ослабляться оставшиеся
связи (
О
Н + ОН – 493 кДж/моль с образованием
атомарного водорода и свободного
радикала ОН, требует затраты энергии
493 кДж/моль. Для отрыва второго атома
водорода необходимо затратить лишь 430
кДж/моль. В процессе последовательной
диссоциации могут ослабляться оставшиеся
связи (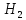 О)
или упрочняться (
О)
или упрочняться (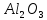 ),
а также может наблюдаться более сложная
зависимость. Так последовательный отрыв
атомов водорода от метана связан с
затратой энергии, равной соответственно
426, 368, 518, 333 кДж/моль.
),
а также может наблюдаться более сложная
зависимость. Так последовательный отрыв
атомов водорода от метана связан с
затратой энергии, равной соответственно
426, 368, 518, 333 кДж/моль.
Однако,
для любого вещества средняя арифметическая
величина совпадает со средней энергией
связи. Так, для

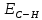 =(426
+ 368 + 518 + 333) : 4 = 412 кДж/моль
=(426
+ 368 + 518 + 333) : 4 = 412 кДж/моль
Длина химической связи
Под длиной связи понимают расстояние между центрами ядер атомов в молекуле, когда силы притяжения уравновешены силами отталкивания и энергия системы минимальна.

 Химическая
связь образуется только в том случае,
если при сближении атомов (двух или
большего числа) полная энергия системы
(сумма кинетической и потенциальной
энергий) понижается. Рассмотрим
зависимость потенциальной энергии Е
системы, состоящей из 2-х атомов водорода
(рис.1) с параллельными (кривая 1) и
антипараллельными (кривая 2) спинами от
расстояния (z)
между ядрами этих атомов.
Химическая
связь образуется только в том случае,
если при сближении атомов (двух или
большего числа) полная энергия системы
(сумма кинетической и потенциальной
энергий) понижается. Рассмотрим
зависимость потенциальной энергии Е
системы, состоящей из 2-х атомов водорода
(рис.1) с параллельными (кривая 1) и
антипараллельными (кривая 2) спинами от
расстояния (z)
между ядрами этих атомов.
Е
800
400 1 отталкивание
 z,
z,

-400 притяжение
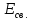
2
Рис.1. Изменение потенциальной энергии в системе из 2-х атомов водорода в зависимости от расстояния между ядрами.
При
совпадающем направлении спинов сближение
атомов приводит к непрерывному возрастанию
энергии системы. В этом случае для
сближения атомов требуется затрата
энергии, так что такой процесс оказывается
энергетически невыгодным, и химическая
связь между атомами не возникает. При
противоположно направленных спинах
сближение атомов до расстояния
 сопровождается уменьшением энергии
системы. При z=
сопровождается уменьшением энергии
системы. При z=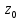 система обладает наименьшей потенциальной
энергией, т. е. находится в наиболее
устойчивом состоянии: дальнейшее
сближение атомов вновь приводит к
возрастанию энергии. Но это не означает,
что в случае противоположно
направленныхспинов атомных электронов
образуется молекула
система обладает наименьшей потенциальной
энергией, т. е. находится в наиболее
устойчивом состоянии: дальнейшее
сближение атомов вновь приводит к
возрастанию энергии. Но это не означает,
что в случае противоположно
направленныхспинов атомных электронов
образуется молекула
 .
.
Образование химической связи между атомами водорода является результатом «перекрывания» электронных облаков, происходящего при сближении взаимодействующих атомов.
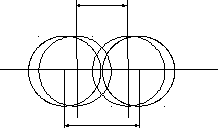 О,74
О,74
1,06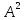
Рис. 2.
Вследствие
такого взаимопроникновения плотность
отрицательного электрического заряда
в межядерном пространстве возрастает.
Положительно заряженные ядра атомов
протягиваются к области перекрывания
электронных облаков, что приводит к
образованию молекулы. При этом длина
химической связи
 =0, 74
=0, 74
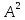 .
.
Метод валентных связей
Для объяснения механизма образования химической связи наиболее широкое распространение получили два метода: метод валентных связей (В.С.) и метод молекулярных орбиталей (М.О.). Метод валентных связей (локализованных электронных пар) исходит из положения, что каждая пара атомов в молекуле удерживается вместе при помощи одной или нескольких общих электронных пар. Так происходит при образовании ковалентной связи. Следовательно, согласно методу В.С. химическая связь локализирована между двумя атомами, т.е. она двухцентровая, двухэлектронная. Изображают такую связь в структурных формулах соединений в виде черточек:

 Н
– Н :N
≡ N:
:О:
Н
– Н :N
≡ N:
:О:

 H
H
H
H
Насыщенность связи
Одно из важнейших свойств химической связи – её насыщаемость. В следствие насыщаемости связи молекулы имеют определённый состав и существует в виде самостоятельных частиц с определённый состав и существует в виде самостоятельных частиц с определённой структурой.
Пребывание двух электронов с противоположными спинами в поле действия двух ядер энергетически выгоднее, чем нахождение каждого электрона в поле своего ядра, поэтому в образовании ковалентной связи принимают участие все одноэлектронные облака:

Например, как видно из электронных формул, атомы кислорода и азота могут соединяться с двумя и тремя, соответственно, одновалентными атомами водорода, атом углерода в возбуждённом состоянии с четырьмя атомами водорода.
Бор в возбуждённом состоянии имеет такую электронную конфигурацию:
![]()
В соответствии с числом непарных электронов атомы В, С, N элементов второго периода могут образовывать, соответственно, три, четыре и три ковалентные связи.
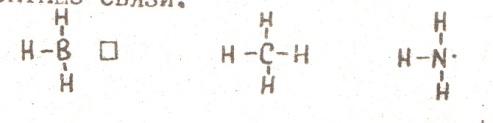
В молекуле BH3 дефицит электронов, атом бора имеет свободную орбиталь. В молекуле NH3 у атома азота имеется неподавленная (несвязывающая) электронная пара. Поэтому молекула BH3 может являться акцептором электронной пары, а молекула NH3 - донором электронной пары. Иными словами, центральные атомы той и другой молекулы способны к образованию четвёртой ковалентной связи по донорно-акцепторному механизму:

Из сопоставления следующих структур:
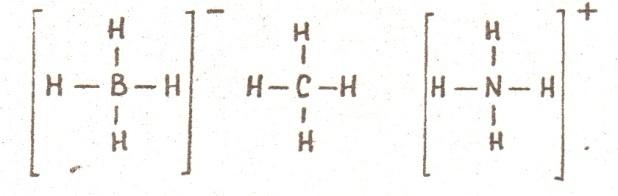
Видно, что атомы бора, углерода и азота в этих соединениях четырёхвалентны. Таким образом, валентность элемента в данном случае определяется числом орбиталей, использованных при образовании химической связи.
Следует отметить, что в ионах BH4- и NH4+ все четыре связи равноценны и неразличимы, следовательно, заряд в них делокализован (рассредоточен) по всему комплексу.
У элементов 2-го периода имеется четыре ковалентные орбитали, поэтому максимальное число ковалентных связей равно 4.
У элементов 3-го периода валентными являются одна s-, три р- и пять d-орбитали, поэтому максимальная валентность этих элементов могла бы быть равной 9. Но наибольший вклад в образование химической связи вносят лишь 6 первых орбиталей внешнего уровня:
![]()
Поэтому максимальная валентность элементов 3-го периода, а также р-элементов больших периодов равна 6, для d-элементов максимальная валентность равна 9.
/Необходимо заметить, что особой чёткости в определении валентности элементов 3-7 периодов пока нет/.
Направленность связи.
Поскольку электронные облака имеют различную форму, их взаимное перекрывание может осуществляться разными способами. В зависимости от способа перекрывания и симметрии образующегося облака различают ϭ, π, δ-связи.

Сигма-связи, (рис. 3) (а) образуется при перекрывании электронных облаков вдоль оси связи. Пи-связи(б) возникают при перекрывании электронных облаков по обе стороны оси связи. Дельта-связи(с) образуются приперекрывании всех четырёх лопастей d-электронных облаков, расположенных в параллельных плоскостях.
Химическая связь образуется лишь в том случае, когда волновые функции перекрывающихся орбиталей имеют одинаковый знак. Исходя из условий симметрии можно сказать, что электроны s-орбиталей могут участвовать лишь в ϭ-связывании, р-электроны в ϭ- и π-связывании, а в d-электроны в ϭ-, π-, δ-связывании. Для f-орбиталей способы перекрывания ещё разнообразнее. Поскольку электронные облака(кроме s-облака) направлены в пространстве, то химические связи, образуемые с их участием пространственно направлены.
Так, например, связи образованные р-орбиталями одного и того же атома, если не принимать во внимание побочных факторов, располагаются под углом в 90 градусов друг к другу. Пространственное расположение ϭ-связей определяет пространственную конфигурацию молекулы. Координационное число центрального атома определяет число ϭ-связей. Координационное число – это количество атомов или группировок, непосредственно связанных с центральным атомом. Например, в соединениях CF4, CO2, H2O координационные числа центральных атомов равны 4, 2, 2.
Гибридизация атомных орбиталей.
Обычно атомы образуют связи за счёт электронов разных энергетических состояний. Например, у атомов Вех /2s1 2p1/, Вх /2s1, 2p2/ и Сх /2s1, 2p3/ в образовании связей одновременно принимают участие как s-, так и р-электроны. Несмотря на различие форм электронных облаков, связи, образованные с их участием, оказываются равноценными и расположенными симметрично. Каким же образом неравноценные по исходному состоянию электроны образуют равноценные химические связи. Ответ на этот вопрос даёт представление о гибридизации валентных орбиталей.
Согласно этому представлению химические связи образуют электроны на «чистых», а «смешанных», т.е. гибридных орбиталей. Иначе говоря, при гибридизации первоначальная форма и энергия орбиталей (электронных облаков) изменяется и образуются орбитали(облака) новой, уже одинаковой формы и энергии.
Характер гибридизации орбиталей центрального атома и их пространственное расположение определяет пространственную структуру молекул и комплексных ионов. При гибридизации одной s- и одной р-орбиталей возникает такое же количество(т.е. две) гибридные sp-орбитали (рис.4)

Так, у атома Вех 2s↑ ↑2p− − при образовании молекулы ВеСI2 возникают две sp-гибридные орбитали(рис.5а), расположенные симметрично под углом в 180 градусов. Форма молекулы – линейна. Гибридные орбитали имеют большую вытянутость по одну сторону ядра, значит они будут в большей степени перекрываться электронными облаками взаимодействующих с ними атомов.
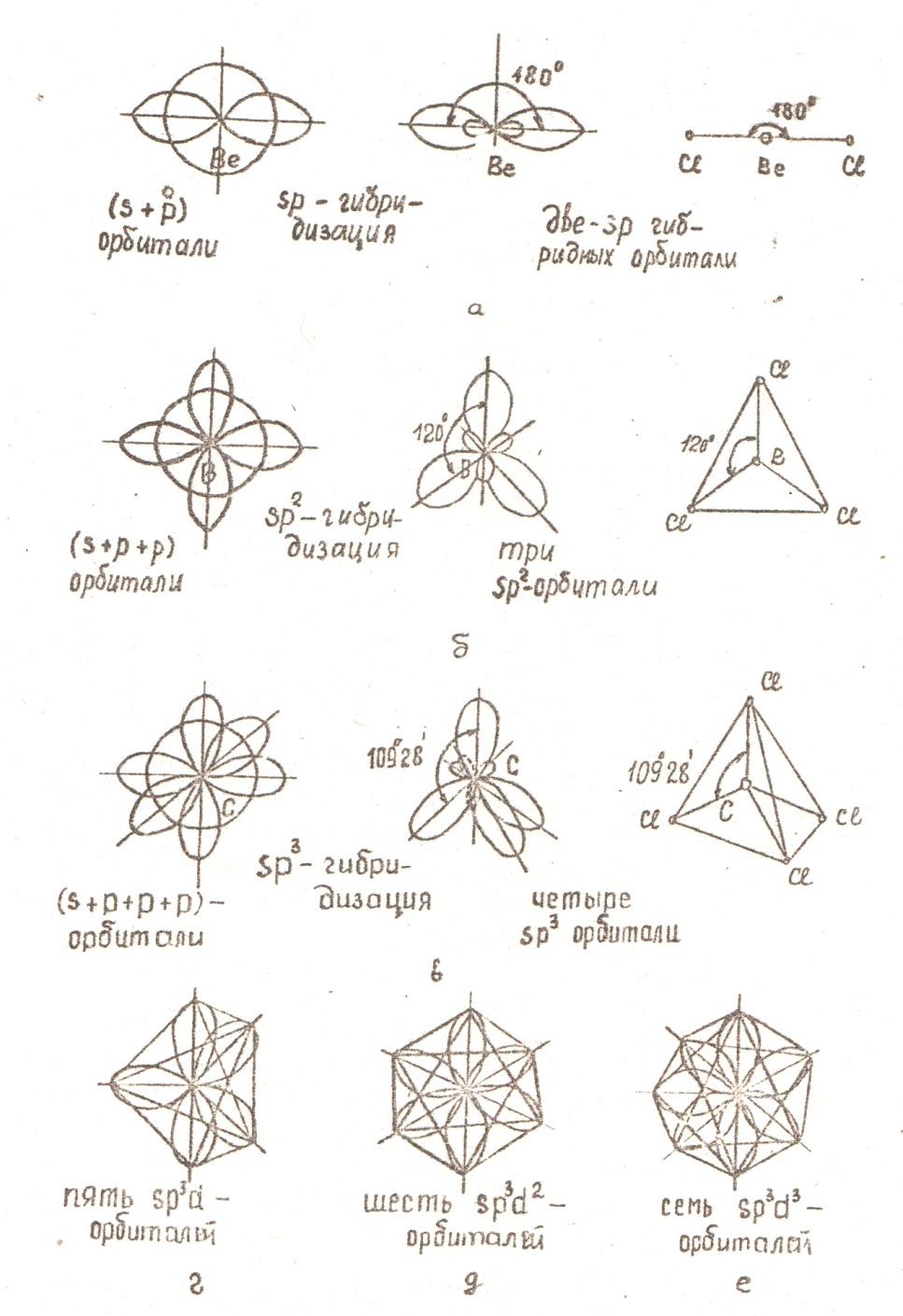
Рис.5 Гибридизация валентных орбиталей.
Следовательно, химическая связь, образуемая с участием электрона гибридной орбитали, должна быть более прочной, чем за счёт участия электронов отдельных s- и р-орбиталей. Sp-гибридизация происходит также при образовании галогенидов цинка, кадмия и ртути (ZnX2, CdX2, HgX2). В возбуждённом состоянии атомы этих элементов имеют по два неспаренных электрона на внешнем уровне, один из которых на s-, другой на р-подуровнях. При возникновении химической связи эти две различные орбитали преобразуются в две одинаковые гибридные (sp-орбитали). Молекулы эти линейные и обе связи с атомами галогена имеют одинаковую длину (X-Zn-X, X-Cd-X и т.д.). Гибридизация одной s- и двух р-орбиталей (sp-гибридизация) имеет место при образовании соединений бора (BCI3 Bx ↑2s ↑↑2p-)
Из одной s- и двух р-орбиталей образуются три эквивалентные sp2-гибридные орбитали, расположенные в одной плоскости под углом 120 градусов друг к другу (рис.5б). Этот вид гибридных орбиталей оказался очень важным для описания двойной связи. Например, молекула этилена С2Н4 по всем данным построена следующим образом: у каждого атома углерода одна р-орбиталь(р2) не гибридизована, а две образуют с s-орбиталью три гибридные орбитали sp2-типа. При образовании молекулы этилена один sp2-электрон от каждого атома углерода идёт на образование ϭ-связи между атомами С-С. Два других sp2-электрона расходуются для связывания атомов водорода(образуются ещё по 2ϭ-связи). Оставшиеся «чистые» орбитали р- образуют π-связи.

Рассмотрим
строение карбонат - иона СО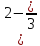 .
.
Атом
углерода Сх
↑2s
↑ ↑
связан с тремя атомами кислорода. В
образовании связи с одним атомом углерода
должны участвовать шесть непарных
электронов от 3-х атомов кислорода.
Четыре из них образуют электронные пары
с непарными атомами углерода
↑
связан с тремя атомами кислорода. В
образовании связи с одним атомом углерода
должны участвовать шесть непарных
электронов от 3-х атомов кислорода.
Четыре из них образуют электронные пары
с непарными атомами углерода
![]()
Но, согласно изложенному выше, химическая связь образоваться лишь в случае полной насыщаемости связи, т.е. оставшиеся 2 непарных электрона кислорода образуют связь за счёт спаривания с двумя «чужими» присоединившимся к данному радикалу электронами (СО3)2-. Координационное число углерода – три. То есть, углерод образует 3ϭ-связи с атомами кислорода, что соответствует sp2-гибридизации атома углерода. Один негибридизованный р-элктрон атома углерода идёт на образование π-связи. Тип гибридизации sp2 говорит о том, что пространственная конфигурация иона [СО3]2- плоский треугольник
![]()
Недостаток такого изображения заключается в том, что π-связь здесь обозначена локализованной. На самом деле все три связи С-О абсолютно равноценны, π-связь делокализована, т.е. равномерно распределена на все 3ϭ-связи. Можно изобразить делокализацию π-связи следующим образом:
![]()
Вследствие
делокализации π-электронного облака
порядок связи С=О составляет примерно
1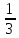 .
.
Гибридизация одной s- и трёх р-орбиталей приводит к sp3-гибридизации. Например, при образовании молекулы CCI4 у атома углерода подвергаются гибридизации 4 орбитали
1s+3p Cx ↑2s ↑↑2p↑ (рис.5,в)
Образованные
четыре гибридные sp3-орбитали
симметрично ориентированы в пространстве
к четырём вершинам тетраэдра под углом
109о28`.
Подобную структуру имеют рассмотренные
выше ионы NH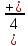 и ВН
и ВН .
Кроме рассмотренных возможны и другие
типы гибридизации валентных орбиталей.
Гибридизация одной s-,
3-х р- и одной d-орбиталей
(PF5
Px
↑3s↑↑↑
↑_ _3d_
_)
приводит к sp3d-гибридизации
(рис.5,г). Форма молекулы (иона) –
тригональная бипирамида. В случае
sp3d2-гибридизации
PF6-
Px
↑3s↑↑↑
↑_ _3d_
_
форма иона – октаэдр (рис.5,д).
.
Кроме рассмотренных возможны и другие
типы гибридизации валентных орбиталей.
Гибридизация одной s-,
3-х р- и одной d-орбиталей
(PF5
Px
↑3s↑↑↑
↑_ _3d_
_)
приводит к sp3d-гибридизации
(рис.5,г). Форма молекулы (иона) –
тригональная бипирамида. В случае
sp3d2-гибридизации
PF6-
Px
↑3s↑↑↑
↑_ _3d_
_
форма иона – октаэдр (рис.5,д).
В случае sp3d3-гибридизации (рис.5,е)
SbBrs3- Sb ↕5s ↑↑5p↑ _ _ _ 5p _ _
форма иона или молекулы – пентагональная бипирамида. Очень часто величины валентных углов в молекулах отличаются от величин валентных углов в правильных геометрических фигурах. Например, в молекулах NH3 и Н2О валентные углы составляют 107,3О и 104,5О (вместо 109,28О). Причиной этого согласно теории валентных связей является наличие центрального атома несвязывающих электронных пар. Искажение валентных углов вызывается взаимным отталкиванием связывающих и несвязывающих электронных пар центрального атома. Облако связывающей электронной пары занимает меньше места, чем облако несвязывающей электронной пары, поэтому в наибольшей степени отталкивание проявляется между несвязывающими парами, несколько меньше – между несвязывающей и связывающей парами, меньше всего отталкиваются электронные облака, образованные связывающими электронными парами. Это можно проиллюстрировать на примере строения молекул СН4, NH3 и Н2О, центральные атомы которых образуют химические связи за счёт электронов sp3-гибридных орбиталей.

У атома углерода на четыре sр3-гибридные орбитали приходится четыре электроны, образуются четыре С-Н-связи.
Форма молекулы – правильный тетраэдр (рис.6,а).
У атома азота на четыре sp3-гибридные орбитали приходится пять электронов. Одна пара электронов – несвязывающая, занимает одну из sp3-гибридных орбиталей (рис.6,б). Валентный угол в аммиаке меньше тетраэдрического.
У атома кислорода на четыре sp3-гибридные орбитали приходится шесть электронов. Здесь две sp3-гибридные орбитали занимают несвязывающие электронные пары. Отталкивающее действие двух несвязывающих электронных пар проявляется в большей степени. Поэтому валенный угол против тетраэдрического искажается ещё сильнее (рис.6, в).
С наличием несвязывающих электронных пар центрального атома связано изменение пространственной конфигурации молекул. Молекула NH3, т.к. одна из вершин тетраэдра занята несвязывающей электронной парой, имеет форму тригональной пирамиды. По тем же причинам форма молекулы воды – угловая.
Для установления пространственной конфигурации молекулы (комплекса) можно воспользоваться выполнением операций в следующей последовательности:
-
Написать электронные формулы и распределить электроны по валентным орбиталям центрального атома и лигандов.
-
Определить степень окисления центрального атома, а следовательно, число электронов, используемыхим на образование связей.
-
Установить наличие у центрального атома несвязывающих электронных пар по разности общего числа его валентных электронов и электронов, используемых в связывании.
-
По величине координационного числа центрального атома определить число ϭ-связывающих электронных пар.
-
По общему числу ϭ-связей и числу несвязывающих электронных пар определить тип гибридизации орбиталей центрального атома. По типу гибридизации определить пространственную конфигурацию молекулы (комплекса).
-
Из сравнения числа орбиталей и электронов центрального атома и лиганда определить возможность дополнительного связывания (возможно по донороно-акцепторному механизму).
-
Составить структурную формулу молекулы (комплекса), отразив делокализацию π-связей.
Пример I. Установить пространственную конфигурацию молекулы диоксида углерода (СО2).
Атомы углерода и кислорода имеют следующие электронные конфигурации:

Степень окисления углерода +4. Следовательно, все четыре валентных электрона центрального атома используются на образование связей, число несвязывающих электронных пар равно 0.
Координационному
числу центрального атома 2 соответствует
2ϭ-связи,
следовательно, в образовании этих связей
принимают участие 2 орбитали (Is+Ip)
атома углерода. Тип гибридизации
валентных орбиталей атома углерода –
Р. Такой тип гибридизации соответствует
линейной форме: О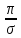 С
С О
О
Неиспользованные в ϭ-связывании два электрона атома углерода и по одному электрону от двух атомов кислорода участвуют в образовании π-связей.
Пример 2. Определить тип гибридизации центрального атома и пространственную конфигурацию молекулы OF2.
Атомы кислорода и фтора имеют электронные конфигурации:

Согласно степени окисления кислорода (центрального атома) +2 на образование связей используется 2 электрона.
Координационное число кислорода равно 2, следовательно, связывающих электронные пар у атома кислорода – две. Всего электронов у кислорода – 6, значит две электронные пары атома кислорода остаются несвязывающими. Несвязывающие электронные пары также участвуют в процессе гибридизации орбиталей центрального атома.
Тип гибридизации валентных орбиталей атома кислорода – sp3.
Пространственная структура молекулы OF2 соответствует тетраэдру, две вершины которого занимают несвязывающие электронные пары, т.е. молекула OF2 имеет угловую форму:

Имея угловую форму, молекула OF2 обладает полярностью. Валентный угол в молекуле меньше тетраэдрического, что объясняется большим эффектом отталкивания между несвязывающими, а также между связывающими электронными парами.
Пример3.
Определить пространственную конфигурацию
молекулы BF3
и комплекса BF .
Объяснить, почему длина связи dBF
и BF3
короче, чем в BF
.
Объяснить, почему длина связи dBF
и BF3
короче, чем в BF .
.
Электронные конфигурации атомов бора и фтора:

показывает, что в соответствии со степенью окисления бора +3, все валентные электроны бора используются на образование ϭ-связей, несвязывающих электронных пар нет.
В молекуле BF3 координационное число бора равно 3. Это отвечает трём ϭ-связывающим электронным парам. Следовательно, тип гибридизации орбиталей атома бора sp2. Молекула BF3 имеет треугольную форму:

После образования 3-х ϭ-связей у атома бора остаётся свободной валентно 2р2-орбиталь, а у атома фтора – неиспользованные 2р2-электронных пары.
Следовательно, между атомами бора и тремя атомами фтора возможно образование одной нелокализованной π-связи по донорно-акцепторному механизму. Это соответствует повышению на одну треть средней кратности связи B-F и сокращению межядерного расстояния.
В
ионе BF атом бора имеет четыре ϭ-связыващие
электронные пары, что соответствует
sp3-гибридизации
валентных орбиталей бора.
атом бора имеет четыре ϭ-связыващие
электронные пары, что соответствует
sp3-гибридизации
валентных орбиталей бора.
Таблица I
Виды гибридизации связей центрального атома А и пространственная конфигурация молекул (комплексов)


У П Р А Ж Н Е Н И Я
1. На рис. 7 изображены схемы перекрытия s- и р-орбиталей. Указать, в каком случае образуется химическая связь и определить её тип.
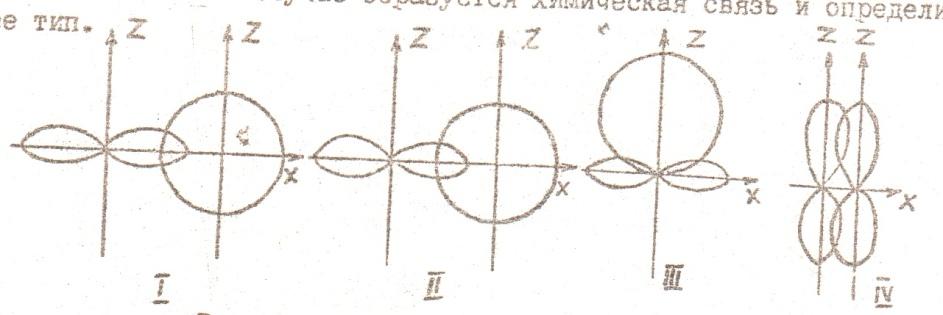
2. Объяснить, почему NH3-пирамида, а BF3-плоская молекула.
3. Каким образом можно представить графически перекрытие А.О. в молекуле СО?
4. Почему не имеет дипольного момента молекула СО2, хотя связь С-О имеет ϻ=0,IID ?
5. Каков тип гибридизации атомов в молекуле С2Н2?
6.
Какие орбитали серы участвуют в
образовании химической связи в молекулах
SF4,
SO2,
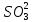 -.
Сколько несвязывающих электронных пар
участвуют в гибридизации?
-.
Сколько несвязывающих электронных пар
участвуют в гибридизации?
7.
Длина химической связи в молекуле SiF4
и SiF имеет различное значение: d1=1,7
A,
dz=1,55
A.
Указать, в какой именно их приведённых
формул длина химической связи короче
и почему?
имеет различное значение: d1=1,7
A,
dz=1,55
A.
Указать, в какой именно их приведённых
формул длина химической связи короче
и почему?
8. Почему строение молекулы ВеН2 можно выразить структурной формулой Н-Ве-Н, в то время, как подобной подход для объяснения строения молекулы ХеF2 не походит? Каковы степени окисления бериллия и ксенона в ВеН2 и ХеН2? Какие орбитали молекул ВеН2 и ХеН2 участвуют в процессе гибридизации?
9. Описать пространственную конфигурацию молекул SF6, CIF5, PF5, CIF3, SF4, SO2, XeF4. Какие из этих молекул полярны, а какие неполярны??
10. Описать строение следующих ионов:
ТеО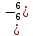 ,
JO
,
JO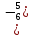 ,
CIO4-,
SiO
,
CIO4-,
SiO .
.
Какой тип гибридизации валентных орбиталей можно приписать центральному атому этих ионов? Какова геометрическая конфигурация их?
Метод молекулярных орбиталей /М.О./
Исследования показали, что в ряде молекул (NO, NO2, CIO2, H2 и др.) а образования химической связи играют роль не электронные пары, а отдельные электроны. Это явление невозможно объяснить с позиции метода молекулярных орбиталей /М.О./.
Согласно методу молекулярных орбиталей, молекула рассматривается как совокупность ядер и электронов и всех ядер. То есть, в отличии от метода валентных связей в методе молекулярных орбиталей молекула рассматривается как единое целое. В основе метода молекулярных орбиталей лежит предположение, что все электроны в молекуле распределяются по соответствующим молекулярным орбиталям. Молекулярные орбитали – многоцентровые и по аналогии с атомными s-, p-, d-, f-орбиталями, обозначаются ϭ-, π-, δ-, φ-.
Описать молекулу по методу М.О. – значит определить тип её орбиталей, их энергию, их энергию и выяснить характер распределения электронов по орбиталям в порядке возрастания их энергия, основываясь на принципе Паули и правиле Хунда, т.е. решить те же задачи, что и при рассмотрении электронных структур атомов. В простейшем приближении молекулярные орбитали можно представить как результат линейной комбинации атомных орбиталей – метод «М.О.-ЛКАО». Согласно «М.О.-ЛКАО» образование молекулярной орбитали можно представить как результат сложения (или вычитания) комбинируемых атомных орбиталей. Пусть электронные орбитали взаимодействующих атомов характеризуется волновыми функциями ψ1, ψ2, ψ3, и т.д. Тогда молекулярная орбиталь, определяющаяся функцией ψ может быть представлена в виде суммы ψ = С1ψ1 + С2ψ2 + С2ψ + …
Коэффициент С указывает на долю участия соответствующих атомных орбиталей в образовании молекулы.
Рассмотрим форму и относительную энергию двухцентровых молекулярных орбиталей, возникающих при линейной комбинации двух Is орбиталей. В 2-х атомных молекул с одинаковыми ядрами (гомоядерных) вклад атомных орбиталей в молекулярные будет одинаковы, т.е. С1=С2. Так как коэффициент С не влияет на вид искомой молекулярной функции ψ и её абсолютные значение, ограничимся нахождением суммы (ψ1 + ψ2).
При сложении атомных волновых функций ψ1 и ψ2 с одинаковыми знаками получается молекулярная волновая функция (рис. 8).

Рис.8. а) схема образования связывающей М.О. из атомных Is-орбиталей;
б) схема образования разрыхляющей М.О. из атомных Is-орбиталей.
Как видно из рисунка, в пространстве между ядрами значение функции ψ больше, чем значение исходных функций ψ1 и ψ2 - \ψ\2 характеризует вероятность нахождения электрона в соответствующей области пространства, т.е. плотность электронного облака. В случае сложения атомных волновых функций с одинаковыми знаками (рис. 8,а), в межядерном пространстве плотность электронного облака |ψ|2 возрастает. Образуется химическая связь. В случае сложения атомных волновых функций с различными знаками (рис. 8,б), в межядерном пространстве плотность облака уменьшается, т.к. в межядерном пространстве ψ=0. В этом случае химическая связь не возникает.
В случае образования химической связи при взаимодействии атомных орбиталей молекулярная орбиталь называется связывающей. В данном случае взаимодействие электронных облаков происходит на оси связи, поэтому молекулярная орбиталь относится к ϭ-типу.
Таким образом, при взаимодействии двух 1s-атомных орбиталей с одинаковыми знаками волновых функций получили связывающую молекулярную орбиталь ϭcz. 1s. При взаимодействии 1s-атомных орбиталей с разными знаками волновых функций получили разрыхляющую молекулярную орбиталь ϭразр.1s.
Переход электронов с атомных орбиталей на молекулярную, сопровождающийся возникновением связи, происходит с выделением энергии. Переход электронов с атомных на разрыхляющую молекулярную орбиталь сопровождается поглощением энергии.
Образование молекулярных орбиталей из атомных обычно отображается поглощением в виде энергетической диаграммы, в котором по вертикали схематически откладывают значение энергии Е (рис.9). Из n числа атомных орбиталей получается такое же число (n) молекулярных.
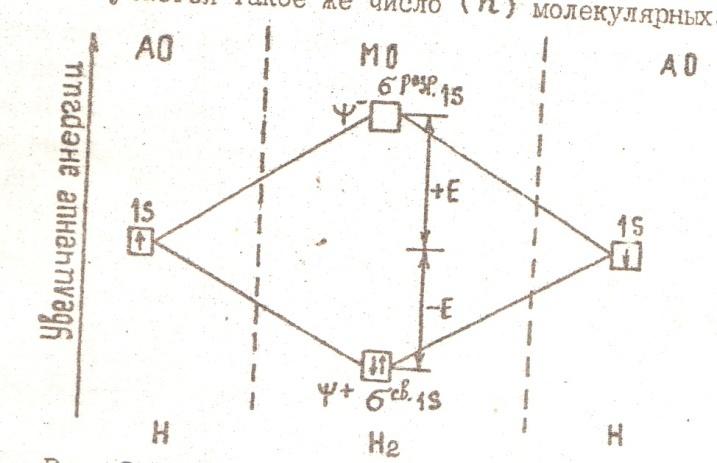
Рис.9. Энергетическая диаграмма уровней энергии атомных и молекулярных орбиталей водорода
В молекуле Н2 – два электрона. Согласно принципу наименьшей энергии и принципу Паули эти два электрона с противоположными спинами заселяют ϭсв.1s- орбиталь. Реакцию образования молекулы водорода из атомов согласно методу МО можно записать:
2Н [1s`] = H2 [(ϭсв.1s)2]
Согласно методу молекулярных орбиталей кратность связи определяется как полуразность связывающих и разрыхляющих электронов:
Кр.связи
=
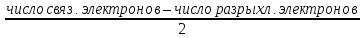
При
образовании связи, естественно, кратность
связи должна быть больше нуля. В молекуле
водорода кратность связи равна 1. Ион
 состоит из двух протонов и одного
электрона. Естественно, что единственный
электрон этого иона должен занимать
энергетически наиболее вгодную позицию,
т.е. ϭсвяз.1s
состоит из двух протонов и одного
электрона. Естественно, что единственный
электрон этого иона должен занимать
энергетически наиболее вгодную позицию,
т.е. ϭсвяз.1s
Таким
образом, электронная формула
 в основном состоянии:
в основном состоянии:
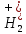 [(ϭсв.1s)1]
[(ϭсв.1s)1]
В системе на двух атомов гелия (He2) четыре электрона. Из них два электрона должны разместиться на связывающей молекулярной орбитали (ϭсв.). Кратность связи равна нулю. Такая молекула не образуется.
Двухатомные гомоядерные молекулы элементов второго периода
У элементов второго периода валентными являются внешние s- и р-орбитали. Чтобы атомные орбитали могли комбинироваться с образованием молекулярных орбиталей, необходимо, чтобы энергии атомных орбиталей были сравнимы по величине, зарядные облака атомных орбиталей обладали бы одинаковыми свойствами симметрии относительно оси молекулы.
Комбинации из 2 s-орбиталей, как и в случае атомных 1 s-орбиталей, приводит к образованию двух молекулярных ϭ-орбиталей ϭсвяз.2s и ϭразр.2s. (рис.10).
Если за линию связи принять ось х, то при комбинации двух 2рх-орбиталей, вытянутых вдоль оси х, образуется ϭсв. 2рх и ϭразр. 2рх орбитали. Так как на оси связи 3-х атомных орбиталей взаимно перпендикулярны, то при комбинации двух 2рz и 2py-орбиталей (двух одинаковых атомов) образуются π-орбитали: πсв. 2рz и πразр. 2рz, πсвяз. 2ру и πразр. 2ру. Так как перекрывание атомных 2ру и 2рz орбиталей происходит одинаковыми способами, то возникающие πсв. 2ру и πсвяз. 2рz орбитали имеют одинаковую энергию, так же как и πразр. 2ру и πразр. 2рz – тоже одинаковы по энергии.
Диаграмма рисунка 10 иллюстрирует порядок размещения электронов по энергетическим орбиталям молекул. Согласно экспериментальным данным, молекулярные орбитали двухатомных молекул элементов конца периода по уровню энергии располагается в следующем порядке (рис.10а): ϭсв.2s < ϭразр.2s < ϭсв.2рх < πсв.2ру = πсв.2рz < πразр.2ру = πразр.2рz < ϭразр.2рx.
Начало периода (рис.10а): ϭсв.2s < ϭразр.2s < πсв.2ру = πсв.2рz < ϭсв.2рх < πразр.2ру = πразр.2рz < ϭразр.2рx.
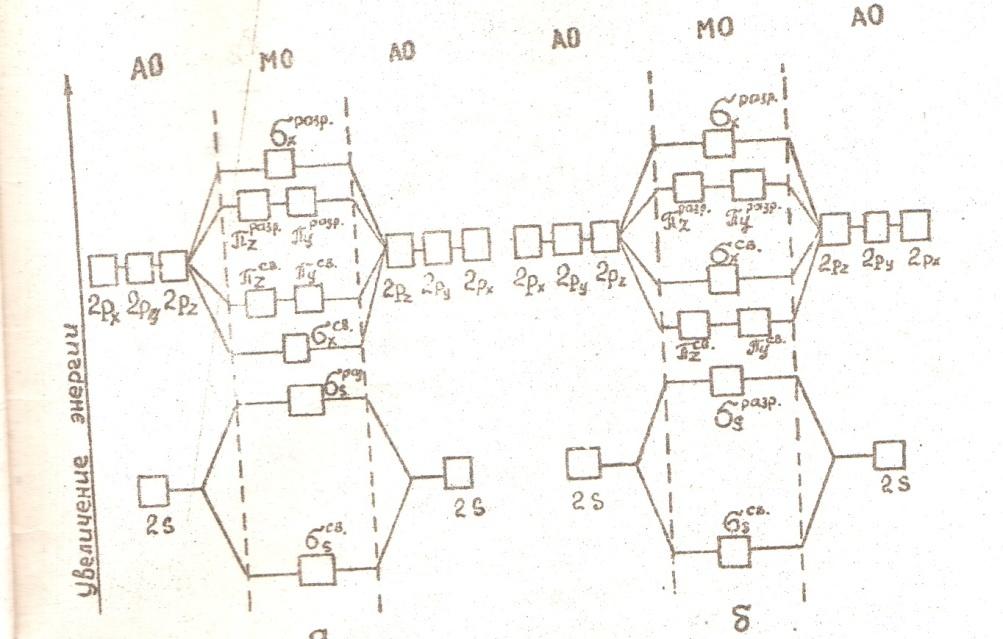
Рис.10. Энергетическая диаграмма уровней 2-х атомных молекул элементов: а) конца второго периода, б) начала второго периода.
Так, например, электронная формула молекулы фтора (F2) будет образовываться их 2-х атомов фтора. Орбиталь внутреннего уровня 1 s2 атомов фтора не вносит существенного вклада в образование химической связи, обозначим её – К. Воспользуемся диаграммой рис.10а и запишем электронную формулу молекулы фтора:
2F
[K
2s2
 ]
= F2
[K
* K
(ϭsсв.)2
(ϭsразр.)2
(ϭxcв.)2
* (πусв.)2
* (πzсв.)2
(πуразр.)]
]
= F2
[K
* K
(ϭsсв.)2
(ϭsразр.)2
(ϭxcв.)2
* (πусв.)2
* (πzсв.)2
(πуразр.)]
Кратность
связи такой молекулы
 = 1
= 1
Ниже зарисованы энергетические диаграммы молекул нескольких элементов начала и конца периода.
По магнитным свойствам различают парамагнитные и диамагнитные вещества. Парамагнитными являются вещества, у которых имеются по два непарных электрона, поэтому они – парамагнитны, молекулы фтора и углерода непарных электронов имеют, следовательно, они диамагнитны.

У П Р А Ж Н Е Н И Я
-
Составить схему молекулярной орбитали молекулы Li2.
Написать электронную формулу и определить кратность химической связи.
-
Будет ли существовать молекула Ве2?
-
Составить схему молекулярной орбитали молекулы N2 и установить кратность связи и магнитную природу её. Будет ли устойчива молекула азота??
-
Начертить схемы молекулярных орбиталей О2,
 ,
,
 .
Объяснить, почему энергия связи в
.
Объяснить, почему энергия связи в
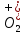 является максимальной в этих 3-х частицах.
является максимальной в этих 3-х частицах. -
Определить кратность химической связи в ионах
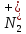 ,
,
 и сделать вывод, какой из ионов более
устойчив и почему.
и сделать вывод, какой из ионов более
устойчив и почему. -
Почему не может существовать молекула Ne2 в основном состоянии?
-
Написать электронные конфигурации ионов и молекул
 ,
Н2,
,
Н2,
 ,
Не.
,
Не.
Каков порядок связи в рассматриваемых системах?
Почему
энергия связи молекулы Н2
больше, чем
 и
и
 ?
?
Какие молекулы и ионы диа-, а какие – парамагнитные?
Лабораторная работа
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ
Целью работы является ознакомление студентов с основными положениями теории валентных связей для объяснения образования и строения комплексных соединений, с их номенклатурой.
Программа коллоквиума
1. Электронные конфигурации элементов d - ряда.
а)Какие орбитали и электроны d - элементов являются валентными ? Чем это объясняется?
б)Почему для d - элементов характерно разнообразие степеней окисления? Как изменяется в ряду d- элементов высшая степень окисления элементов ?
2.Электронные конфигурации ионов, отвечающие различным степеням
окисления d- элементов,
II. Комплексные соединения.
1. Основные положения теории валентных связей для объяснения
образования и строения комплексных соединений.
а) Объяснить особую склонность d- элементов к образованию комплексных соединений, тип гибридизации.
б) Комплексообразователь, лиганды, координационное число, устойчивость комплексных соединений, константа нестойкости. Магнитные свойства комплексных соединений, образование соединений с комплексным катионом и комплексным анионом.
2. Номенклатура комплексных соединений.
3. Получение и свойства комплексных соединений.
а) Получение комплексных соединений с помощью реакции замещения лигандов во внутренней сфере.
б) Получение комплексных соединений с помощью окислительно-. восстановительных реакций.
в)Установление координационной формулы комплексной соли по реакциям, характерным для ионов внешней сферы.
Основополагающие представления о комплексных соединениях ввел в науку Альфред Вернер (1898 г.). В развитии химии комплексных соединений большую роль сыграли труды Л.А.Чугаева и его учеников.
В определении понятия "комплексное соединение" нет полного единства. Это обусловлено разнообразием комплексных соединений и разнообразной гаммой их характерных свойств. Комплексными называются соединения, в узлах кристаллов которых находится комплексы, способные к самостоятельному существованию в растворе. Следует подчеркнуть, что такое определение, конечно, далеко не исчерпывающее и оно применимо лишь в известных пределах.
Согласно Вернеру, в большинстве комплексных соединений различают внутреннюю и внешнюю сферы. Например, в комплексных: соединения К2[BeF4] и [Zn(NH3)4] Cl2 внутреннюю сферу составляют группировки атомов (комплексы) [BeF4]-2 и [Zn(NH3)4]+2 ,а внешнюю сферу - соответственно К+ и С1-, Центральный атом или ион внутренней сферы называется комплексообразователем, координированные вокруг него молекулы или ионы противоположного знака наминаются лигандами (адцендами). В формулах комплексных соединений внутреннюю сферу (комплекс) часто заключают в квадратные скобки. Число, показывающее, сколько лигандов удерживает комплексообразователь, называется координационным. Чаще всего оно равно 6 или 4, реже -2.
Координационные числа зависят от объемов комплексообразоваятеля и лигандов и их зарядов.
Чем больше объем комплексообразователя и меньше объем лигандов, тем больше лигандов может разместиться вокруг комплексообразователя и наоборот. Так, Al+3 может удержать шесть ионов F - с малым радиусом: Na [AIF6] и только четыре более объемистых лиганда ( Вг-1 или J-)
Увеличение заряда комплексообразователя и понижение заряда лигандов способствует повышению координационного числа комплексообразователя. Так, у Сu+ и Аu+ координационные числа 2, а у Cu+2 и Аu+2 координационные числа 4.
Мерой устойчивости комплекса служит ее константа диссоциации, которая в этом случае называется константой нестойкости комплекса и обозначается через Кн.
(MeLn)+m Me+m + nL
Кн
= ![]()
![]()
![]()
![]()
![]()
![]()
где Me - металл, L - лиганд.

Диссоциация комплексных ионов обычно незначительна и величина Кн - мала. Очевидно, что чем больше константа нестойкости, тем менее устойчиво комплексное соединение.
Так, среди однотипных соединений
[Aq(NO2)2]-1 [Ag(S203)2)]-3 [Ag(NH3)2]+1 [Ag(CN)2]-1
KH 1,3*10-3 1*10-13 6,8*10-8 1*10-21
Устойчивость комплекса возрастает при переходе от [Ag(NO2)2]-[Ag(CN)2]
Заряд комплексного иона равен алгебраической сумме зарядов комплексообразователя и лигандов или заряду ионов, находящихся во внешней сфере, взятых с противоположным знаком. Например:
внешняя сфера K2[BeF4] внутренняя сфера
где ион Be+2- комплекеообразователь, лиганды- кислотные остатки фтористоводородной кислоты Координационное число комплексов образователя равно 4, суммарный заряд лигандов равен -4„следовательно , заряд комплексного иона +2 - 4 = -2. Зная заряд комплексного иона и заряд лигандов, можно определить валентность комплексообразователя. Например, в соединении состава K3[Fe(CN)6] заряд комплексного иона равен -3 (K3 [Fe(CN)6 ]-3. Тогда, обозначив валентность комплексообразователя через X, находим: X +(-.6) = - 3, X = +3, CN-- кислотный остаток цианистоводородной кислоты, координационное число равно 6.
По характеру электрического заряда различают катионные, анионные и нейтральные комплексы. Катионный комплекс можно рассматривать, как образованный в результате координации вокруг положительного иона нейтральных молекул.
Рассмотрим процесс образования комплексного соединения, в котором роль лигандов играют нейтральные молекулы аммиака NН3 , а центральным атомом является положительный ион какого-либо металла, например, меди, взятой в виде хлорида или другой соли. Молекулы аммиака, имеющие по одной не поделенной паре электронов, являются донорами, а ион меди - акцептором.
При взаимодействии возникают четыре донорно-акцепторные связи, и получается комплексное соединение, относящееся к амминам:


H̤ +2
H:N̤̤̤:H
□ H̤ H̤ H̤
□ Cu+2 □ + H :N̤: = H :N̤ : Cṳ : N̤: H = [Cu(NH3)4]Cl2
□ H H H
H:N̤:H
H
Аналогичным образом можно показать, что при взаимодействии иона Cr+3 с шестью молекулами воды образуется комплексный ион


□ □ □ ОН2
□ Cr+3 □ + 6: ОН2 → ОН2 Cr ОН2
□ ОН2 ОН2 ОН2
В этом случае, акцептором является центральный атом - хром, а донорами электронной пары - молекулы воды.
В анионном комплексе роль комплексообразователя играет атом положительной степени окисления, а лигандами являются атомы отрицательной степени окисления.
Нейтральные комплексы образуются при координации вокруг атома молекул , а также при одновременной координации вокруг положительного иона - комплексообразователя отрицательных ионов и молекул: [Fe(С0)5] , [Pt(NH3)2Cl2]
Электронейтральные комплексы, следовательно, являются комплексными соединениями без внешней сферы.
Роль комплексообразователя может играть любой элемент периодической системы. В соответствии со своей химической природой неметаллические элементы обычно дают анионные комплексы, в которых роль лигандов играют атомы наиболее электроотрицательных элементов, например: К[PF6] , К3[Р04] .
Что же касается типичных металлических элементов, то способность к образованию комплексных соединений у них выражена слабо. Имеющиеся немногочисленные комплексные ионы являются катионами, например: Ga(NH3)8Cl2 ,[S(H2 O)6]Cl2.
Склонность к комплексообразованию сильно выражена у элементов, относящихся к переходной группе периодической системы. Это обусловлено наличием свободных орбиталей.
При комплексообразовании путем присоединения лигандов, электроны комплексообразующего атома металла спариваются, оставляя отдельные энергетические ячейки, которые могут быть заняты свободными электронными парами координированных групп лигандов.
В процессе комплексообразования у элементов IV периода обнаруживается тенденция к созданию электронной конфигурации, сходной с электронной конфигурацией инертного газа криптона, завершающего этот период. Его электронная конфигурация может быть записана следующим образом: Is22s 22p63S2ЗрбЗd104s24p6 Атом железа с порядковым номером 26" должен иметь"" конфигурацию
Is22s22p63s23p63d64s2 Для заполнения электронами своей оболочки до оболочки криптона иону Fe+3 недостает 12 электронов, из которых четыре заполняют орбитали Зd-слоя до Зd 10 ,два -орбиталь 4s и шесть- 4р -орбитали. Следовательно, ион Fer'3 может явиться акцептором 6 электронных пар, и его координационное число должно равняться 6.
Номенклатура комплексных соединений
В 1958 году на Менделеевском съезде теоретической и прикладной химии была принята новая номенклатура комплексных соединений.
Название комплексного катиона начинают с указания внутренней сферы, причем в, следующей последовательности: лиганды (если они смешанные, то сначала нейтральные молекулы, а затем- анионы)„Если одинаковых лигандов во внутренней сфере комплекса больше одного, то их количество отмечают при помощи греческих числительных (2-ди , 3-три, 4-тетра, 5„цента ,6-гекса, 7-гептаs 8-окта). Ниже перечислены наиболее часто встречающиеся лиганды;
F- - фторо- S-2 тио- ОН- чпидроксо—
С1-. хлоро- СО-23- карбонатов- NH3 аммин-
Вr--бромо- CN-- циано- H2O акво
I- -иодо- CNS- родано- СО -карбонил-.
NO-2 нитро- PO-34 фосфато
NO-3-нитрато- NH2 амидо-
После названия лиганда называют центральный ион (ион-комплексообразователь); при этом в основу кладут русские незнания элементов в родительном падеже, а затем - латинские названия внешней .сферы.
[Со(NH3)4С03]С1 -тетраамин-карбонато-кобальта (Ш)xлорид,
[Al(H 2O)6] CI3 - гексаакваалюминия хлорид
Соль содержит комплексный анион
Вначале также называют лиганды, начиная с нейтральных молекул с введением в наименование (если это нужно) греческих числительных» После этого называют комплексообразователь, используя латинское название элемента с прибавлением суффикса - ат. Студень окисления центрального иона (если это необходимо) также отмечается римскими цифрами в скобках. Наконец, называют катион, образующий внешнюю сферу комплексного соединения (используется русское название элемента в родительном падеже)
K[CO(NH3)2 (NO2)4] - диамминтетранитрокобальтат калия
K3 [Fe(CN)6] - гексацианоферрат (Ш) калия
K2[PtCl6]- гексахлороплатинат (IУ) калия
Наименование нейтральных комплексов составляется из названий лигандов (в последовательности, указанной для комплексных, катионов) и русского названия центрального элемента в именительном падеже. При этом валентность комплексообразователя в названии не указывается,
[Pt(NH3)2Cl4] - диамминтетрахлороплатина
[Ni(CO)4]- тетракарбрнилникель
[CO(NH3)3(NO3)3] триамминтринитрокобальт
Для объяснения образования и свойств комплексных соединений в настоящее время применяется метод валентных связей ( ВС)теория кристаллического поля (ТКП) и метод молекулярных орбиталей (МО).
Метод валентных Связей. Согласно этому методу образование комплексов осуществляется за счет донорно-акцепторного взаимодействия чаще всего неподеленных электронных пар лигандов и свободных орбиталей комплексообралопателя.
Разберем
случай, когда координационное число
комплексообразователя равно 4. Так,
двухзарядный катион
Zn+2
, отдав два 4s
-электрона, имеет на
внешнем
уровне Зd10электронов
Для достижения электронной конфигурации криптона катион должен принять.8 электронов. Если роль донора играют четыре нейтральные молекулы аммиака, которые и дают эти восемь электронов, то образующийся комплекс несет два положительных заряда.
Рассмотрим процесс образования этого комплексного соединения. Здесь центральным атомом является положительный ион металла,Zn+2 взятый в виде растворимой соли. Молекулы аммиака, имеющие по одной неподеленной паре электронов, являются, донорами, а ион цинка - акцептором. При взаимодействии возникают четыре донорно-акцепторные связи за счет четырех свободных орбиталей иона Zn+2 и четырех молекул аммиака, представляющих каждый по одной неподеленной паре электронов и заполняющих 4 орбитали.
Получается комплексное соединение:
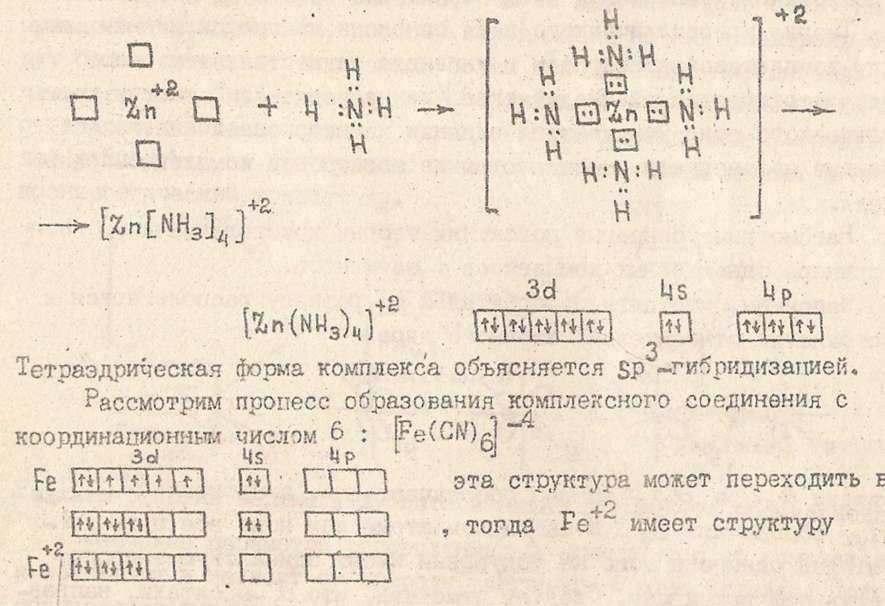
Ион Fe+2 играет роль акцептора и представлявет для образования связи 6 орбиталей с d2sр3 -гибридизацией, Роль лигандов играют ионы CN- они имеют по одной неподеленной паре электронов атома азота и углерода и являются донорами. При взаимодействии возникают шесть донорно-акцепторных связей:
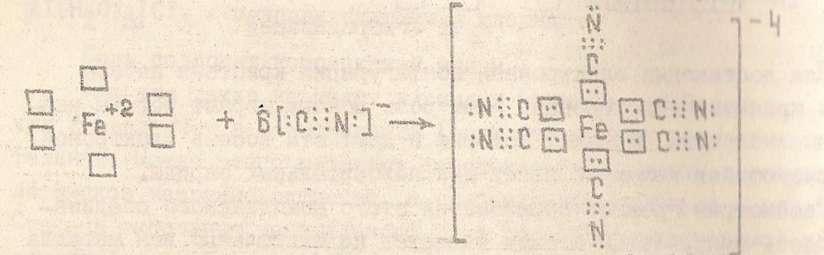
Октаэдрическая формула молекулы объясняется d2sp3 гибридизацией.
Теория кристаллического поля
В 1930 году Ван-Флек и Бэте выдвинули теорию кристаллического поля, которая получила распространение среди химиков гораздо позже (1951 г.), когда на ее основе удалось истолковать спектры поглощения комплексных соединений.
Теория кристаллического поля основана на предположении, кто между комплексообразователем и лигандом осуществляется чисто электростатическое взаимодействие (ионная связь). В теории кристаллического поля учитывается влияние электростатического поля лигандов на энергетическое состояние электронов комплексообразователя.
Рассмотрим основные положения теории кристаллического поля на примере одноядерных комплексов d-элементов.
Напомним, что пять d-орбиталей по-разному располагайся в пространстве относительно атомного ядра.
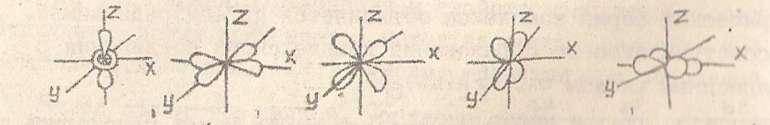
Орбитали dx и d xy обычно обозначают d j а орбитали (dx и dyz обозначают de . В свободном атоме или ионе все пять d- орбиталей одного и того же подуровня имеют одинаковую энергию, то есть они вырождены. Следует отметить, что d j орбитали, направленные вдоль осей, участвуют в образовании € -связей, а d j орбитали, локализованные между осями, являются несвязывающими или участвуют в образовании P связей.
Рассмотрим типичный октаэдрический комплекс ML6 где L - лиганда. В нем электрическое поле, создаваемое неподеленными 6~ парами шести лигандов, которые, окружают ион Мп+, направлено вдоль осей х, у и z. :

о - центральный атом • - лиганда
Представим
себе, что свободный ион по мере приближения
к нему лигандов постепенно начинает
испытывать влияние поля. Пока лиганды
находятся далеко от металла, можно
считать, что это поле сферически
симметрично и под его воздействием
энергия всех электронов постепенно
увеличивается. Но по мере приближения
лигандов становится заметно, что их
поле направлено вдоль осей и на некоторые
электроны действует сильнее, чем на
другие. Вдоль осей направлены d
j-орбитали,
поэтому находящиеся на них электроны
будут более сильно отталкиваться
полем неподеленных пар лигандов, чем
электроны, находящиеся на de-орбиталях.
Под влиянием поля октаэдрически
расположенных лигандов пять вырожденных
сгорбит а- лей распадутся на две группы:
высокоэнергетический дублет и
низкоэнергетический триплет de.
Разница энергий верхнего дублета и нижнего триплета обозначается ∆.
Значение параметра расщепления ∆ изменяется от комплекса к комплексу и зависит от природы лигандов и иона металла, Поле, создаваемое лигандами, зависит от их природы и особенно от легкости, с которой электроны лиганда возмущаются ионом. На практике лиганды можно расположить в порядке уменьшения влияния их кристаллического поля:
C N ~ > NH3 > H2O > F- > CI- > Br >I
Этот порядок по существу не зависит от иона металла, к которому они присоединены.
Может показаться, что этот ряд, часто называемый спектро-химическим рядом, вначале несколько необычен, так как можно было бы ожидать, что F- будет создавать большее поле, чем CN- Однако нужно помнить, что электроны иона фтора крепче удерживаются ядром и не легко возмущаются ионом металла, как электроны более "мягкого" цианид-иона.
На величину расщепления уровней энергии кристаллическим полем влияет степень окисления центрального атома и тип имеющихся у него d-электронов. С увеличением степени окисления d- элемента с возрастания заряда иона) увеличивается, так как лиганды ближе подходят к центральному иону и, следовательно, вызывают большее расщепление d-уровня. В подгруппах d-элементов при переходе от четвертого к пятому и в особенности к шестому периоду ∆ однотипных комплексов заметно возрастает. Это объясняется тем, что 4d 5d орбитали простираются в пространстве дальше от ядра, чем 3-орбитали. Это отвечает более сильному отталкиванию электронов и лигандов и соответственно большему расщеплению 4 d- и 5 d-уровней по сравнению с 3d- уровнем.
Магнитная восприимчивость. Как известно, атомы, содержащие неспаренные электроны, парамагнитны. В сущности неспаренный электрон ведет себя как маленький магнит, то есть "втягивается" в приложенное поле. Если на орбитали два электрона, их спины должны быть противоположно направлены, и их равные противоположно направленные магнитные моменты будут компенсировать друг друга. Когда все орбитали имеют по два электрона, атом диамагнитен. Атом с неспаренным электроном парамагнитен. Магнитный момент Mm, связанный с неспаренным электроном, является векторной величиной, он может быть измерен на опыте, а также вычислен по формуле:
Mm
= ![]()
![]() где n-число
неспаренных электронов
где n-число
неспаренных электронов
Теория кристаллического поля достаточно просто и наглядно объясняет магнитные свойства комплексов, их спектры и ряд других свойств. Для понимания этих свойств необходимо знать характер распределения электронов по d-орбиталям иона, находящегося в поле лигандов. Последнее зависит от соотношения величины энергии расщепления и энергии отталкивания электронов друг от друга.
Когда поле лигандов велико, велик параметр расщепления ∆ электроны сначала полностью заполняют орбитали с меньшей энергией, а уж затем орбитали с большей энергией.
Когда поле лигандов мало, будет мал ∆ пять d-орбиталей последовательно заполняются сначала по одному, а затем по второму электрону.
В качестве примера рассмотрим характер распределения 3 d электронов иона Со+3 при образовании октаэдрических комплексов [CoF6]3-
и [Co(NH3)6]3+

Рассчитано, что энергия отталкивания электронов одной и той же орбитали для иона Со+3равна 251 кДж/моль, энергия расщепления ∆1 его 3d орбиталей в октаэдрическом поле ионов составляет 156 кДж/моль, а в поле молекул H3N ∆2 = 265 кДж/моль.
Таким образом, в поле иона F- значение ∆ невелико, поэтому число непарных электронов на орбиталях расщепленных уровней Со3+такое же, как в свободном ионе. Но в поле, создаваемом молекулами H3N, энергия расщепления большая и энергетически более выгодно, когда d -электроны иона Со3+располагаются только на нижних -d-орбиталях.
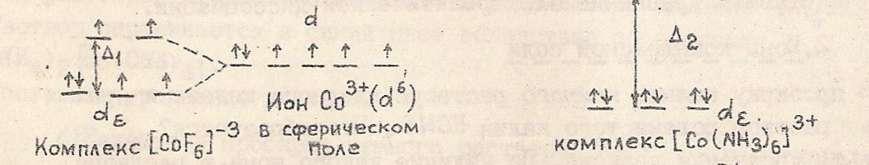
По характеру распределения электронов по орбиталям Со3+ ион [CoF6]3- является высокоспиновым (четыре непарных электрона), а ион [Co(NH3)6]3+ низкоспиновым комплексом (непарные электроны отсутствуют).
В соответствии с характером распределения электронов по d-орбиталям комплекс [Co(NH3)6]3+ диамагнитен, а комплекс[СоF6]3+„ парамагнитен:
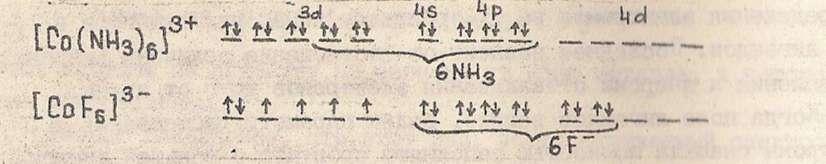
Величину параметра расщепления обычно определяют по спектрам поглощения соединений. Кванты света, возбуждающие переход электронов с нижних d - орбиталей на верхние, соответствуют видимой области спектра, и значения ∆ лежат в пределах 1 эв<∆<4 эв. Этим объясняется тот факт, что соединения d-элементов обычно окрашены.
Один из недостатков простой теории кристаллического поля состоит в том, что все действующие силы считаются чисто электростатическими. Перераспределение электронов (образование ковалентных связей) между ионом металла и лигандами не рассматривается, за исключением случая, когда оно возникает из-за поляризации неподеленных пар электронов ионом металла.
Экспериментальная часть
1.Ионы двойной соли
В три пробирки налить по 2 мл раствора KAI(SO4)2, в одну добавить раствор кислого виннокислого натрия Na 4C 4H 4O 6, в другую - несколько капель раствора NaOH, в третью - раствор соли бария BaCl2. Составить ионные уравнения всех трех реакций. На присутствие каких ионов в исследуемом растворе указывают эти реакции? Составить уравнение электролитической диссоциации.
2 Ионы комплексной соли
В пробирку налить немного раствора хлорного железа и прибавить раствор роданистого калия KCNS . Что образуется? Составьте уравнения реакций. На наличие какого иона в растворе указывает эта реакция?
Провести аналогичную реакцию, взяв вместо раствора FeCI3, раствор красной кровяной соли К3 [Fe(CN)6] , Появляется ли красное окрашивание? Есть ли в растворе красной кровяной соли ионы
трехвалентного железа Fe4. Составить уравнение электролитической диссоциации.
3. Комплексные соединения в реакциях обмена
а) К разбавленному подкисленному раствору FeCI3 добавить разный объем раствора желтой кровяной соли К3 [Fe(CN)6]. Что образуется? Составить уравнение реакции.
б) К свежеприготовленному раствору FeSO4 прибавить растворы красной и желтой кровяных солей. С какой из солей получается си¬нее окрашивание? Написать уравнение реакции в молекулярной и ионной формах.
в) К раствору CuSO4 прибавить раствор желтой кровяной соли. Что образуется? Написать уравнение реакции.
г) К раствору KCI прибавить раствор комплексной соли Na2[Co(NO2)6] . Выделяется желтый кристаллический осадок
KNa2[Co(NO2)6]. Составить уравнение реакции.
4, Образование аммиакатов меди и цинка
Налейте в пробирки 2 мл раствора CuSO4 и 2 мл ZnSO4, подействуйте на него раствором NaOH. Составьте уравнение реакции и отметьте цвет осадка. Понемногу добавляйте в пробирку концентри-рованного раствора аммиака. Наблюдайте за растворением осадка и изменением окраски вследствие образования ионов [Cu(NH3)4]+2 и [Zn(NH3)4]+2 Составьте уравнение реакции.
5.Соединения с комплексным отрицательным ионом.
а) На часовой стекло поместить по 3-4 капли концентрированных растворов хлористого кобальта и роданистого аммония Раствор окрашивается в синий цвет вследствие образования (NH4)2[Сo(CNS)4].
Составьте уравнение реакции.
б) Налейте в пробирку немного раствора Hg(NO3)2 и несколько капель раствора KI. Наблюдайте образование осадка.
Напишите уравнение реакции.
ХИМИЧЕСКАЯ ТЕРМОДИНАМИКА
Курс общей химии предполагает знакомство студентов с некоторыми основными положениями, химической термодинамики. Между тем, в современных учебниках по общей химии данный материал изложен явно недостаточно. В то же время студент 1 курса встречает существенные трудности при самостоятельном изучении учебных пособий по химической термодинамике, содержащих к тому же вопросы, не предусмотренные программой для технических вузов.
Настоящий раздел призван помочь студентам получить общие представления о ряде наиболее важных закономерностей химической термодинамики и на этой основе научить их анализировать простые химические реакции.
1. Основные понятия химической термодинамики. Термодинамическая система и термодинамический процесс. Внутренняя энергия, теплота, работа.
2. Первый закон термодинамики. Адиабатный, изотермный, изохорный и изобарный процессы. Энтальпия. Стандартная энтальпия образования. Энтальпия химической реакции.
3. Термохимия. Основные законы термохимии. Термохимические расчеты.
4. Термодинамическая вероятность и энтропия.
5. Второй закон термодинамики. Энергия Гиббса. Стандартный изобарный потенциал образования. Направление процесса, энтальпийный и энтропийный факторы.
6. Константа химического равновесия. Связь константы химического равновесия с изобарном потенциалом, энтальпией и энтропией процесса.
7. Изобарный потенциал и направленность окислительно-восста-новительных реакций.
1 Основные понятия химической термодинамики
Химическая термодинамика является одним из разделов физической химии. Химическая термодинамика изучает различные химические и физико-химические процессы путем исследования изменений энергетических состояний участвующих тел. Термодинамической системой называют тело или группу взаимодействующих тел. мысленно выделяемых в пространстве. Остальная часть пространства называется окружающей средой.
Система считается изолированной, если она лишена возможности обмениваться с окружающей средой массой и энергией. Система, которая обменивается с Окружающей средой только энергией, называется замкнутой.
Системы разделяются на два больших класса - гомогенные и гетерогенные. Гомогенной называется физически однородная система имеющая одинаковые физические свойства во всех своих частях, например, смеси различных: газов, растворы как жидкие, так и твердые Гетерогенной называется система, состоящая из нескольких однородных частей с разными физическими свойствами.
Система всегда находится в том или ином состоянии, которое характеризуется всей совокупностью ее физических и химических свойств: объемом V , давлением Р, температурой Т, плотностью d и т.д. Такие свойства называются функциями состояния системы. При некоторых определенных сочетаниях значений функций состояния система находится в состоянии равновесия, то есть в таком состоянии, которое остается неизменным во времени при отсутствии изменений окружающей среды.
Свойства неизолированной системы могут изменяться, при этом естественно изменяется и состояние системы, что и представляет собой термодинамический процесс. Если в результате процесса система через ряд промежуточных состояний возвращается к исходному состоянию, то такой процесс называется циклом.
2. Первый закон термодинамики в применении к химическим реакциям.
Неотъемлемым свойством любой системы является энергия, которая представляет собой общую меру различных форм движения материи. Основным термодинамическим понятием является внутренняя энергия системы U, которую рассматривают как совокупность всех видов энергии, связанных со всевозможными движениями и взаимодействиями внутри системы. Это энергия молекул и энергия электронов, энергия взаимодействия атомов внутри молекулы, энергия межмолекулярного взаимодействия и т.д. Запас внутренней энергии зависит только от состояния системы, то есть внутренняя энергия является функцией состояния. Это означает, что изменение внутренней энергии .не зависит от характера термодинамического процесса, а
определяется разностью внутренних энергий системы в конечном U- и начальным U1 состояниях: U = U2 – U1
В состав внутренней энергии не входят потенциальная и кинетическая энергии системы в целом, поскольку Они зависят не только от внутренних свойств самой системы, но и от свойств тел окружающей среды.
Энергию взаимодействия атомов в молекуле называют химической энергией. Изменение этой энергии имеет место при химических превращениях и обусловлено в первую очередь различиями в энергии разорванных и образованных химических связей в процессе реакции.
Величина энергии отдельной химической связи очень мала. Например, чтобы разорвать связь в одной молекуле водорода достаточно потратить энергию в количестве 7,22 –10-19 Дж. Однако химическая термодинамика имеет дело с системами, содержащими большое число частиц, а, следовательно, и с достаточно заметными количествами энергии. Так, энергия химической связи, приходящаяся на I моль водорода (2г), составляет 4,35 *105 Дж.
Совершая какой-либо процесс, система может обмениваться с окружающей средой энергией как в форме теплоты Q, так и в форме работы А. (Первый закон термодинамики (является частным случаем закона сохранения энергии ) утверждает, что убыль внутренней энергии системы расходуется на выделение теплоты и совершение
работы: -∆U= - Q+А (2)
В отличии от внутренней энергии теплота и работа зависят от способа осуществления процесса и не является функциями состояния (нельзя сказать, что данная система характеризуется определенной теплотой или работой). Теплота или тепловой эффект процесса - это количественная характеристика энергии, которую система в ходе процесса получила от окружающей среды (отдала окружающей среде) в форме кинетической энергии теплового, то есть хаотического движения частиц(атомов, молекул, электронов и т.п.). Работа характеризует обмен энергией в форме кинетической энергии направленного, упорядоченного движения частиц.
Тепловым эффектом сопровождается подавляющее большинство химических реакций. Реакции, протекающие с выделением тепла, называются экзотермическими, а с поглощением – эндотермическими .Обычно тепловой эффект относится к 1 молю образовавшегося или сгоревшего вещества и измеряют в килоджоулях (кДж).
Под работой в химических процессах подразумевается в основном работа против сил внешнего давления р. В первом приближении она выражается так: А = р∆V , (3)
где V = V2 – V1 -разность объемов продуктов реакции и объемов исходных: веществ V1 .
При разных процессах перехода из состояния 1 в состояние 2 система обменивается с окружающей средой различными количествами работы и теплоты. Но независимо от характера процесса разность между теплотой и работой остается величиной постоянной и равна изменению внутренней энергии:
Qi - Ai = U2 – U1 = const. (4)
В применении к процессам, ограниченным определенными условиями, уравнение (2) упрощается и процесс можно охарактеризовать одной из величин, входящих в уравнение (2):
1. При адиабатном процессе, то есть процессе в случае изолированной системы Q = 0. Однако система может совершать работу за счет внутренней энергии, например, расширение нагретого газа, связанное с его охлаждением:
Q=∆U + А = 0, А =-∆U (5)
2. При изотермном процессе Т = const Для системы из идеальных газов ∆U = 0, так как в этом случае запас внутренней энергии зависит только от температуры. Все тепло, получаемое системой, переходит в работу:
QT= А . (6)
3. Пря изохорном процессе V-const работа расширения невозможна А = 0. Тоща при условии отсутствия других форм работы вся поглощенная теплота расходуется на увеличение внутренней энергии
QV= ∆U. (7)
В этом случае теплота процесса равна изменению термодинамической функции состояния U , Это важно при исследовании химических реагирующих систем. Так, экспериментальное определение теплоты реакции горения обычно осуществляют при постоянном объеме.
4. При изобарном процессе Р = const, Так как большинство химических реакций проводят при постоянном давлении, данное условие наиболее часто встречается в химии. При этом процесс связан с изменением объема и совершением работы расширения (или сжатия)
А = p(-V2-V1) и изменением внутренней энергии:
Q = (-U2 –U1) + p(V2 – V1) = (U2+PV2) - (U1+ pV1) (8)
Так как и p и V являются функциями состояния (или свойствами) системы, сумма U+pV также является свойством системы и обозначается через Н: Н = U + pV. (9)
Н имеет размерность энергии и называется энтальпией, которая складывается из внутренней энергии pV «объемной энергии» pV. Абсолютное значение энтальпии, также как и внутренней энергии, знать невозможно. Однако, разность энтальпии в двух состояниях может быть найдена. Для изобарного процесса эта разность равна тепловому эффекту, в чем и проявляется наиболее ясно физический смысл энтальпии:
Qp = H2 - H1 = ∆Н, (10)
где Н - изменение энтальпии или просто энтальпия химической реакции.
Для экзотермических реакций энтальпия самих молекул понижается, и изменение энтальпии реакции имеет отрицательный знак (∆Н < 0). В случае эндотермических реакций изменение энтальпии положительно (∆Н > 0). Таким образом, энтальпия реакции равна по величине и противоположна по знаку тепловому эффекту реакции при постоянном давлении. Например:
∆Н = - 100 кДж/моль
СН4 + С12 — CH3CI + HCI Qp = 100 кДж/моль
Энтальпия реакции может быть представлена как разность энергии диссоциации разрывающихся и образующихся связей. Для экзотермических реакций энергия образующихся связей превышает энергию разрывающихся связей:

CH3 – H + Cl – Cl (670) CH3 – Cl + H – Cl (770)
H=670-770=100 (к/Дж/моль)
В случае эндотермических реакций образующиеся связи, наоборот, должны быть менее прочны. В результате такие реакции протекают частично, либо совсем не реализуются, например:

CH3 – H + I – I (578) CH3 – I + H – I (519)
∆H=578-519=59 (к/Дж/моль)
Поскольку энтальпия зависит от температуры (в меньшей степени и от давления), сопоставлять можно только энтальпии реакций, отнесенные к одинаковым условиям. В качестве последних принимаются так называемые стандартные состояния веществ, то есть их наиболее стабильные формы при Т = 298,15°К и р = 101 325 Па (1атм), например, йод - твердый, углерод-графит, кислород - газообразный, вода - жидкая (хотя, иногда применяют в качестве стандартного и парообразное состояние воды) и т.д. Например, в реакции:
C(граф.) + O2(г) → CO2 (г). ∆H˚298 = 393,5 кДк/моль (1)
стандартным состоянием углерода является графит (наиболее стабильная форма при 298°К), а кислород и двуокись углерода находятся при давлении 1 атм. Символ H˚298 означает стандартную энтальпию реакции, отнесенную к 1 молю продукта.
Запись химических реакций с указанием теплового эффекта (или энтальпии) называют термохимическим уравнением. При этом также указывается агрегатное состояние принимающих участие в реакции веществ: (к) - кристаллическое, (ж) - жидкое, (г) - газообразное, (р) - раствор, например:
H 2(Г) + 1/2 O 2(г) H2O(ж) ∆H˚298 = - 286,0 кДж/моль (2)
Расчеты энтальпий реакций на основе абсолютных энтальпий реагентов(∆H˚298 = H˚прод. - H˚исх.) невозможны вследствие неопределенности последних. Проблема решается допущением, что энтальпия образования простого вещества в его стандартном состоянии равна нулю.
Тогда энтальпии реакций (1) и (2) равны энтальпиям образования СО2 и H2О, соответственно, из простых веществ:
∆H˚298 = ∆H˚обр298
Энтальпия реакции, при которой в стандартных условиях образуется I моль сложного вещества из простых веществ, называется стандартной энтальпией образования данного вещества и обозначается ∆H˚298 (часто один из индексов опускается).
Стандартные энтальпии образования определены для многих сложных веществ.
В таблице 1 приведены данные для некоторых соединений.
Стандартные энтальпии образования соединений при 298,15˚K из простых веществ (∆H˚298, кДж/моль)
|
Вещество |
∆Н298о обр. кДж/моль |
Вещество |
∆Н298о обр. кДж/моль |
|
AgBг (к) |
-100.7 |
NaNO3(k) |
-467.0 |
|
AgС1 (к) |
-127.2 |
NaOH (к) |
-426.6 |
|
AgF (к) |
-206.0 |
Na2C03(K) |
-1131.5 |
|
АgN03(к) |
-124,6 |
NaHS04(K) |
-288.0 |
|
А1С13(к) |
-704.6 |
Na2S04(K) |
-1388.I |
|
AIF3 (к) |
-1511.4 |
03 (г) |
+142.4 |
|
А1203(к) |
-1676.8 |
РС13(ж) |
-319.9 |
|
BaS04(K) |
-1474.2 |
РС15(к) |
-434.6 |
|
CaC03(K) |
-1207.7 |
PH3 (г) |
+ 22.9 |
|
СаС12(к) |
-796.3 |
PbCI2(к) |
-360.9 - |
|
СаС2 (к) |
- 59.9 |
Рb0(к) красный |
-219.4 |
|
СаО (К) |
-635.5 |
Рb0(к) желтый |
-217.8 |
|
Са(0Н)2(к) |
-986.8 |
S02(г) |
-297.1 |
|
HCI (г) |
- 92.4 |
S03 (г) |
-396.I |
|
СuС12(к) |
-215.7 |
H2S (г) |
- 20.9 |
|
Сu(0Н)2(к) |
.-444;6 |
H2S04(ж) |
-814.8 |
|
Сu SО4(к) |
-771.4 |
SnCI2(к) |
-331.2 |
|
FeCI2(к) |
-342.0 |
Sn С14(ж) |
-529.2 |
|
FeCI3(к) |
-399.7. |
SnCI4(г) |
-489.4 |
|
FеС0Н)2(к) |
-562.0 |
С(графит) |
0 |
|
Fе(0Н)3(к) |
-827.2 |
С (алмаз) |
+ 1.8 |
|
FeS04 (к) |
-929.5 |
СН4 (г) |
- 74.9 |
|
FeO (K) |
-265.0 |
C2H2(г) |
+226.2 |
|
Fe2O3(к) |
-822.7 |
C2H4(г) |
+ 52.5 |
|
MgCI2(K) |
-641.8 |
C2H6(г) |
+ 84.8 |
|
MgO (к) |
-602. |
CH30H (ж) |
-239.6 |
|
Mg(0H)2(K) |
-925.3 |
C2H50H (Ж) |
-277.1 |
|
MgS04(K) |
-1262.6 |
СН3С0СН3(ж) |
-248.3 |
|
MnO (K) |
-385.4 |
CCI4 |
-135.4 |
|
МnO2 (к) |
-521.8 |
CHCI3 (ж) |
-132.7 |
|
NH3 (г) |
- 45.8 |
CH3C00H (ж) |
484.4 |
|
NH4CI (к) |
-314.4 |
CH3СOOC2H5 (ж) |
-486.6 |
|
NH40H (ж) |
-361.5 |
|
|
|
HN03 (ж) |
-174.2 |
|
|
|
NO (Г) |
+ 90.3 |
|
|
|
N02(p) |
+ 33.5 |
|
|
|
N203(г) |
+ 83.3 |
|
|
|
N205(к) |
- 42.7 |
|
|
|
H20 (ж) |
- 286.0 |
|
|
|
H20 (г) |
-242.0 |
|
|
|
Н202(ж) |
-187.9 |
|
|
|
H202(г) |
-136.2 |
|
|
|
NaBr (к) |
-361.6 |
|
|
|
NaCI (к) |
-411.4 |
|
|
|
NaF (к) |
-574.0 |
|
|
ТЕРМОХИМИЯ
Поскольку мы ограничимся рассмотрением реакций, протекающих при постоянном давлении, энтальпию реакции будем отождествлять с тепловым эффектом.
Раздел химии, изучающий энергию химических реакций, называется термохимией. Термохимия, исторически сложившаяся раньше термодинамики, в настоящее время претерпела некоторые изменения и стала разделом химической термодинамики, В основе термохимических расчетов лежат следующие основные законы термохимии:
1.Закон Лавуазье-Лапласа( 1780-1784 ). Тепловой эффект образования данного соединения в точности равен, но противоположен по знаку тепловому эффекту его разложения. Так, если при образовании I моля СО2 из углерода и кислорода выделяется 393,5 кДж теплоты, то при разложении на углерод и кислород столько же теплоты поглощается.
2.Закон Гесса (1840). Тепловой эффект химической реакции при постоянном давлении (также при постоянном объеме) не зависит от пути хода реакции, то есть от промежуточных стадий, и определяется только родом начальных и конечных веществ и их состоянием.
Термодинамическое обоснование закона состоит в том, что энтальпия - функция состояния и поэтому она зависит от конечного и начального состояний, но не зависит от пути, связывающего их. Закон Гесса дает возможность рассчитать тепловые эффекты реакций в тех случаях, когда экспериментально их определить очень трудно или вообще невозможно.
Вычислим тепловой эффект реакции:
С(граф) + ½О2(г) → СО(г) ∆Нх = ?
который экспериментально измерить нельзя, так как всегда образуется некоторое количество СО2. Однако известны тепловые эффекты следующих реакций:
С(граф.) + О2(г) → СО2(г) ∆Н1 = -393'5 кДж/моль
CО(г),+ ½О2(г) → СО2(г) ∆Н2= -283 кДж/моль
Используя данные уравнения, составим схему двух путей образования СО2:
∆Н1

С + О2 СО2 По закону Гесса:


∆Нх ∆Н2 ∆Н1 = ∆Нх + ∆Н2, отсюда
∆Нх = ∆Н1 - ∆Н2 = -393,5 + 283,8 = -110,5(кДж\моль)
СО + ½О2
Из закона Гесса вытекает следствие: тепловой эффект химической реакции равен сумме теплоты образования продуктов реакции за вычетом суммы теплоты образования исходных веществ.
Для реакции: аА + вВ + ... → дД + еЕ ...
тепловой эффект определяется равенством:
∆Н = [д(∆Нобр)Д + e(∆Hoбp)E + …] - [а(∆Нобр)А +в(∆Нобр)В + …] (12)
Так, для реакции:
СН3С00Н(ж) + С2Н5ОН(ж) → СН3СООС2Н5(ж) + Н2О(ж)
можем записать:
Н=[(∆Нобр)СН3СООС2H5 + (∆Нобр)Н2] - [(∆Hобр)CH3COOH +(∆Hобр)С2Н5ОН]
Применив справочные данные, находим ∆Н = -11,3 кДк/моль. Если известны стандартные энтальпии сгорания исходных веществ (которые также, как и стандартные энтальпии образования табулированы для многих веществ), то тепловой эффект (энтальпию) реакции можно рассчитать по уравнению, вытекающему из закона Гесса:
∆Н =[а(∆Нсгор)А + в(∆Нсгор)В +…] - [Д(∆Нсгор)Д +e(∆Hcгop)Е +…] (13)
Термохимические расчеты позволяют анализировать широкий круг различных физико-химических процессов. Для этой цели привлекают данные по энтальпиям растворения (∆Н°раств), энтальпиям плавления (∆Н°плав) и испарения (∆Н°исп), энтальпиям связей (ДН°) соединений, энтальпиям образования ионов (∆Н°ион) и т.д.
ЭНТРОПИЯ
Первый закон термодинамики позволяет рассчитать энергетические балансы процессов, но ничего не говорит о направлении, в котором может протекать тот или иной процесс. Так, первому закону не противоречило бы разложение поваренной соли на металлический натрий и газообразный хлор в стандартных условиях за счет охлаждения среды (однако этого не происходит).
Возможность реализации процессов связана с термодинамической вероятностью состояния W . Величина W представляет число различных микросостояний, которыми может быть осуществлено макросостояние. Если макросостояние вещества характеризуется, например, такими его свойствами, как температура, давление, объем и др., то микросостояние - это характеристика каждой частицы вещества (ее положение в пространстве, скорость, направление перемещения). Так как термодинамические системы состоят из огромного числа частиц, то данному микросостоянию отвечает колоссальное число различных микросостояний. Согласно второму закону термодинамики, самопроизвольно могут протекать только те процессы, в которых изолированная система из менее вероятного состояния (меньшее значение W ) переходит в более вероятное состояние (большее значение W)
Величины W колоссальны. Даже для системы из 10 частиц W порядка 104. Для I моля вещества (6,203 * 1023 молекул или атомов) W невообразимо велико. В термодинамике вместо термодинамической вероятности пользуются функцией, называемой энтропией и связанной с вероятностью соотношением (уравнение Больцмана):
S=KLnW
где к=1,38*10-23 Дж/град - константа Больцмана.
По отношению к I молю вещества энтропия выражается в Дж/моль-град.
С точки зрения статистической термодинамики энтропия является мерой беспорядочности молекулярного состояния системы. Для кристалла с идеальной кристаллической решеткой чистого вещества при абсолютном нуле ( t= - 273,15°С), который представляет пример предельной упорядочности, энтропия равна нулю (постулат Планка) . В соответствии с уравнением макросостояние идеальных кристаллов при абсолютном нуле осуществлялось бы только одним микросостоянием W= I. Это означает, что ионы (атомы или молекулы) совершают в узлах кристаллической решетки одинаковые колебательные движения с так называемой нулевой энергией. При нагревании идеальный порядок нарушается - появляются частицы, колеблющиеся а различными энергиями, что ведет к увеличению числа микро- состояний и увеличению энтропии. Резким увеличением энтропии сопровождаются фазовые переходы "твердое тело - жидкость","жидкость - (газ) пар", связанные с разрушением кристаллической структуры(плавление с образованием в процессе испарения системы слабо взаимодействующих хаотически движущихся частиц. Таким образом, газообразному состоянию свойственна наибольшая энтропия. Рост энтропии имеет место при расширении газа, растворении кристалла. Все процессы, связанные с увеличением упорядочности, например, охлаждение» сжатие, кристаллизация и. т.д. сопровождаются уменьшением энтропии. Например, если при Т = 500°К и Р=1 атм. SNH3=212 Дж/моль.* град, то при Т =500°К и Р=300 1атм. SNH3=146 Дж/моль. град
Для различных веществ сравнивают энтропий, отнесенные к стандартным условиям. При этом газы считают идеальными, а для растворов принимают их состояние при концентрации равной единице, предполагая, что раствор обладает свойствами бесконечно разбавленного раствора. Такая энтропия называется стандартной и обозначается S 298o. Стандартные энтропии для различных веществ также приведены в справочниках. Тот факт, что вещество в рассматриваемых условий: можно охарактеризовать энтропией, является следствием того, что энтропия представляет собой свойство системы и ее изменение не зависит от пути течения процесса. Для химической реакции:
аА + вВ + …→ дД + еЕ + …..
изменение энтропии равно сумме энтропий продуктов реакции за вычетом суммы энтропий исходных веществ:
∆S = ( ДSД + еSЕ + ….) - ( аSА + вSВ + …) (15)
Направление протекания химического процесса
1. Энергия Гиббса
Для процессов, происходящих в природе, известны две движущие силы: первая - стремление перейти в состояние с наибольшей энергией и выделить тепло при таком переходе, т.е. понизить энтальпию, и вторая - стремление перейти в наиболее вероятное состояние с максимально допустимой в данных условиях степенью беспорядка, максимальной энтропией, т.е. повысить энтропию. Если в процессе степень беспорядка не изменяется (∆S = 0), то направление процесса определяется изменением энтальпии и процесс происходит самопроизвольно в направлении уменьшения энтальпии
(∆H < 0).
Если в процессе не происходит энергетических изменений (∆Н = 0), то фактором, определяющим направление реакции, является энтропия и процесс пойдет самопроизвольно в направлении, при котором степень беспорядка возрастает, то есть в сторону увеличения энтропии.
Для количественного сопоставления этих противодействующих факторов необходимо их выразить в одинаковых: единицах.
Это возможно сделать, если помножить ∆S на Т-1(град )
Таким образом, влияние энтальпийного фактора -представляется величиной ∆Н, а влияние энтропийного фактора - величиной Т*∆S , Предположим, что энтальпийный и энтропийный факторы компенсируют друг друга. Тогда имеет место равенство:
∆Н = Т*∆S, которое определяет состояние равновесия системы. Оно относится к равновесию между кипящей жидкостью и ее насыщенным паром и к равновесию между плавящимися кристаллами и отвердевающей жидкостью, и к другим видам фазовых превращений индивидуальных веществ. Иными словами, выше написанное уравнение характеризуется состоянием системы, когда скорости протекающих в ней противоположных процессов(например, испарения и конденсации, прямой и обратной химической реакции) становятся равными. Из этого уравнения возможно определить в равновесном процессе:
S = (16)
В общем случае суммарный эффект, отображающий влияние энтальпийного и энтропийного факторов в процессах, протекающих при посто¬янной температуре и давлении, определяется изменением изобарно - изотермического потенциала, или просто изобарного потенциала:
∆G = ∆Н - Т∙∆S. (17)
G - называют еще свободной энергией Гиббса в честь американского ученого Гиббса - одного из основателей современной химической термодинамики.
Значение выражается в -кДж/моль или ккал/моль. Характер изменения изобарного потенциала позволяет судить о принципиальной возможности или невозможности осуществления процесса. Условием принципиальной возможности процесса является неравенство: ∆G <0. Иными словами, если G в процессе реакции уменьшается, то процесс возможен и, начавшись, он протекает самопроизвольно. Увеличение же изобарного потенциала ∆G>0 свидетельствует о невозможности осуществления процесса в данных условиях. Если же ∆G = 0, то
система находится в состоянии химического равновесия. При расчетах широко используется значение∆G 298о образ. стандартного изобарно-изотермического потенциала образования, т.е. изменение изобарного потенциала при протекании реакции образования одного моля соединения из простых веществ при стандартных условиях. Подобно энтальпии образования изобарный потенциал образования простых веществ принимается равным нулю. Значения для многих простых веществ определены и сведены в таблицы. Стандартные изобарные потенциалы некоторых веществ приведены в таблице 2.
|
Вещество |
∆Go 298 обр. кДж/моль |
So 298 кДж/моль* град |
Вещество |
∆Go 298 обр. кДж/моль |
So 298 кДж/моль* град |
|
1 |
2 |
3 |
4 |
5 |
6 |
|
AgBr (к) AgCl (к) AgF (к) AgNO3 (к) AlCl3 (к) AlF3 (к) Al2O3 (к) BaSo4 (к) CaSo3 (к) CaCl2 (к) CaC2 (к) CaO (к) Ca(OH)2(к) HCl (г) CuCl (к) Cu(OH)2 (к) CuSO4 (к) FeCl2 (к) FeCl3 (к) Fe(OH)2 (к) Fe(OH)3 (к) Fe(SO)4 (к) FeO (к) Fe2O3 (к) MgCl2 (к) MgO (к) Mg(OH)2 (к) MgSO4 (к) MnO (к) MnO2 (к) NH3 (г) NH4Cl (к) NH4OH (ж) HNO3 (ж) NO (г) NO2 (г) N2O3 (г) N2O5 (к) H2O (ж) H2O (г) H2O2 (г) NaBr (к) NaCl (к) |
- 97.3 - 109.8 - 188.0 - 33.6 - 629.0 - 1432.1 - 1583.3 - 136З.2 - 1128.6 - 1011.5 - 64.9 - 604.4 - 899.2 - 95.4 - 171.5 - 359.6 - 662.2 - 302,6 - 334.2 -480.1 - 700.1 - 825.5 - 244.5 - 740.8 -592.2 - 569.8 - 834.2 -1148.3 - 363.6 - 467.0 - 16,3 - 203.3 - 254.4 - 80.9 + 80.6 + 51.6 + 140.6 + 114.2 -237.4 -228.8 -105.6 - 349.5 - 384.3 |
107.2 96.2 83.7 141.0 109.4 66.5 50.9 132.3 88.8 104.7 70.0 39.8 83.4 186.9 108.2 84.0 109.3 118.1 142.4 88 105 121.0 60.8 87.5 89.7 27.0 63,2 91.4 61.6 53.2 192.8 95.9 165.7 155.7 210.7 240.3 307.3 178.4 70.0 188.8 233.0 86.9 72.2
|
NaF (к) NaNO3 (к) NaOH (к) Na2CO3 (к) NaHSO4 (к) O3 (к) PCl3 (к) PCl5 (к) PH3 (к) PbCl2 (к) PbO (к) красн. PbO (к) желт. SO2 (г) SO3 (г) H2S (к) H2SO4 (ж) SnCl2 (к) SnCl4 (ж) SnCl4 (г)
C (графит) C (алмаз) CH4 (г) C2H2 (г) C2H4 (г) C2H6 (г) C3OH (ж) C2H5OH (ж) CH3COOCH3 (ж) CCl4 (ж) CHCl3 (ж) CH3COOH (ж) |
- 543.7 - 366.1 - 380.4 - 1048.8 — + 162.8 - 272.6 — + 25 .4 - 315.6 -189.2 - 188.3 - 300.4 - 371.4 - 33.8 - 690.8 — - 458.0 - 449.9
— — - 50.8 + 208.6 + 68.4 - 33.0 - 167.2 - 174.3 -155.5 - 64.7 - 71.9 - 389.6 |
51.3 116,6 64.5 138.9 — 239.0 217.3 199.3 210.3 136.1 66.2 68.9 248.2 256.9 193.3 157.0 — 259.2 365.1
— — 186.4 200.9 219.4 229.6 126.7 161.1 200.5 214.6 203.1 159.9 |
Изменение изобарного потенциала, как и изменение энтальпии и энтропии системы, не зависит от пути процесса. Поэтому, изменение изобарного потенциала в результате реакции равноправности между суммой изобарных потенциалов образования продуктов реакции и суммой изобарных потенциалов исходных веществ.
Для реакции аА + вВ + ... = дД+ еЕ + ... изменение стандартного изобарного потенциала определяется уравнением:
∆Go = [д(∆Go обр.)Д + е(∆Go обр.)Е+…]-[а(∆Go обр.)А + в(∆Go обр.)В +…] (18)
или в общем случае:
∆Go = Ʃ∆Goобр.прод.— Ʃ∆Goобр.исходных (19)
Пользуясь значениями стандартных изобарных потенциалов образования (табл.2), можно подсчитать ∆Go многих реакций, например:
NО(г) + ½O2(г) → NО(г), ∆Go298 = ?
∆Go298 = ∆Goобр.NO2 – (∆Goобр.NO + ½∆Goобр.O2)
По значениям изменения стандартного изобарного потенциала можно определить возможность протекания реакции в стандартных условиях, а также грубо оценить. ее возможность в других условиях.
Для большинства реакций можно принять, что, если ∆Go298 < 0, то процесс принципиально осуществим не только в стандартных, но и в других реальных условиях. Если же ∆Go298 > 0, то процесс в реальных условиях не пойдет.
Энтальпийный и энтропийный факторы и направление процесса.
Уравнение (18)позволяет рассмотреть вопрос о•направленности химических процессов в зависимости от характера изменений энтальпии и энтропии.
1.Если ∆Н < 0(уменьшение энтальпии, реакция с выделением тепла ) и ∆S > 0 (увеличение энтропии и степени беспорядка), то ∆G<0. Это значит, что экзотермическая реакция с увеличением энтропии возможна при всех температурах. Процессы, протекающие с уменьшением энтальпии и увеличением энтропии, необратимы. К необратимым процессам относится, например, разложение бертолетовой соли:
КСIO3(к) → КСl(к) + 3\2O2(г)
∆Нo298 = -44,7 кДж/моль, ∆Sо298 = 247,2 Дж/град ∙ моль
В данном случае ∆Hо298 < О, а ∆Sо298 > О, следовательно, возможна лишь прямая реакция. На это же указывает и высокое отрицательное значение ∆Gо298 = -118 кДж/моль.
2. Если ∆Н > 0(увеличение энтальпии, реакция с поглощением тепла) и ∆S < 0 (уменьшение энтропии, увеличение степени порядка), то в этом случае ∆G > 0 всегда. Это значит, что эндотермическая реакция с уменьшением энтропии не возможна ни при каких температурах.
3. В остальных случаях:
знак ∆G зависит от соотношения энтальпийного ∆Н и энтропийного (Т∙∆S) факторов. Реакция будет протекать только в случае, когда ∆G < 0.
Рассмотрим две следующие реакции:
1) 2А1(к) + 3/2 O2(г) → А12O3(к)
∆Нo298 = - 1676,8 кДж/моль
∆So298 = - 313,6 Дж/град ∙ моль
∆Gо298 = -1583,3 кДж/моль
½С(графит) +| ½СО2(г) → СО(г)
∆Нo1700 = + 87 кДж/моль
∆So1700 = + 82 Дж/град ∙ моль
∆G1700 = - 52 кДж/моль
Первая реакция экзотермическая, протекает с уменьшением объема. Возможность протекания этой реакции (∆G < 0) определяется энтальпийным фактором, который перекрывает противодействие энтропийного фактора.
Вторая реакция эндотермическая, протекает с увеличением объема. Возможность протекания ее (∆G < 0), наоборот, определяется энтропийным фактором. При высокой температуре энтропийный фактор (стремление к разъединению частиц) перекрывает энтальпийный фактор и реакция, начавшись, далее может протекать самопроизвольно.
Большое влияние энтропийного фактора на направление процессов может быть показано на примерах процессов растворения кристаллических веществ в жидкостях. Если растворение сопровождается выделением тепла, возможность процесса (∆G < 0) определяется уменьшением Н (∆Н < 0) и увеличением S(∆S > 0), если поглощением, то возможность процесса определяется только увеличением S (∆S > 0).
Изменение температуры оказывает влияние на величину энтропийного фактора, а, следовательно, и величину ∆G, что следует из уравнения ∆G = ∆Н - Т∙ ∆S.
Влияние температуры на ∆G определяется знаком и величиной ∆S При ∆S > 0 повышение температуры будет способствовать смещению ∆G в область отрицательных значений. При ∆S < 0 повышение температуры смещает в область положительных значений.
На рис.3 показана зависимость ∆G от температуры для ряда реакций, л Afr, кДж/моль

При ∆S > 0 кривые идут вниз, при ∆S < 0 - вверх. Иначе говоря, для реакций , протекающих с увеличением энтропии, например, 2С(графит) + О2(г) → 2СО(г), ∆S > 0 повышение температуры приводит к увеличению отрицательного значения ∆G. Следовательно, высокотемпературный режим благоприятствует протеканию процесса.Для реакций, протекающих с уменьшением энтропии, например, 2 Hg(ж) + О2(г) = 2HgO(кр.), ∆S < 0 с повышением температуры ∆G смещается в сторону положительных значений. Следовательно, в этом случае высокотемпературный режим препятствует протеканию процесса. При условиях, когда ∆G положительно, реакция будет протекать в обратном направлении. Как видно из рис. 3, изменение знака ∆G для рассматриваемой реакции достигается при 500°К. Следовательно, выше этой температуры реакция пойдет в обратном направлении. Для грубой оценки того, в каком направлении может протекать та или иная реакция при низких и высоких температурах, можно воспользоваться приближенными уравнениями изобарного потенциала реакции. При низких температурах множитель Т в уравнении ∆G= ∆Н - Т∙ ∆S мал и абсолютное произведение тоже мало. В этом случае для реакций, имеющих значительный тепловой эффект, ∆Н > Т∙ ∆S и вторым членом уравнения можно пренебречь. При этом получим ∆G ≈ ∆H. При достаточно высоких температурах (множитель Т велик) имеем обратное соотношение: ∆Н < Т∙ ∆S. Пренебрегая теперь первым членом в выражении изобарного потенциала, полупим ∆G ≈ - Т∙ ∆S. Эти приближенные равенства показывают, что при низких температурах критерием направления самопроизвольного протекания реакции в первом приближении может служить знак теплового эффекта реакции, а при высоких -знак изменения энтропии. То есть при низких температурах самопроизвольно протекают экзотермические реакции, при высоких – знак изменения энтропии. То есть при низких температурах самопроизвольно протекают экзотермические реакции, при высоких – реакции, сопровождающиеся увеличением энтропии.
Константа химического равновесия
Если химическая реакция достигает состояния динамического равновесия (состояние, при котором прямая и обратная реакции протекают с одинаковой скоростью), то это состояние полностью описывается температурой, концентрацией (давлением в случае газофазовой реакции) и химическим составом системы. Зная равновесную концентрацию Сi (парциональное давление Pi), химический состав, можно подсчитать для данной температуры константу равновесия К. Для реакции аА + вВ + ... = дД + еЕ константа равновесия определяется следующим образом:
КС
= ![]()
![]() ∙
∙ ![]()
![]() (20) КР
=
(20) КР
= ![]()
![]() ∙
∙ ![]()
![]() (21)
(21)
(реакция в растворе) (реакция в газовой фазе)
Высоким значениям К соответствует большое количество продуктов реакции в равновесной смеси (большая часть исходных веществ прореагировала).Малым значениям К соответствует малое количество продуктов. По константе равновесия судят о степени прохождения химической реакции.
Изменение изобарного потенциала связано с константой равновесия уравнением:
∆G = - 2,303 RTlgK (22)
При Т = 298,15 К_ ∆G = - 5.71lgK 298(кДж/моль) (23)
С помощью уравнений (22) и (23) можно вычислить К для реакции, протекающей при температуре Т, если известно ∆G. С учетом уравнений (18) и (23) нетрудно дополнить соотношение:
2,303
lgK
= ![]()
![]() +
+ ![]()
![]() (24)
(24)
Это уравнение применяют для двух различных целей.
Если известно изменение энтальпии реакции (∆Н) и ее константа равновесия при какой-либо одной температуре, то .можно вычислить значение константы равновесия этой реакции при любой другой температуре. И наоборот, если известны значения K1 и K2 при двух различных температурах это позволяет найти изменение энтальпии для рассматриваемой реакции.
Величина К весьма чувствительна к значениям ∆Н и ∆S . При низких температурах, как это видно из уравнения (24), константа равновесия для однотипных реакций будет определяться в первую очередь энтальпией реакции, при высоких температурах - энтропией реакции.
Изобарный потенциал - мера количественной, оценки направленности окислительно-восстановительных реакции
Важным примером термодинамики в химии является установление соотношения между изменением изобарного потенциала в окислительно-восстановительной реакции и электрохимическими потенциалами соответствующих "полуреакций" окисления и восстановления. Такого рода процессы протекают, в частности, в гальваническом элементе. Для таких же окислительно-восстановительных реакций изменение изобарного потенциала определяется произведением протекающего через гальванический элемент электрического заряда n ∙ F (n - количество электронов, участвующих в электрохимической реакции, F =96500 Кл - число Фарадея) на электродвижущую силу элемента Е:
∆G = - nFE. (26)
Уравнение (26) указывает, что возможны только те окислительно-восстановительные процессы, которые обеспечивают положительное значение Е. Электродвижущая сила - это разность потенциалов гальванического элемента при бесконечно медленном течении реакции (когда сила во внешней грани гальванического элемента равна нулю).
Величина Е рассчитывается по разности электродных потенциалов окисления и восстановления :
Е = γокисл – γвосст (27)
Для стандартных условий (концентрация ионов или молекул, участвующих в реакции, равны единице) электродный потенциал называется стандартным и обозначается . Значения стандартных электродных потенциалов различных полуреакций найдены опытным путем и приведены в справочниках. Из двух полуреакций окислительные свойства проявляет та из них, потенциал которой более положителен. Например:
Ag+ + e- → Ag γo = + 0,80 В
No3- + 4H+ + 3e- → NO + 2H2O γo = + 0,96 В
Азотная кислота окисляет серебро:
3Ag + 4HNO3 → 3AgNO3 + NO + 2H2O
Стандартные электродные потенциалы связаны с константой равновесия. С учётом уравнений (22) и (26) можем записать:
Ео
=
2,303 ![]() lgK (28)
lgK (28)
Где Ео = электродвижущая сила при стандартных условиях.
Рассчитаем ∆Go (изменение изобарного потенциала при стандартных условиях) и К протекающий в медно-цинковом гальваническом элементе реакции:
CuSO4 + Zn → ZnSO4 + Cu
На электродах имеют место следующие полуреакции:
Катод: Cu2+ + 2e → Cu
Анод: Zn – 2e → Zn2+
Тогда ∆G = -nFFo = - nF (γoокисл – γoвосст) = - 2 ∙ 96500 [0,337 – (-0,763)] = - 212,3 (кДж/моль)
LgK
= ![]() (γoокисл
– γoвосст)
=
(γoокисл
– γoвосст)
= ![]() = 37,2
= 37,2
Откуда К ≥ 1037
Отрицательная величина ∆Go и большое значение К свидетельствуют о том, что реакция протекает самопроизвольно, так как она обладает большой электродвижущей силой. И наоборот, если для обратимой реакции К имеет очень низкое значение, скажем 10-20, величина ∆Go положительна и реакция не может идти самопроизвольно. Так, по изменению изобарного потенциала можно предсказать способность реакции к самопроизвольному протеканию, оценивая ее по величине константы равновесия. Это особенно важно, так как позволяет еще до эксперимента выяснить, способна ли реакция выполнять полезную работу.