

2006_-Byelorussian_Pharmacopoeia_Volume_1
.pdfР, пробирку закрывают, встряхивают и оставляют на 60 мин. Затем
прибавляют 1 мл реактива железа (III) хлорида и сульфаминовой кислоты Р
и оставляют на 15 мин.
Параллельно с теми же количествами реактивов и в этих же условиях готовят эталон, используя вместо 0,5 мл разведенной испытуемой вакцины 0,5 мл
раствора формальдегида Р, разведенного до содержания формальдегида
(СН2О) 5 мкг/мл.
Измеряют оптическую плотность (2.2.25) полученных растворов на спектрофотометре при длине волны 628 нм, используя в качестве
компенсационного раствора контрольный раствор.
Оптическая плотность испытуемого раствора не должна превышать
оптическую плотность эталона.
Если испытуемая вакцина представляет собой эмульсию, водную фазу
отделяют следующим способом. К вакцине прибавляют равный объем изопропилмиристата Р и перемешивают. К трем объемам полученной
смеси прибавляют два объема 1 М раствора кислоты хлористоводородной,
три объема хлороформа Р и четыре объема раствора 9 г/л натрия хлорида Р. Тщательно перемешивают и центрифугируют с ускорением 15000 g в течение 60 мин. Водный слой отбирают и измеряют его объем. Водный слой используют для испытания на формальдегид, как указано выше.
Вычисляют концентрацию формальдегида в эталоне с учетом разведения вакцины в процессе разделения слоев и готовят эталон с соответствующей
концентрацией.
Если описанная процедура не позволяет достичь разделения слоев, прибавляют раствор 100 г/л полисорбата 20 Р в растворе 9 г/л натрия хлорида Р и повторяют процедуру, центрифугируя с ускорением 22500 g.
2.4.19. ЩЕЛОЧНЫЕ ПРИМЕСИ В ЖИРНЫХ МАСЛАХ
В пробирку помещают 10 мл свежеперегнанного ацетона Р и 0,3 мл воды Р,
прибавляют 0,05 мл раствора 0,4 г/л бромфенолового синего Р в спирте Р; при необходимости раствор нейтрализуют 0,01 М раствором кислоты
хлористоводородной или 0,01 М раствором натрия гидроксида, затем
прибавляют 10 мл испытуемого масла, встряхивают и оставляют до разделения слоев.
Для изменения окраски верхнего слоя в желтую должно быть израсходовано
не более 0,1 мл 0,01 М раствора кислоты хлористоводородной.
2.4.20. АНТИОКСИДАНТЫ В ЖИРНЫХ МАСЛАХ
Испытание проводят методом тонкослойной хроматографии (2.2.27),
используя пластинки, покрытые тонким слоем силикагеля G Р. Перед
применением пластинки высушивают при температуре 130°С в течение 2 ч. Испытуемый раствор (а). 20 г испытуемого образца, взятого из среднего
слоя масла помещают в делительную воронку, прибавляют 50 мл петролейного эфира Р и энергично встряхивают с двумя порциями по 30 мл метанола (75 %
об/об). После четкого разделения слоев нижние, метанольные, слои
объединяют и выпаривают при пониженном давлении и возможно более
низкой температуре в атмосфере азота. Остаток растворяют в 5 мл хлороформа,
свободного от этанола Р. Хранят в плотно укупоренном сосуде.
Испытуемый раствор (b). Слой петролейного эфира (верхний слой), полученный при приготовлении испытуемого раствора (а), осторожно
выпаривают до сухого остатка. Прибавляют 0,5 г пирогаллола Р, растворенного
в 100 мл этанола Р, затем 15 мл свежеприготовленного раствора 330 г/л
натрия гидроксида Р и кипятят с обратным холодильником в течение 30 мин. Охлаждают, прибавляют 250 мл воды Р и извлекают неомыляемые вещества
тремя порциями по 100 мл петролейного эфира Р. Объединенные
извлечения петролейного эфира промывают водой Р до отсутствия щелочной
реакции и выпаривают до сухого остатка. Остаток растворяют в 5 мл
хлороформа, свободного от этанола Р. Хранят в хорошо укупоренном сосуде.
А. НЕПОЛИГИДРОКСИАНТИОКСИДАНТЫ Пластинку помещают в хроматографическую камеру с хлороформом,
свободным от этанола, Р. Когда фронт растворителя пройдет около 12 см от нижнего края пластинки, пластинку вынимают из камеры, высушивают в течение 20 мин на воздухе, затем в течение 20 мин в эксикаторе под вакуумом.
На линию старта хроматографической пластинки, подготовленной, как
указано в ы ш е , в с т а р т о в у ю т о ч к у № 1 ( с м . Рис. 2.4.20.-1) наносят испытуемый раствор (а) в виде пятна диаметром не более 5 мм. Объем наносимого испытуемого раствора (а) зависит от его концентрации и составляет
обычно от 2 мкл до 10 мкл. В стартовые точки №№2 и 3 (см. Рис. 2.4.20.-1) наносят по 2 мкл окрашенного раствора, содержащего по 0.1 г/л диметилового желтого Р,
судана красного GР и индофенолового синего Р в бензоле Р. На пластинке
отмечают длину пробега (10 см) в двух направлениях. В первый раз пластинку
хроматографируют в камере с хлороформом, свободным от этанола Р.
Пластинку высушивают на воздухе в течение 10 мин, затем поворачивают на 90° и хроматографируют в камере с бензолом Р. Пластинку высушивают на воздухе в течение 5 мин и опрыскивают раствором 200 г/л кислоты фосфорномолибденовой
Р в этаноле Р до устойчивого окрашивания пластинки в желтый цвет. Через 2
мин начинают проявляться синие пятна. В течение первых 5-10 мин пластинку обрабатывают парами аммиака до тех пор, пока фон не станет чисто белым. На пластинке остаются синие, светло-фиолетовые или зеленоватые пятна.
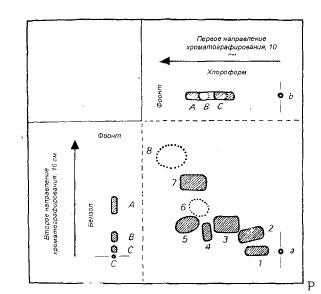
Рисунок 2.4.20.-1.Типичные хроматограммы антиоксидантов
(Методы А и С)
а ― стартовая точка №1+пятно галлатов+пятно
нордигидрогваяретовой кислоты b ― стартовая точка №2
с ― стартовая точка №3 1 ― гваяковая смола
2― 3-(1,1 – Диметилэтил)-4-метоксифенол
3― 2-(1,1 – Диметилэтил)-4-метоксифенол
4― 2.2.5,7,8 – Пентаметил-6-хроманол
5― тетраэтилтиурам дисульфид
6― а-Токоферол
7― дибутилгидроксианизол
8― бутилгидроксианизол
А ― жёлтый В ― красный С ― синий
Хроматограмму оценивают, сравнивая с Рис. 2.4.20.-1. Если на линии старта обнаруживаются синие пятна, проводят разделение и идентификацию
полигидроксиантиоксидантов (метод В).
В. ПОЛИГИДРОКСИАНТИОКСИДАНТЫ На линию старта хроматографической пластинки наносят 1 мкл, 2 мкл, 4 мкл
и 6 мкл испытуемого раствора (а), а также от 1 мкл до 2 мкл окрашенного
раствора, приготовленного, как указанно в методе А. Пластинку помещают в камеру со смесью растворителей: кислота уксусная ледяная Р-бензол Р-
петролейный эфир Р (30:60:60). Когда фронт растворителей пройдёт около 13 см от линии старта, пластинку вынимают из камеры, высушивают на воздухе,
опрыскивают раствором 200 г/л кислоты фосфорномолибденовой Р в этаноле Р и продолжают проявление по методике, описанной для
идентификации неполигидроксиантиоксидантов.

Хроматограмму оценивают, сравнивая положение пятен на хроматограмме
испытуемого раствора с Рис 2.4.20.-2.
Рисунок 2.4.20.-2. Типичные хроматограммы полигидроксиантиоксидантов (метод В)
1 ― окрашенный раствор
2 ― бутилированный гидроксианизол
3 ― гваяковая смола
4 ― нордигидрогваяретовая кислота
5 ― метилгаллат
6 ― этилгаллат
7 ― пропилгаллат
8 ― октилгаллат
9 ― добецилгаллат
А ― жёлтый В ― красный С ― синий
С. АНТИОКСИДАНТЫ, НЕИЗВЛЕКАЕМЫЕ МЕТАНОЛОМ
Испытание проводят методом тонкослойной хроматографии на другой пластинке. Хроматографирование проводят по методике, описанной для неполигидроксиантиоксидантов, используя вместо испытуемого раствора (а) испытуемый раствор (b). Пластинку опрыскивают раствором 10 г/л
дихлорхинонхлоримида Р в этаноле Р. Пятна проявляются в течение 15 мин.
Хроматограмму оценивают, сравнивая с Рис. 2.4.20 – 1. Зоны, отмеченные пунктиром, соответствуют α-токоферолу и бутилированному гидрокситолуолу. Пятна
β- и γ-токоферола находятся в зоне, соответствующей 2-(1,1-диметилэтил)-4- метоксифенолу.
Если в испытуемом веществе в существенном количестве присутствует бутилированный гидрокситолуол, его определяют по методу А.
2.4.21. ПОСТОРОННИЕ МАСЛА В ЖИРНЫХ МАСЛАХ МЕТОДОМ ТОНКОСЛОЙНОЙ ХРОМАТОГРАФИИ
Испытание проводят методом тонкослойной хроматографии (2.2.27) с
использованием в качестве тонкого слоя кизельгура G Р. Пластинку
пропитывают, поместив в камеру, содержащую смесь вазелинового масла Р
и петролейного эфира Р (10:90) в таком количестве, чтобы нижний край пластинки погрузился на 5 мм ниже поверхности жидкости. Когда смесь для
пропитывания поднимется не менее чем на 12 см от нижнего края пластинки, ее вынимают из камеры и дают растворителю испариться в течение 5
мин. Последующее хроматографирование проводят в том же направлении, что и пропитывание.
Приготовление смеси жирных кислот. К 2 г масла прибавляют 30 мл 0,5 М
раствора калия гидроксида спиртового и нагревают с обратным холодильником в течение 45 мин. Прибавляют 50 мл воды Р, оставляют до
охлаждения, переносят в делительную воронку и встряхивают с тремя порциями эфира Р, по 50 мл каждая. Эфирные слои отделяют, водный слой
подкисляют кислотой хлористоводородной Р и вновь встряхивают с тремя порциями эфира Р, по 50 мл каждая. Все эфирные слои объединяют и встряхивают с тремя порциями воды Р, по 10 мл каждая, отбрасывая
промывные воды. Объединенные эфирные слои высушивают над натрия сульфатом безводным Р и фильтруют. Затем эфир выпаривают на
водяной бане, а из остатка готовят испытуемый раствор или раствор сравнения. Жирные кислоты также можно извлечь из раствора,
полученного в ходе определения неомыляемых веществ.
Испытуемый раствор. 40 мг смеси жирных кислот, полученной из испытуемого масла, растворяют в 4 мл хлороформа Р.
Раствор сравнения. 40 мг смеси жирных кислот, полученной из смеси 19 объемов
кукурузного масла Р и 1 объема рапсового масла Р, растворяют в 4 мл хлороформа Р.
На линию старта хроматографической пластинки наносят по 3 мкл каждого раствора. Помещают пластинку в камеру со смесью растворителей: вода Р - кислота уксусная ледяная Р (10:90). Когда фронт растворителей пройдет 8 см от линии старта, пластинку вынимают из камеры, высушивают при температуре 110°С в течение 10 мин и оставляют до охлаждения. Затем, если нет других указаний в частной статье, пластинку помещают в закрытую
плотно подогнанной крышкой хроматографическую камеру, предварительно
насыщенную парами йода (на дно камеры в выпарительной чашке помещен йод Р). Через некоторое время проявляются коричневые или
желтовато-коричневые пятна. Пластинку вынимают из камеры и оставляют
на несколько минут. Когда коричневый фон исчезнет, пластинку опрыскивают раствором крахмала Р. Появляющиеся синие пятна могут
приобретать коричневую окраску при высушивании и вновь становятся
синими после опрыскивания водой Р.
На хроматограмме испытуемого раствора всегда должны обнаруживаться пятно с Rf около 0,5 (олеиновая кислота) и пятно с Rf около 0,65 (линолевая кислота), соответствующие пятнам на хроматограмме раствора сравнения.
На хроматограммах некоторых масел может обнаруживаться пятно с Rf около 0,75 (линоленовая кислота). Путем сравнения пятен на хроматограмме
испытуемого раствора с пятнами на хроматограмме раствора сравнения убеждаются в отсутствии на хроматограмме испытуемого раствора пятна
с Rf около 0,25 (эруковая кислота).
2.4.22. ПОСТОРОННИЕ ЖИРНЫЕ КИСЛОТЫ В МАСЛАХ МЕТОДОМ ГАЗОВОЙ ХРОМАТОГРАФИИ
Испытание на посторонние жирные кислоты проводят путем перевода жирных кислот, содержащихся в испытуемом масле, в метиловые эфиры.
МЕТОД А
Этот метод неприменим для масел, содержащих глицериды жирных кислот с эпокси-, гидроксиэпокси-, циклопропиловыми или
циклопропениловыми группами,а также для масел, в составе которых
большая часть жирных кислот имеет длину цепи менее восьми атомов
углерода, и для масел с кислотным числом более 2,0.
Испытание проводят методом газовой хроматографии (2.2.28).
Испытуемый раствор. Испытуемое масло высушивают перед метилированием, если это указано в частной статье. 1,0 г масла помещают в
круглодонную колбу вместимостью 25 мл со шлифом, снабженную обратным холодильником и газоотводной трубкой. В колбу прибавляют 10 мл метанола
безводного Р, 0,2 мл раствора 60 г/л калия гидроксида Р в метаноле Р,
присоединяют обратный холодильник и, пропуская через смесь азот Р со
скоростью около 50 мл/мин, встряхивают и нагревают до кипения. Когда раствор станет прозрачным (обычно через 10 мин), продолжают нагревание
втечение последующих 5 мин. Затем колбу охлаждают под проточной водой и содержимое переносят в делительную воронку. Колбу промывают 5 мл гептана Р, переносят смывы в ту же делительную воронку и встряхивают. Прибавляют 10 мл раствора 200 г/л натрия хлорида Р и энергично
встряхивают. Оставляют до расслоения, затем переносят органический слой
всосуд, содержащий натрия сульфат безводный Р и через некоторое время фильтруют.
#Допускается применение других методик перевода содержащихся в испытуемом масле жирных кислот в метиловые эфиры, указанных в частной статье.
Раствор сравнения (а). Готовят 0,50 г смеси веществ, применяемых для калибровки (калибровочной смеси), состава, приведенного в одной из Табл. 2.4.22, как указано в частной статье (если в частной статье не указан определенный раствор, готовят смесь, состав которой приведен в Табл.
2.4.22.-1). Смесь растворяют в гептане Р и доводят объем раствора этим
же растворителем до 50,0 мл.
Табл. 2.4.22.-1
Приготовление смеси веществ, применяемых для калибровки*
|
|
|
|
Состав |
|
|
|
|
|
(в процентах м/м) |
|
Вещество для |
Эквивалент |
Изотерми- |
Линейный |
||
калибровки |
длины |
|
ческий |
градиент |
|
|
цепи** |
|
режим |
температуры |
|
|
(1) |
|
(2) |
|
|
Метиллаурат Р |
12.0 |
|
12.0 |
5 |
10 |
Метилмиристат Р |
14.0 |
|
14.0 |
5 |
15 |
Метилпальмитат Р |
16.0 |
|
16.0 |
10 |
15 |
Метилстеарат Р |
18.0 |
|
18.0 |
20 |
20 |
Метиларахидат Р |
20.0 |
|
20.0 |
40 |
20 |
Метилолеат Р |
18.6 |
|
18.3 |
20 |
20 |
Табл. 2.4.22.-2
Приготовление смеси веществ, применяемых для калибровки*
|
|
|
Состав (в |
|
|
|
|
|
процентах м/м.) |
||
Вещество для |
Эквивалент |
Изотерми- |
|
Линейный |
|
калибровки |
длины |
ческий |
|
градиент |
|
|
цепи** |
режим |
|
температуры |
|
|
(1) |
(2) |
(2) |
|
|
Метилкапроат Р |
6.0 |
6.0 |
5 |
|
10 |
Метилкаприлат Р |
8.0 |
8.0 |
5 |
|
35 |
Метилкапрат Р |
10.0 |
10.0 |
10 |
|
35 |
Метиллаурат Р |
12.0 |
12.0 |
20 |
|
10 |
Метилмиристат Р |
14.0 |
14.0 |
40 |
|
10 |
Табл. 2.4.22.-3
Приготовление смеси веществ, применяемых для калибровки*
|
|
|
Состав (в |
|
|
|
|
|
процентах м/м) |
||
Вещество для |
Эквивалент |
Изотерми- |
|
Линейный |
|
калибровки |
длины |
ческий |
|
градиент |
|
|
цепи** |
режим |
|
температуры |
|
|
(1) |
(2) |
|
|
|
Метилмиристат Р |
14.0 |
14.0 |
5 |
|
15 |
Метилпальмитат Р |
16.0 |
16.0 |
10 |
|
15 |
Метилстеарат Р |
18.0 |
18.0 |
15 |
|
20 |
Метиларахидат Р |
20.0 |
20.0 |
20 |
|
15 |
Метилолеат Р |
18.6 |
18.3 |
20 |
|
15 |
Метилэйкозаноат Р |
20.2 |
20.2 |
10 |
|
10 |
Метилбегенат Р |
22.0 |
22.0 |
10 |
|
5 |
Метиллигноиерат Р |
24.0 |
24.0 |
10 |
|
5 |
*Для газовой хроматографии с применением капиллярной колонки и делением потока рекомендуется прибавлять к смеси веществ, применяемых для калибровки, компоненты с большей длиной цепи.
**Эти значения, вычисленные с использованием калибровочной кривой, даны в качестве примера для колонки, заполненной полиэтиленгликоль сукцинатом Р (1)
и макроголом 20 000 Р (2).
Раствор сравнения (b). 1.0 мл раствора сравнения (а) доводят гептаном Р до 10.0 мл.
Хроматографируют на газовом хроматографе с пламенно-ионизационным детектором в следующих условиях:
-колонка стеклянная или из нержавеющей стали длиной от 2 м до 3 м и
внутренним диаметром от 2 мм до 4 мм, заполненная диатомитом носителем для газовой хроматографии Р с размером частиц от 125
мкм до 200 мкм, на который нанесено от 5% до 15%
полиэтиленгликольсукцината Р или полиэтиленгликольадипината Р;
-газ-носитель – азот для хроматографии Р;
-скорость газа-носителя – 25 мл/мин;
-температура колонки – 180°С;
-температура инжектора и детектора – 200оС.
При необходимости или если указано в частной статье, температуру
колонки увеличивают от 120°С до 200°С со скоростью 5°С в мин.
Хроматографировать возможно также на газовом хроматографе с пламенно–ионизационным детектором в следующих условиях:
-колонка капиллярная стеклянная или кварцевая длиной от 10 м до 30 м и
внутренним диаметром от 0.2 мм до 0.8 мм, внутренняя поверхность которой покрыта слоем поли[(цианопропил)(метил)][(фенил) (метил)]силоксана Р или
макрогола 20 000 Р толщиной от 0,1 мкм до 0,5 мкм или другой подходящей неподвижной фазой;
-газ-носитель – гелий для хроматографии Р или водород для хроматографии Р;
-скорость газа-носителя – 1,3 мл/мин (для колонки с внутренним диаметром
0,32 мм);
-деление потока – 1:100 или менее в зависимости от внутреннего диаметра применяемой колонки (в случае использования колонки с
внутренним диаметром 0,32 мм деление потока должно составлять 1:50);
-температура колонки – от 160°С до 200°С, в зависимости от длины
колонки и используемой неподвижной фазы (для колонки длиной 30 м, покрытой слоем макрогола 20 000 Р, температура должна составлять 200°С);
-температура инжектора и детектора – 250°С.
При необходимости или если указано в частной статье, температуру
колонки увеличивают от 170°С до 230°С со скоростью 3°С в мин (для колонки, покрытой слоем макрогола 20 000 Р)
Хроматографируют 0,5 мкл раствора сравнения (а). Чувствительность системы регулируют таким образом, чтобы высота основного пика на полученной хроматограмме составляла от 50% до 70% шкалы регистрирующего устройства.
Определяют времена удерживания жирных кислот, входящих в состав калибровочной смеси. Хроматографируют 1 мкл раствора сравнения (b) и
рассчитывают отношение сигнал/шум для пика, соответствующего
метилмиристату.
Хроматографируют от 0,5 мкл до 1,0 мкл испытуемого раствора. Время хроматографирования должно в 2,5 раза превышать время удерживания
метилолеата. Хроматограмму оценивают, как описанно ниже.
При использовании калибровочных смесей №1 или №3 хроматографическая система считается пригодной, если выполняются
следующие условия:
-на хроматограмме раствора сравнения (а) число теоретических тарелок (n) (2.2.28), вычисленное для пика, соответствующего метилстеарату, составляет не менее 2000 для набивной колонки и не менее 30 000 для капиллярной колонки;
-на хроматограмме раствора сравнения (а) коэффициент разделения (Rs) (2.2.28) пиков, соответствующих метилолеату и метилстеарату, составляет не менее 1,25 для набивной колонки и не менее 1,8 для капиллярной колонки;
-на хроматограмме раствора сравнения (b) отношение сигнал/шум (2.2.28) для пика метилмиристата составляет не менее 5.
При использовании калибровочной смеси №2 хроматографическая система считается пригодной, если выполняются следующие условия:
-на хроматограмме раствора сравнения (а) число теоретических тарелок (n) (2.2.28), вычисленное для пика, соответствующего метилкапрату, составляет
не менее 1500 для набивной колонки и не менее 15 000 для капиллярной колонки;
-на хроматограмме раствора сравнения (а) коэффициент разделения (Rs) (2.2.28) пиков, соответствующих метилкаприлату и метилкапрату, составляет не менее 2 для набивной колонки и не менее 4 для капиллярной колонки;
- на хроматограмме раствора сравнения (b) отношение сигнал/шум (2.2.28)
для пика метилкапроата составляет не менее 5.
ОЦЕНКА ХРОМАТОГРАММ
Следует избегать условий хроматографирования, которые могут дать неразделенные пики (наличие компонентов с небольшим различием между временами удерживания, например, линоленовая и арахидоновая кислоты.
Качественный анализ. Строят калибровочную кривую, используя
хроматограммы раствора сравнения (а) и данные Таблиц 2.4.22.
а) Для хроматограмм, полученных в изотермическом режиме, вычисляют
логарифмы приведенных времен удерживания как функцию эквивалента числа атомов углерода в жирных кислотах. Калибровочная кривая насыщенных кислот представляет собой прямую линию. Логарифмы
приведённых времен удерживания ненасыщенных кислот расположены на этой линии как точки, соответствующие не целым значениям
«эквивалента длины цепи». Идентификацию компонентов жирных кислот испытуемого масла проводят, рассчитываая логарифмы приведённых времён удерживания пиков, полученных на хроматограмме испытуемого
раствора, и устанавливая по калибровочной кривой «эквивалентны длины цепи».
#Допускается идентификация жирных кислот испытуемого масла путем
сравнения времен удерживания пиков на хроматограмме испытуемого
раствора с временами удерживания пиков на хроматограмме раствора сравнения или на стандартной хроматограмме, описанной в частной статье.
#Приведенное время удерживания - разница между временем
удерживания пика вещества и временем удерживания несорбирующегося (в
условиях определения) вещества.
б) Для хроматограмм, полученных с использованием линейного градиента температуры, определяют времена удерживания, находящиеся в зависимости от числа атомов углерода в жирных кислотах, и
идентифицируют жирные кислоты, входящие в состав испытуемого масла,
путем сравнения с калибровочной кривой.
Количественный анализ. Обычно используют метод внутренней
нормализации; при этом сумму площадей всех пиков на хроматограмме,
кроме пиков, относящихся к растворителю, принимают за 100%. Рекомендуется применение электронного интегратора. Содержание
каждого компонента вычисляют как отношение площади соответствующего пика к сумме площадей всех пиков. Пики, площадь которых составляет
менее 0.05% от суммы площадей всех пиков, не учитывают, если нет других
указаний в частной статье.
Вопределенных случаях, т.е. при наличии жирных кислот с 12 или менее
атомами углерода, в частной статье должен быть указан поправочный коэффициент для преобразования площадей пиков в проценты (м/м).
МЕТОД В
Этот метод неприменим для масел, содержащих глицериды жирных
кислот с эпокси-, гидроксиэпокси-, циклопропиловыми и
циклопропениловыми группами и для масел с кислотным числом более
2,0.
Испытуемый раствор. 0,100 г испытуемого вещества помещают в центрифужную пробирку с завинчивающейся крышкой, растворяют в 1 мл

гептана Р и 1 мл диметилкарбоната Р и энергично перемешивают при
умеренном нагревании (от 50°С до 60°С). К еще теплому раствору прибавляют 1 мл раствора 12 г/л натрия Р в метаноле безводном Р, приготовленного с
необходимыми предосторожностями, и энергично перемешивают в течение 5 мин. Прибавляют, 3 мл воды дистиллированной Р и энергично перемешивают в
течение 30 с. Центрифугируют в течение 15 мин с ускорением 1500 g. Хроматографируют 1 мкл органического слоя.
Растворы сравнения и оценка хроматограмм. Если в частной статье нет других указаний, поступают, как указано в Методе А.
Хроматографируют на газовом хроматографе с пламенно-ионизационным детектором в следующих условиях:
-кварцевая колонка длиной 30 м, с внутренним диаметром 0,25 мм, покрытая слоем макрогола 20 000 Р толщиной 0,25 мкм;
-газ-носитель – гелий для хроматографии Р;
-скорость газа-носителя – 0,9 мл/мин;
-деление потока – 1:100;
-температура колонки и скорость подъема температуры – по следующей программе:
|
|
|
|
|
|
Время |
Температура |
Скорость подъема |
Примечания |
|
(мин) |
(°С) |
температуры |
|
|
|
|
(оС/мин) |
|
|
|
|
|
|
Колонка |
0-15 |
100 |
- |
Изотермический режим |
|
15-36 |
100→225 |
10 |
Линейный градиент |
|
|
|
|
температуры |
|
|
|
|
|
|
36-61 |
225 |
- |
Изотермический режим |
Инжектор |
|
250 |
|
|
|
|
|
|
|
Детектор |
|
250 |
|
|
МЕТОД С
Этот метод неприменим для масел, содержащих глицериды жирных
кислот с эпокси-, гидроперекисными, альдегидными, кетоновыми,
циклопропиловыми и циклопропениловыми группами и сопряженными полиненасыщенными и ацетиленовыми компонентами из-за частичного или полного разрушения этих групп.
Испытуемый раствор. 0,10 г испытуемого вещества помещают в коническую колбу вместимостью 25 мл, растворяют в 2 мл раствора 20 г/л натрия гидроксида
Р в метаноле Р и кипятят с обратным холодильником в течение 30 мин. Затем через холодильник прибавляют 2,0 мл раствора бора фторида в метаноле Р и кипятят еще 30 мин, после чего прибавляют через холодильник 4,0 мл гептана Р и кипятят 5 мин. Охлаждают, прибавляют 10,0 мл раствора натрия хлорида
насыщенного Р, встряхивают в течение 15 с и прибавляют такой объем
раствора натрия хлорида насыщенного Р, чтобы верхний слой поднялся к
горлу колбы. Отбирают 2,0 мл верхнего слоя, помещают в делительную воронку, промывают тремя порциями воды Р, по 2 мл каждая, и высушивают над натрия сульфатом безводным Р.
Растворы сравнения, хроматографическая методика и оценка хроматограмм. Если в частной статье нет других указаний, поступают как указано в методе А.