

2007_-Byelorussian_Pharmacopoeia_Volume_2
.pdfДикалия фосфат |
111 |
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A.1 г испытуемого образца суспендируют 50 мл воды Р, кипятят в течение 1 мин и охлаждают. К 1 мл полученного раствора прибавляют 0,05 мл раствора йода Р1. Появляется окраска от темно-синей до красновато-коричневой, исчезающая при нагревании.
B.5 мл вязкого раствора, полученного в ис-
пытании на подлинность А, центрифугируют. К верхнему слою прибавляют 2 мл раствора натрия гидроксида Р и, по каплям, при встряхи-
вании, 0,5 мл раствора меди сульфата Р и ки-
пятят. Образуется красный осадок.
C. Испытуемый образец очень легко растворим в кипящей воде Р с образованием вязкого раствора.
Испытания
pH (2.2.3). От 2,0 до 8,0. 5,0 г испытуемого образца растворяют в 100 мл воды, свободной от углерода диоксида, Р. Измеряют рН получен-
ного раствора.
Хлориды. Не более 0,2 %. 2,5 г испытуемого образца растворяют в 50 мл кипящей воды Р, разводят водой Р до 100 мл и фильтруют. 1 мл фильтрата разводят до 15 мл, прибавляют 1 мл
кислоты азотной разведенной Р, прибавляют полученную смесь в один прием к 1 мл раст-
вора серебра нитрата Р2 и выдерживают в те-
чение 5 мин в защищенном от света месте. При поперечном просмотре на черном фоне опалесценция полученного раствора по интенсивности не должна превышать опалесценцию раствора сравнения, приготовленного параллельно с использованием смеси из 10 мл эталонного раст-
вора хлорида (5 ppm Cl) Р и 5 мл воды Р.
Восстанавливающие сахара. Не более
10 % в пересчете на глюкозу. К 2,0 г испытуемого образца (в пересчете на сухое вещество) прибавляют 100 мл воды Р, встряхивают в течение 30 мин, разводят водой Р до 200,0 мл и филь-
труют. К 10,0 мл раствора медно-тартратного щелочного Р прибавляют 20,0 мл фильтрата, перемешивают и нагревают на плитке, отрегулированной таким образом, чтобы раствор был доведен до кипения в течение 3 мин. Кипятят 2 мин и немедленно охлаждают. Прибавляют 5 мл раст-
вора 300 г/л калия йодида Р и 10 мл 1 М раст-
вора кислоты серной, перемешивают и сразу же титруют 0,1 М раствором натрия тиосуль-
фата, используя в качестве индикатора раствор крахмала Р, добавляемый в конце титрования. Процедуру повторяют, начиная со слов «К 10,0 мл…», используя вместо фильтрата 20,0 мл раствора 1,0 г/л глюкозы Р. Параллельно проводят контрольный опыт. Значение (Vк − Vд) не должно превышать значения (Vк − Vг), где Vд, Vг
и Vк — объемы, в мл, 0,1 М раствора натрия тиосульфата, пошедшие на титрование декстрина, глюкозы и в контрольном опыте, соответственно.
Тяжелые металлы (2.4.8, метод С). Не бо-
лее 0,002 % (20 ppm). 1,0 г испытуемого образца выдерживает испытание на тяжелые металлы. Эталон готовят с использованием 2 мл эталон-
ного раствора свинца (10 ppm Pb) Р.
Потерявмассепривысушивании(2.2.32).
Не более 13,0 %. 1,000 г испытуемого образца сушат при температуре от 130°С до 135°С в течение 90 мин.
Сульфатная зола (2.4.14, метод А). Не бо-
лее 0,5 %. Определение проводят из 1,0 г испытуемого образца.
#Остаточные количества органических растворителей (2.4.24). Испытуемый образец должен выдерживать требования статьи (5.4).
#Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Декстрин в условиях испытаний не обладает антимикробным действием.
Дикалия фосфат (# калия гидрофосфат)
Dikalii phosphas
DIPOTASSIUM PHOSPHATE |
|
K2HPO4 |
М.м. 174,2 |
ОПРЕДЕЛЕНИЕ
Дикалия фосфат содержит не менее 98,0 % и не более 101,0 % К2HPO4 в пересчете на сухое вещество.
ОПИСАНИЕ (СВОЙСТВА)
Порошок белого цвета или бесцветные кристаллы. Очень гигроскопичен.
Очень легко растворим в воде, очень мало растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Раствор S, приготовленный как указано в разделе «Испытания», имеет слабощелочную реакцию (2.2.4).
В. Раствор S дает реакцию (b) на фосфаты
(2.3.1).
С. Раствор S дает реакцию (а) на калий
(2.3.1).
ИСПЫТАНИЯ
Раствор S. 5,0 г испытуемого образца раст-
воряют в воде дистиллированной Р и дово-
дят объем раствора этим же растворителем до
50 мл.
Прозрачность (2.2.1). Раствор S должен быть прозрачным.
Цветность (2.2.2, метод II). Раствор S должен быть бесцветным.
Восстанавливающие вещества. Смесь из 5 мл раствора S, 5 мл кислоты серной разведенной Р и 0,25 мл 0,02 М раствора калия

112 |
Государственная фармакопея Республики Беларусь |
перманганата нагревают на водяной бане в течение 5 мин. Раствор должен сохранить слабокрасную окраску.
Калияфосфат.Рассчитываютсоотношение (Х) из количества миллилитров 1 М раствора кислоты хлористоводородной (10,0 мл) и 1 М раствора натрия гидроксида (n1, мл и n2, мл),
израсходованных при количественном определении, по формуле:
X= n2 -10 10-n1
Полученное соотношение не должно превы-
шать 0,025 (2,5 %).
Хлориды (2.4.4). Не более 0,02 % (200 ppm).
К 2,5 мл раствора S прибавляют 10 мл кислоты азотной разведенной Р и доводят водой Р до объема 15 мл. Полученный раствор должен выдерживать испытание на хлориды.
Сульфаты (2.4.13). Не более 0,1 %. К 1,5 мл раствора S прибавляют 2 мл кислоты хлори-
стоводородной разведенной Р и доводят водой дистиллированной Р до объема 15 мл. Получен-
ный раствор должен выдерживать испытание на сульфаты.
Мышьяк(2.4.2,методА).Неболее0,0002 % (2 ppm). 5 мл раствора S должны выдерживать испытание на мышьяк.
Железо (2.4.9). Не более 0,001 % (10 ppm). 10 мл раствора S должны выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод А). Не бо-
лее 0,001 % (10 ppm). 2,0 г испытуемого образца растворяют в 8 мл воды Р, подкисляют 6 мл кис-
лоты хлористоводородной разведенной Р до значения рН от 3 до 4 и доводят объем раствора водой Р до 20 мл. 12 мл полученного раствора должны выдерживать испытание на тяжелые металлы. Эталон готовят с использованием эта-
лонного раствора свинца (1 ppm Рb) Р.
Натрий. Не более 0,1 %, если субстанция предназначена для производства лекарственных средств для парентерального применения. Атомно-эмиссионная спектрометрия (2.2.22, ме-
тод 1).
Испытуемый раствор: 1,00 г испытуемого образца растворяют в воде Р и доводят этим же растворителем до 100,0 мл.
Раствор сравнения. Готовят соответствую-
щими разведениями эталонного раствора нат рия (200 ppm Na) Р водой Р.
Интенсивность эмиссии измеряют при длине волны 589 нм.
Потерявмассепривысушивании(2.2.32).
Не более 2,0 %. 1,000 г испытуемого образца сушат при температуре от 125°С до 130°С.
Стерильность (2.6.1). Если субстанция предназначена для производства лекарственных средств для парентерального применения без дополнительнойпроцедурыстерилизации,онадолжна выдерживать испытание на стерильность.
Бактериальные эндотоксины (2.6.14). Ме-
нее1,1МЕ/мг,еслисубстанцияпредназначенадля производства лекарственных средств для парентерального применения без дополнительной процедуры удаления бактериальных эндотоксинов.
#Остаточные количества органических растворителей (2.4.24). Испытуемый образец должен выдерживать требования статьи (5.4).
#Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Дикалия фосфат в условиях испытаний не обладает антимикробным действием.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,800 г испытуемого образца (m, г) раство-
ряют в 40 мл воды, свободной от углерода диоксида, Р; прибавляют 10,0 мл 1 М раствора кислоты хлористоводородной и титруют 1 М раствором натрия гидроксида потенциометри-
чески (2.2.20) до первого скачка потенциала на кривой титрования (n1, мл). Продолжают титрование до второго скачка потенциала на кривой титрования (n2, мл — общий объем 1 М раст-
вора натрия гидроксида, израсходованного при титровании).
Содержание К2HPO4 (Х) в процентах рассчитывают по формуле:
Х =1742×(10-n1) , m×(100-d)
где d — потеря в массе при высушивании, в процентах.
ХРАНЕНИЕ В воздухонепроницаемом контейнере.
МАРКИРОВКА
При необходимости указывают:
––субстанция пригодна для производства лекарственных средств для парентерального применения;
––субстанция стерильна;
––субстанция не содержит бактериальных эндотоксинов.
Диметилсульфоксид (# Димексид)
Dimethylis sulfoxidum
DIMETHYL SULFOXIDE
O
|
|
|
|
|
S |
||
H3C |
|
|
CH3 |
C2H6OS |
|
|
М.м. 78,1 |
ОПРЕДЕЛЕНИЕ
Диметилсульфоксид представляет собой сульфинилбисметан.
Диметилсульфоксид |
113 |
ОПИСАНИЕ (свойства)
Бесцветная жидкость или бесцветные кристаллы. Гигроскопичен.
Смешивается с водой и с 96 % спиртом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: С. Вторая идентификация: А, В, D.
А. Испытуемый образец выдерживает испытание «Относительная плотность», как указано в разделе «Испытания».
В. Испытуемый образец выдерживает испытание «Показатель преломления», как указано в разделе «Испытания».
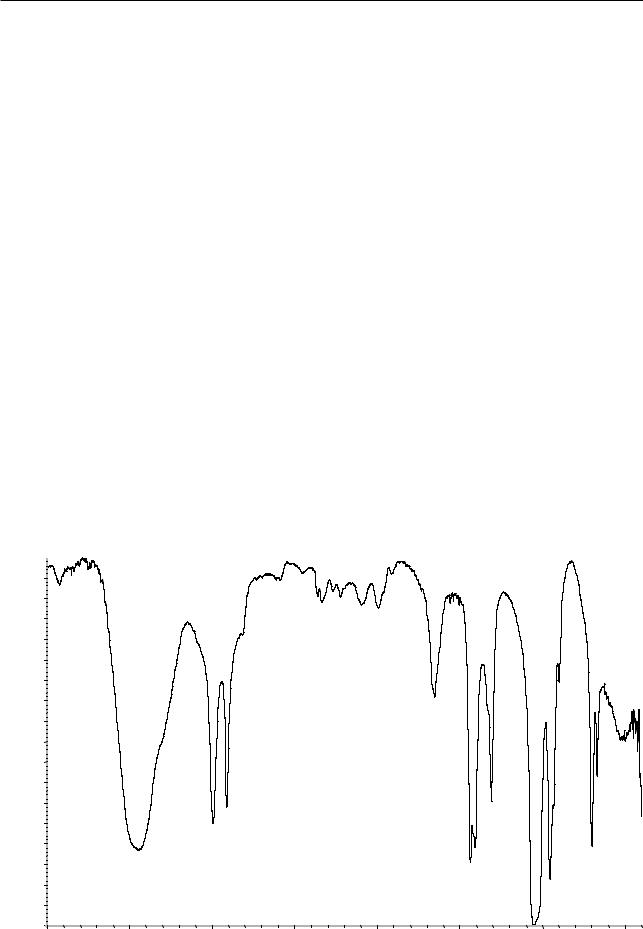
С. Инфракрасный спектр пропускания (2.2.24) испытуемого образца соответствует спектру ФСО диметилсульфоксида # или спект ру, представленному на рисунке 1.
D. 50 мг никеля хлорида Р растворяют в 5 мл испытуемого образца. Появляется зеленоватожелтое окрашивание. Полученный раствор нагревают на водяной бане при температуре 50°С. Появляется синевато-зеленое или зеленое окрашивание, которое при охлаждении раствора переходит в желто-зеленое.
ИСПЫТАНИЯ
Кислотность. 50,0 г испытуемого образца растворяют в 100 мл воды, свободной от углерода диоксида, Р, прибавляют 0,1 мл раствора фенолфталеина Р1. При прибавлении не бо-
лее 5,0 мл 0,01 М раствора натрия гидроксида
окраска раствора должна измениться.
Относительная плотность (2.2.5). От 1,100
до 1,104.
Показатель преломления (2.2.6). От 1,478
до 1,479.
Температура затвердевания (2.2.18). Не более 18,3°С.
Оптическая плотность (2.2.25). Через ис-
пытуемый образец пропускают азот Р в течение 15 мин. Измеряют оптическую плотность, используя в качестве раствора сравнения воду. Оптическая плотность при длине волны 275 нм не должна превышать 0,30. Оптическая плотность не должна превышать 0,20 как при длине волны 285 нм, так и при 295 нм. В интервале от 270 нм до 350 нм спектр поглощения испытуемого образца не должен иметь максимумы поглощения.
Сопутствующие примеси. Газовая хрома-
тография (2.2.28).
Раствор внутреннего стандарта. 0,125 г дибензила Р растворяют в ацетоне Р и дово-
дят объем раствора этим же растворителем до
50 мл.
Испытуемый раствор (а). 5,0 г испытуе-
мого образца растворяют в ацетоне Р и доводят объем раствора этим же растворителем до
10,0 мл.
Испытуемый раствор (b). 5,0 г испытуемо-
го образца растворяют в ацетоне Р, прибавляют 1,0 мл раствора внутреннего стандарта и доводят ацетоном Р до объема 10,0 мл.
Раствор сравнения. 50,0 мг испытуемого образца и 50 мг диметилсульфона Р раство-
П а
95
90
85
80
75
70
65
60
55
50
45
40
35
30
25
20
15
10
4000 |
3500 |
3000 |
2500 |
2000 |
1500 |
1000 |
500 |
|
|
|
В ( -1) |
|
|
|
|
Рисунок 1. Инфракрасный спектр пропускания ФСО диметилсульфоксида.

114 |
Государственная фармакопея Республики Беларусь |
ряют в ацетоне Р, прибавляют 10,0 мл раствора внутреннего стандарта и доводят ацетоном Р до объема 100,0 мл.
Условия хроматографирования:
––колонка стеклянная длиной 1,5 м и внут ренним диаметром 4 мм, заполненная диатоми-
том для газовой хроматографии Р (125—180
мкм), импрегнированным 10 % (м/м) полиэти-
ленгликольадипинатом Р;
––газ-носитель: азот для хроматографии Р;
––скорость газа-носителя: 30 мл/мин;
––температура колонки: 165°С;
––температура блока ввода пробы и детектора: 190°С;
––детектор: пламенно-ионизационный;
––объем вводимой пробы: 1 мкл;
––время хроматографирования: 4-кратное время удерживания по отношению к времени удерживания диметилсульфоксида, которое составляет около 5 мин.
Порядок выхода пиков: раствор сравнения:
диметилсульфоксид, диметилсульфон и ди бензил.
Чувствительность системы: регулируют таким образом, чтобы высота трех пиков, следующих за пиком растворителя, составляла не менее 70 % шкалы регистрирующего устройства.
Пригодность хроматографической системы:
––разрешение: не менее 3 между пиками диметилсульфоксида и диметилсульфона.
На хроматограмме испытуемого раствора (а) подтверждают отсутствие пика с таким же временем удерживания, как у внутреннего стандарта.
На хроматограмме раствора сравнения рассчитывают отношение (R) площади пика диметилсульфоксида к площади пика дибензила. На хроматограмме испытуемого раствора (b) рассчитывают отношение суммы площадей всех дополнительных пиков, кроме пиков диметилсульфоксида, дибензила и ацетона, к площади пика дибензила. Это отношение не должно пре-
вышать R (0,1 %).
Вода (2.5.12). Не более 0,2 %. Определение проводят из 10,0 г испытуемого образца.
#Остаточные количества органических растворителей (2.4.24). Испытуемый образец должен выдерживать требования статьи (5.4).
#Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Диметилсульфоксид в условиях испытания обладает антимикробным действием. Посев на питательные среды № 1 и № 2 проводят из разведения 1:10, на питательные среды № 11 и № 8 — из разведения 1:50.
ХРАНЕНИЕ
В воздухонепроницаемом стеклянном контейнере в защищенном от света месте.
Динатрия фосфат дигидрат (# Натрия гидрофосфат дигидрат)
Dinatrii phosphas dihydricus
DISODIUM PHOSPHATE DIHYDRATE
Na2HPO4 · 2H2O |
М.м. 178,0 |
ОПРЕДЕЛЕНИЕ
Динатрия фосфат дигидрат содержит не ме-
нее 98,0 % и не более 101,0 % Na2HPO4 в пере-
счете на сухое вещество.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок либо бесц ветные кристаллы.
Растворим в воде, практически нерастворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Раствор S, приготовленный как указано в разделе «Испытания», имеет слабощелочную реакцию (2.2.4).
В. Испытуемый образец выдерживает испытание «Потеря в массе при высушивании», как указано в разделе «Испытания».
С. Раствор S дает реакцию (b) на фосфаты
(2.3.1).
D. Раствор S дает реакцию (а) на натрий
(2.3.1).
ИСПЫТАНИЯ
Раствор S. 5,0 г испытуемого образца раст-
воряют в воде дистиллированной Р и дово-
дят объем раствора этим же растворителем до
100 мл.
Прозрачность (2.2.1). Раствор S должен быть прозрачным.
Цветность (2.2.2, метод II). Раствор S должен быть бесцветным.
Восстанавливающие вещества. К 5 мл раствора S прибавляют 5 мл кислоты серной разведенной Р, 0,25 мл 0,02 М раствора калия перманганата и нагревают на водяной бане в течение 5 мин. Раствор должен сохранять слаборозовое окрашивание.
Натрия фосфат. Рассчитывают соотношение (Х) из количества миллилитров 1 М раст-
вора кислоты хлористоводородной (25 мл)
и1 М раствора натрия гидроксида (n1, мл
иn2, мл), израсходованных при количественном определении, по формуле:
X= n2 -25. 25-n1
Полученное соотношение не должно превы-
шать 0,025.
Динатрия эдетат |
115 |
Хлориды (2.4.4). Не более 0,04 % (400 ppm).
К 2,5 мл раствора S прибавляют 10 мл кислоты азотной разведенной Р и доводят водой Р до объема 15 мл. Полученный раствор должен выдерживать испытание на хлориды.
Сульфаты (2.4.13). Не более 0,1 %. К 3 мл раствораSприбавляют2млкислотыхлористо водородной разведенной Р и доводят водой дис-
тиллированной Р до объема 15 мл. Полученный раствор должен выдерживать испытание на сульфаты.
Мышьяк(2.4.2,методА).Неболее0,0004 % (4 ppm). 5 мл раствора S должны выдерживать испытание на мышьяк.
Тяжелые металлы (2.4.8, метод А). Не бо-
лее 0,002 % (20 ppm). 12 мл раствора S должны выдерживать испытание на тяжелые металлы. Эталон готовят с использованием эталонного раствора свинца (1 ppm Рb) Р.
Железо (2.4.9). Не более 0,004 % (40 ppm). 5 мл раствора S доводят водой Р до объема 10 мл. Полученный раствор должен выдерживать испытание на железо.
Потерявмассепривысушивании(2.2.32).
От 19,5 % до 21,0 %. 1,000 г испытуемого образца сушат при температуре 130°С.
#Остаточные количества органических растворителей (2.4.24). Испытуемый образец должен выдерживать требования статьи (5.4).
#Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Динатрия фосфат дигидрат в условиях испытаний не обладает антимикробным действием.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
2,000 г испытуемого образца (m, г) растворяют в 50 мл воды Р, прибавляют 25,0 мл 1 М
раствора кислоты хлористоводородной и титруют 1 М раствором натрия гидроксида по-
тенциометрически (2.2.20) до первого скачка потенциала на кривой титрования (n1, мл). Продолжают титрование до второго скачка потенциала на кривой титрования (n2,мл— общий объем
1 М раствора натрия гидроксида, израсходо-
ванного при титровании).
Содержание Na2HPO4 (Х), в процентах, вычисляют по формуле:
Х =1420×(25-n1), m×(100-d)
где:
d — потеря в массе при высушивании, в процентах.
Динатрия эдетат (# Динатриевая соль
этилендиаминтетрауксусной кислоты, Трилон Б)
Dinatrii edetas
DISODIUM EDETATE
|
|
CO2H |
|
N |
CO2Na |
NaO2C |
N |
2H2O |
HO2C |
|
|
C10H14N2Na2O8 · 2H2O |
М.м. 372,2 |
|
ОПРЕДЕЛЕНИЕ
Динатрия эдетат содержит не менее 98,5 % и не более 101,0 % динатрия дигидро (этилендинитрило)тетраацетата дигидрата.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Растворим в воде, практически нерастворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: A, B, D.
Вторая идентификация: B, C, D.
А. Инфракрасный спектр пропускания (2.2.24) испытуемого образца, полученный в дисках, соответствует спектру ФСО динатрия эдетата # или спектру, представленному на рисунке 1.
В. 2 г испытуемого образца растворяют
в25 мл воды Р, прибавляют 6 мл раствора свин-
ца нитрата Р, перемешивают и прибавляют
3 мл раствора калия йодида Р. Не должен обра-
зовываться осадок. Полученный раствор подще-
лачивают раствором аммиака разведенным Р2 по красной лакмусовой бумаге Р и прибавляют 3 мл раствора аммония оксалата Р. Осадок не образуется.
С. 0,5 г испытуемого образца растворяют
в10 мл воды Р, прибавляют 0,5 мл раствора кальция хлорида Р, подщелачивают раствором аммиака разведенным Р2 по красной лакмусовой бумаге Р и прибавляют 3 мл раствора ам-
мония оксалата Р. Осадок не образуется.
D. Испытуемый образец дает реакции (а)
и(b) на натрий (2.3.1).
ИСПЫТАНИЯ
Раствор S. 5,0 г испытуемого образца раст-
воряют в воде, свободной от углерода диокси-
да, Р и доводят объем этим же растворителем до 100 мл.

116 |
Государственная фармакопея Республики Беларусь |
|
|
П а
21 |
|
|
|
|
|
|
|
20 |
|
|
|
|
|
|
|
19 |
|
|
|
|
|
|
|
18 |
|
|
|
|
|
|
|
17 |
|
|
|
|
|
|
|
16 |
|
|
|
|
|
|
|
15 |
|
|
|
|
|
|
|
14 |
|
|
|
|
|
|
|
13 |
|
|
|
|
|
|
|
12 |
|
|
|
|
|
|
|
11 |
|
|
|
|
|
|
|
10 |
|
|
|
|
|
|
|
9 |
|
|
|
|
|
|
|
8 |
|
|
|
|
|
|
|
7 |
|
|
|
|
|
|
|
6 |
|
|
|
|
|
|
|
4000 |
3500 |
3000 |
2500 |
2000 |
1500 |
1000 |
500 |
|
|
|
В ( -1) |
|
|
|
|
Рисунок 1. Инфракрасный спектр пропускания ФСО динатрия эдетата, полученный в дисках с калия бромидом Р.
Прозрачность (2.2.1). Раствор S должен быть прозрачным.
Цветность (2.2.2, метод II). Раствор S должен быть бесцветным.
рН (2.2.3). От 4,0 до 5,5. Измеряют рН раствора S.
Примесь А. Не более 0,1 %. Жидкостная хроматография (2.2.29). Испытание проводят в защищенном от света месте.
Растворяющая смесь. 10,0 г железа сульфатапентагидратаРрастворяютв20мл0,5М раствора кислоты серной и прибавляют 780 мл
воды Р. Доводят рН раствора до значения 2,0
при помощи 1 М раствора натрия гидроксида
и доводят водой Р до 1000 мл.
Испытуемый раствор. 0,100 г испытуемо-
го образца растворяют в растворяющей смеси и доводят до 25,0 мл этим же растворителем.
Раствор сравнения. 40,0 мг нитрилотри уксусной кислоты Р растворяют в растворяющей смеси и доводят до 100,0 мл этим же растворителем. К 1,0 мл полученного раствора прибавляют 0,1 мл испытуемого раствора и доводят до 100,0 мл растворяющей смесью.
Условия хроматографирования:
––колонкадлиной0,10мивнутреннимдиаме- тром 4,6 мм, заполненная сферическим углеро-
дом графитированным для хроматографии Р1
(размер частиц 5 мкм) с удельной площадью поверхности 120 м2/г и размером пор 25 нм;
––подвижная фаза: 50,0 мг железа сульфата пентагидрата Р растворяют в 50 мл 0,5 М раствора кислоты серной и прибавляют 750 мл
воды Р. Доводят рН раствора до значения 1,5
при помощи 0,5 М раствора кислоты серной или 1 М раствора натрия гидроксида, прибав-
ляют 20 мл этиленгликоля Р и доводят до объе-
ма 1000 мл водой Р;
––скорость подвижной фазы: 1 мл/мин;
––спектрофотометрический детектор,
длина волны 273 нм;
––объем вводимой пробы: 20 мкл; растворы фильтруют и вводят немедленно;
––время хроматографирования: 4-кратное время удерживания по отношению к времени удерживания железного комплекса примеси А.
Время удерживания: железного комплекса примеси А — около 5 мин, железного комплекса кислоты эдетатовой — около 10 мин.
Пригодность хроматографической систе-
мы: раствор сравнения:
––разрешение между пиками железного комплекса примеси А и железного комплекса кислоты эдетатовой не менее 7;
––отношение сигнал/шум для пика примеси А не менее 50.
Площадь пика примеси А на хроматограмме испытуемого раствора не должна превышать площадь соответствующего пика на хроматограмме раствора сравнения (0,1 %).

# Диэтаноламин |
117 |
Железо (2.4.9). Не более 0,008 % (80 ppm). 2,5 мл раствора S доводят водой Р до объема 10 мл. Полученный раствор должен выдерживать испытания на железо. К каждому раствору перед прибавлением кислоты тиогликолевой Р добавляют 0,25 г кальция хлорида Р.
#Хлориды (2.4.4). Не более 0,01 % (100 ppm). К 10 мл раствора S прибавляют 3 мл
кислоты азотной разведенной Р и фильтруют.
Объем фильтрата доводят водой Р до 15 мл. Полученный раствор должен выдерживать испытание на хлориды.
#Сульфаты (2.4.13). Не более 0,06 % (600 ppm). 5 мл раствора S доводят водой дистиллированной Р до объема 15 мл. Полученный раствор должен выдерживать испытание на сульфаты.
Тяжелые металлы (2.4.8, метод F). Не более 0,002 % (20 ppm). 1,0 г испытуемого образца должны выдерживать испытание на тяжелые металлы. Эталон готовят с использова-
нием 2 мл эталонного раствора свинца (10 ppm Pb) Р.
#Остаточные количества органических растворителей (2.4.24). Испытуемый образец должен выдерживать требования статьи (5.4).
#Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Динатрия эдетат в условиях испытаний обладает антимикробным действием в от-
ношении Escherichia coli, Staphylococcus aureus, Pseudomonas aeruginosa. Методы устранения антимикробного действия неэффективны. Посев на среды № 1 и № 2 проводят методом мембранной фильтрации.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,500 г испытуемого образца растворяют в воде Р и доводят объем раствора этим же растворителем до 300 мл. К полученному рас-
твору прибавляют 2 г гексаметилентетрамина Р, 2 мл кислоты хлористоводородной разведенной Р и титруют 0,1 М раствором свинца нитрата, используя около 50 мг индикаторной смеси ксиленового оранжевого Р.
1 мл 0,1 М раствора свинца нитрата соот-
ветствует 37,22 мг C10H14N2Na2O8·2H2O.
ХРАНЕНИЕ В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А.
CO2H
N
HO2C CO2H
CO2H
А. Нитрилотриуксусная кислота.
# Диэтаноламин
|
H |
|
N |
HO |
OH |
C4H11NO2 |
М.м. 105,1 |
ОПРЕДЕЛЕНИЕ
Диэтаноламин представляет собой смесь этаноламинов, содержащую в основном диэтанол амин.Содержитнеменее98,5 %инеболее101,0 % этаноламинов в пересчете на безводный C4H11NO2.
ОПИСАНИЕ (свойства)
Прозрачная бесцветная или слегка желтоватая вязкая жидкость либо кристаллы, расплывающиеся на воздухе.
Смешивается с водой, ацетоном и 96 % спиртом.
Относительная плотность: около 1,09. Температура плавления кристаллов: около
28°С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
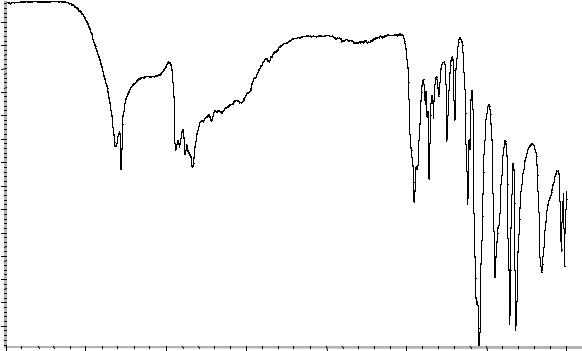
Инфракрасный спектр пропускания (2.2.24) испытуемого образца должен соответствовать спектру ФСО диэтаноламина или спектру, представленному на рисунке 1.
ИСПЫТАНИЯ
Раствор S. 5,0 г испытуемого образца раст-
воряют в воде, свободной от углерода диокси-
да, Р и доводят объем раствора этим же растворителем до 100 мл.
рН (2.2.3). От 10,0 до 11,5. Измеряют рН раствора S.
Показательпреломления(2.2.6).От1,4770
до 1,4790.
Температура затвердевания (2.2.17). Не ниже 25,7°С.
Вода (2.5.12). Не более 0,15 %. Определение проводят из 20,000 г испытуемого образца. В качестве растворителя используют смесь из
25 мл кислоты уксусной ледяной Р и 40 мл метанола Р.
Остаточные количества органических растворителей (2.4.24). Испытуемый образец должен выдерживать требования статьи (5.4).
Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Диэтаноламин в условиях испытания обладает антимикробным действием. Посев на питательные среды № 2 и № 8 проводят из разведения 1:10, на питательные среды № 1
и № 11 — 1:20.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
3,000 г испытуемого образца растворяют в
50 мл воды Р и титруют 1 М раствором кислоты хлористоводородной до розового окрашивания,
118 |
Государственная фармакопея Республики Беларусь |
|
|
П а
95 |
|
|
|
|
|
|
|
90 |
|
|
|
|
|
|
|
85 |
|
|
|
|
|
|
|
80 |
|
|
|
|
|
|
|
75 |
|
|
|
|
|
|
|
70 |
|
|
|
|
|
|
|
65 |
|
|
|
|
|
|
|
60 |
|
|
|
|
|
|
|
55 |
|
|
|
|
|
|
|
50 |
|
|
|
|
|
|
|
45 |
|
|
|
|
|
|
|
40 |
|
|
|
|
|
|
|
35 |
|
|
|
|
|
|
|
30 |
|
|
|
|
|
|
|
4000 |
3500 |
3000 |
2500 |
2000 |
1500 |
1000 |
500 |
|
|
|
В ( -1) |
|
|
|
|
Рисунок 1. Инфракрасный спектр пропускания ФСО диэтаноламина.
используя в качестве индикатора 0,1 мл раст-
вора метилового оранжевого Р.
1 мл 1 М раствора кислоты хлористоводо-
родной соответствует 105,14 мг C4H11NO2.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защищенном от света месте.
Желатин
Gelatina
GELATIN
ОПРЕДЕЛЕНИЕ
Желатин представляет собой очищенный белок, полученный неполным кислотным (тип А), или неполным щелочным (тип В), или ферментным гидролизом животного коллагена (включая рыбий и птичий); может также быть смесью различных типов.
# Производитель должен гарантировать соответствие желатина требованиям статьи 5.2.8.
В результате гидролиза получают гелеобразующий или не образующий геля желатин. Требования данной статьи распространяются на оба сорта продукта.
Желатин, описанный в данной статье, непригоден для парентерального применения или других специальных целей.
ОПИСАНИЕ (СВОЙСТВА)
Твердое вещество от светло-желтого до слегка желтовато-коричневого цвета, обычно
в виде полупрозрачных листков, кусков, гранул или порошка.
Практически нерастворим в обычных органических растворителях; набухает в холодной воде и при нагревании образует коллоидный раствор, который дает при охлаждении более или менее твердый гель.
Изоэлектрическая точка — важный параметр качества для применения желатина в различных целях: для желатина типа А обычно находится в пределах рН 6,0 и рН 9,5 и для типа В — в пределах рН 4,7 и рН 5,6.
Различные формы водных растворов желатина могут различаться по прозрачности и окраске. Для конкретного применения обычно используют подходящую спецификацию по испытаниям «Прозрачность» и «Цветность».
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 2 мл раствора S, приготовленного, как указано в разделе «Испытания», прибавляют
0,05 мл раствора меди сульфата Р. Переме-
шивают и прибавляют 0,5 мл раствора натрия гидроксида разведенного Р. Появляется фиоле-
товое окрашивание.
В.0,5гиспытуемогообразцапомещаютвпробирку, прибавляют 10 мл воды Р. Оставляют на 10мин,затемнагреваютпри60°Свтечение15мин ивыдерживаютвертикальновтечение6чпри0°С. Переворачивают пробирку. Для негелеобразующих сортов содержимое вытекает сразу, а для гелеобразующих сортов — не вытекает сразу.
ИСПЫТАНИЯ
Раствор S. 1,00 г испытуемого образца растворяют в воде, свободной от углерода ди-

Желатин |
119 |
оксида, Р при температуре около 55°С, доводят объем этим же растворителем до 100 мл и при этой температуре проводят испытания.
рН (2.2.3). От 3,8 до 7,6. Измеряют рН раствора S.
Удельнаяэлектропроводность(2.2.38).Не более 1 мС.см-1 в 1,0 % растворе при (30±1,0)°С.
Серы диоксид (2.5.29). Не более 0,005 % (50 ppm).
Пероксиды. Не более 0,001 % (10 ppm).
Определение проводят с использованием пе-
роксидных тест-полосок Р.
Органическийокислительно-восстановитель ный индикатор тест-полоски при окислении приобретает синее окрашивание. Интенсивность окраски пропорциональна количеству пероксида и для определения его концентрации может сравниваться с цветовой шкалой, прилагаемой к тест-полоскам.
Пригодность испытания. Погружают тестполоску на 1 с в эталонный раствор водорода пероксида (10 ppm H2O2) Р. Вынимают тестполоску, стряхивают избыток жидкости и через 15 с сравнивают с цветовой шкалой. Цвет тестполоски должен соответствовать концентрации 10 ppm по шкале. В противном случае испытание является недействительным.
Испытание. 20,0±0,1 г испытуемого образца помещают в лабораторный стакан и прибавляют 80,0±0,2 мл воды Р. Перемешивают до полного увлажнения желатина и оставляют образец при комнатной температуре на 1—3 ч. Накрывают стакан часовым стеклом. Помещают стакан
вводяную баню с температурой (65±2)°С на 20±5 мин для растворения образца. Перемешивают содержимое стакана стеклянной палочкой до получения однородного раствора. Погружают тест-полоску на 1 с в испытуемый раствор. Вынимают тест-полоску, стряхивают избыток жидкости и через 15 с сравнивают с цветовой шкалой. Концентрацию, определенную по цветовой шкале, умножают на 5 для расчета содержания пероксида в испытуемом образце в ppm.
Прочность студня (число Блюм). От 80 %
до 120 % от указанного номинального числа. Сила(прочность)студнявыражаетсявмассе
вграммах, необходимой для такой силы, которая при приложении на плунжер диаметром 12,7 мм дает погружение 4 мм в студень с концентрацией 6,67 % (м/м), выдержанный при 10°С.
Оборудование. Текстурный анализатор, или желометр, состоит из:
––цилиндрического плунжера диаметром 12,7±0,1 мм с ровной напорной поверхностью с круглыми краями;
––бутыли с внутренним диаметром 59±1 мм и высотой 85 мм;
Прибор настраивают согласно инструкции производителя. Параметры: расстояние 4 мм, скорость 0,5 мм/с.
Методика. Проводят испытание дважды. По 7,5 г испытуемого образца помещают в каждую бутыль. Прибавляют 105 мл воды Р. Каждую бутыль накрывают часовым стеклом и оставляют на 1—4 ч. Нагревают на водяной бане в течение 15 мин при температуре (65±2)°С, помешивая стеклянной палочкой. Убеждаются, что раствор однородный и что весь конденсат в бутылях инкорпорирован. Охлаждают при комнатной температуре в течение 15 мин и помещают бутыли в термостат с контролируемой температурой (10,0±0,1)°С и приспособлением, удерживающим платформу, на которой стоят бутыли, в строго горизонтальном положении. Закрывают бутыли резиновыми пробками и оставляют на 17±1 ч. Вынимают бутыли, быстро протирают насухо. Последовательно устанавливают бутыли на платформу прибора, настроив последний так, чтобы плунжер соприкасался с поверхностью студня без усилий. Проводят измерение. Результат представляют как среднее значение двух измерений.
Железо. Не более 0,003 % (30 ppm). Атомно-
абсорбционная спектрометрия (2.2.23, метод 1).
Испытуемый раствор. 5,00 г испытуемого образца помещают в коническую колбу, прибав-
ляют 10 мл кислоты хлористоводородной Р. За-
крывают колбу и помещают на водяную баню при температуре от 75°С до 80°С на 2 ч. Охлаждают и доводят содержимое колбы до 100,0 г водой Р.
Раствор сравнения. Готовят с использова-
нием эталонного раствора железа (8 ppm Fe)
Р, при необходимости разводят водой Р.
Длина волны: 248,3 нм.
Хром. Не более 0,001 % (10 ppm). Атомно-
абсорбционная спектрометрия (2.2.23, метод 1).
Испытуемый раствор. Готовят, как описано в испытании на железо.
Раствор сравнения. Готовят с использова-
нием эталонного раствора хрома (100 ppm Cr)
Р, при необходимости разводят водой Р.
Длина волны: 357,9 нм.
Цинк. Не более 0,003 % (30 ppm). Атомно-
абсорбционная спектрометрия (2.2.23, мeтод 1).
Испытуемый раствор. Готовят, как описано в испытании на железо.
Раствор сравнения. Готовят с использова-
нием эталонного раствора цинка (10 ppm Zn)
Р, при необходимости разводят водой Р.
Длина волны: 213,9 нм.
Потерявмассепривысушивании(2.2.32).
Не более 15,0 %. 1,000 г испытуемого образца сушат при температуре от 100°С до 105°С # в течение 18 ч.
Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Желатин в условиях испытаний не обладает антимикробным действием.
ХРАНЕНИЕ
В защищенном от влаги месте # при температуре не выше 25°С.
120 |
Государственная фармакопея Республики Беларусь |
|
|
МАРКИРОВКА
Указывают:
––силу геля (число Блюм) или это негеле образующий сорт.
# ФУНКЦИОНАЛЬНО ОБУСЛОВЛЕННЫЕ ХАРАКТЕРИСТИКИ
Если предполагается использование желатина при производстве твердых и мягких капсул, рекомендуется проводить следующие дополнительные испытания:
pH 6,67 % (м/м) раствора желатина (2.2.3).
Определяют рН 6,67 % (м/м) раствора желатина при температуре 60°С.
рН 6,67 % (м/м) раствора желатина типов А и В с разным числом Блюм (Bl) должен быть следующим:
––желатин типа А (из свиных шкур) 270 Bl —
от 5,0 до 5,7;
––желатин типа В (из говяжьих шкур и говяжьих костей) 250 Bl — от 5,0 до 6,0;
––желатин типа А (из свиных шкур) 175 Bl —
от 5,0 до 5,7;
––желатин типа В (из говяжьих шкур и говяжьих костей) 150 Bl — от 5,5 до 6,0.
Размер частиц. 100,0 г испытуемого образца просеивают в течение 3 мин со скоростью 1 удар в секунду.
Не менее 90 % порошка должно пройти через сито с размером отверстий 5,0 мм и остаться на сите с размером отверстий 0,5 мм.
Прозрачность (2.2.1). Раствор S, полученный из желатина типов А и В, по степени мутности не должен превышать эталон IV.
Цветность (2.2.2, метод II). Окраска раствора S должна быть не интенсивнее эталона
Y(Ж)4.
Продолжительность растворения. Не бо-
лее 25 мин. 10,0 г испытуемого образца, взвешенного с точностью до 0,01 г, помещают в лабораторный стакан, прибавляют 100 мл воды Р
стемпературой от 15°С до 18°С и оставляют набухать при этой же температуре 30 мин. Затем стакан с содержимым помещают в термостат
стемпературой (40±1)°С и выдерживают при помешивании до полного растворения желатина.
Время, прошедшее с момента доведения температуры в стакане до 40°С до полного раст ворения желатина, принимают за продолжительность растворения.
Динамическая вязкость. Динамическая вязкость 6,67 % (м/м) раствора желатина при 60°С в пересчете на желатин с содержанием влаги 11,5 % должна быть следующая:
––желатин типа А (из свиных шкур) 270 Bl — от 4,2 до 4,8 мПа.с;
––желатин типа В (из говяжьих шкур и говяжьих костей) 250 Bl — от 4,2 до 4,8 мПа.с;
––желатин типа А (из свиных шкур) 175 Bl — от 2,7 до 3,2 мПа.с;
––желатин типа В (из говяжьих шкур и говяжьих костей) 150 Bl — от 3,5 до 4,0 мПа.с.
Вязкость 6,67 % (м/м) раствора желатина определяется при температуре 60°С измерением времени истечения 100 мл раствора через стандартную пипетку.
Вязкость желатина определяется как динамическая вязкость 6,67 % (м/м) раствора желатина в воде при температуре 60°С, выраженная в мПа.с.
Для калибровки при температуре 60°С используют два стандартных калибровочных масла с вязкостью в пределах 2—10 сСт (1 сСт = 0,01 Ст). Вязкость масел должна различаться как минимум в два раза.
Оборудование:
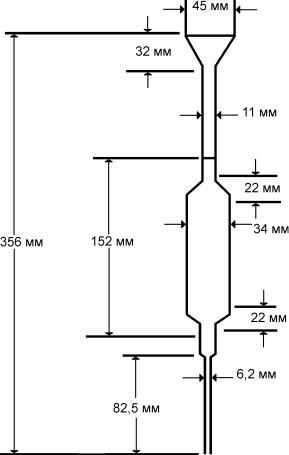
1. Пипетка вместимостью 100 мл с определенным диаметром входного и выходного отверстия и отметкой нижнего уровня на стекле (рисунок 1).
Рисунок 1. Пипетка для определения вязкости.
Могут использоваться другие аналогичные типы вискозиметра (например, U-образный вискозиметр — рисунок 2).
2.Термостатическая баня для пипетки с возможностью для термостатического перемешивания и температурой (60±0,1)°С.
3.Точный термометр (ртутный или электронный) с ценой деления 0,1°С, с длинным тонким корпусом для измерения температуры внутри пипетки.