

Gosudarstvennaya_farmakopeya_RB
.pdfБаза данных пригодна только для исходного прибора, либо для подобного
прибора при условии определения того, что перенесенная на прибор база данных остаётся пригодной.
Методика. Подготовку и исследование пробы проводят таким же образом,
как и при создании базы данных. Для облегчения сравнения спектров могут быть
использованы подходящие математические преобразования lg (1/T) или lg (1/R) спектра для образца и спектральной библиотеки сравнения, например, получение второй производной.
Сравнение преобразований спектров исследуемого образца и спектральной
библиотеки сравнения включает использование подходящей методики хемометрической классификации.
2.2.41. КРУГОВОЙ ДИХРОИЗМ
Круговым дихроизмом называют разность оптических плотностей в пределах
полосы поглощения для света с левой и правой круговой поляризацией, присущую оптически активным веществам.
Прямое измерение даёт величину:
|
|
A = AL − AR |
|
|
|
A |
– |
оптическая плотность кругового дихроизма, |
|||
AL |
– |
оптическая плотность для света с левой круговой поляризацией, |
|||
AR |
– |
оптическая плотность для света с правой круговой поляризацией. |
|||
|
Круговой дихроизм рассчитывают по уравнению: |
||||
|
|
ε = ε L − ε R = |
|
A |
|
|
|
c |
× l |
||
|
|
|
|||
ε |
– |
молярный круговой дихроизм или молярный дифференциальный |
|||
дихроичный коэффициент поглощения, выраженный в л×моль-1 ×см-1, |
|||||
εL |
– |
молярный коэффициент поглощения (2.2.25) для света с левой круговой |
|||
поляризацией, |
|
|
|
||
εR |
– |
молярный коэффициент поглощения для света с правой круговой |
|||
поляризацией, |
|
|
|
||
c |
– |
концентрация вещества в исследуемом растворе в моль×л−1, |
|||
l |
– |
длина оптического пути в сантиметрах. |
|||
Для характеристики кругового дихроизма могут быть также использованы следующие единицы:
Фактор диссимметрии:

g = εε
ε |
– |
молярный коэффициент поглощения (2.2.25). |
Молярная эллиптичность:
Некоторые типы приборов непосредственно показывают величину эллиптичности Θ, выраженную в градусах. При использовании таких приборов
молярная эллиптичность [Θ] может быть рассчитана по следующему уравнению:
|
[Θ] = |
Θ × M |
|
|
c × l ×10 |
||
|
|
||
[Θ] – |
молярная эллиптичность, выраженная в градус×см2×децимоль−1, |
||
Θ– величина эллиптичности, показываемая прибором,
M |
– |
относительная молекулярная масса исследуемого вещества, |
c |
– |
концентрация исследуемого вещества в растворе в г/мл, |
l |
– |
длина оптического пути в сантиметрах. |
Молярная эллиптичность также связана с молярным круговым дихроизмом следующим уравнением:
[Θ] = 2,303 ε 4500π ≈ 3300 ε
Молярная эллиптичность часто используется при анализе белков и нуклеиновых кислот. В этом случае молярная концентрация, выраженная в
единицах мономерных остатков, рассчитывается, исходя из выражения:
молекулярная масса число аминокислот
Для белков среднее значение относительной молекулярной массы
мономерного остатка составляет от 100 до 120 (в среднем 115), для нуклеиновых кислот (в виде натриевой соли) – около 330.
Прибор. Источником света (S) является ксеноновая лампа (Рис. 2.2.41.-1); свет проходит через двойной монохроматор (M), снабжённый кварцевыми
призмами (P1, P2).
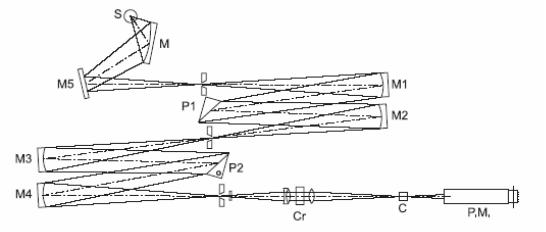
Рис. 2.2.41.-1. - Оптическая схема дихрографа
Линейный луч из первого монохроматора расщепляется на два компонента, поляризуемые под правым углом вторым монохроматором. Дополнительный луч устраняется выходной щелью монохроматора.
Поляризованный и монохроматический свет проходит через двоякопреломляющий модулятор (Cr): в результате образуется переменный свет с круговой поляризацией.
Затем луч проходит через исследуемый образец (C) и попадает на фотоумножитель (PM), за которым следует усилитель, производящий два
электрических сигнала: один постоянного тока Vc, а другой переменного тока с
частотой модуляции Vac характерной для исследуемого образца. Фаза является показателем кругового дихроизма. Отношение Vac/Vc пропорционально
дифференциальной оптической плотности A, которая создаётся сигналом.
Дихрограф обычно позволяет проводить измерения в диапазоне длин волн от 170 до 800 нм.
Градуировка прибора. Точность шкалы оптической плотности.
Растворяют 10,0 мг изоандростерона R в диоксане R и доводят до 10,0 мл этим же растворителем. Снимают спектр кругового дихроизма раствора в диапазоне от 280 нм до 360 нм. Величина ε, измеренная в максимуме при 304 нм, равна + 3,3.
Также может быть использован раствор (1S)-(+)-10-камфорсульфоновой кислоты
R.
Линейность модуляции. Растворяют 10,0 мг 1S)-(+)-10-камфорсульфоновой
кислоты R в воде R и доводят этим же растворителем до 10,0 мл. Определяют точную концентрацию камфорсульфоновой кислоты в растворе методом УФ-
спектрофотометрии (2.2.25), используя величину удельного показателя поглощения
при 285 нм, равную 1,49.
Снимают спектр кругового дихроизма раствора в диапазоне от 185 нм до 340
нм. Величина ε, измеренная в максимуме при 290,5 нм, равна от + 2,2 до + 2,5, а
измеренная в максимуме при 192,5 нм – от – 4,3 до – 5.
Может быть использован (1S)-(+)- или его оптический изомер -(1R)-(+)-
камфорсульфонат аммония R.
2.2.42. Плотность твердых тел
Плотность твердых тел соответствует их средней массе единицы объема и
обычно выражается в граммах на сантиметр кубический (г/см3), хотя
международной единицей является килограмм на метр кубический (1 г/см3=1000
кг/м3).
Вотличие от газов и жидкостей, плотность которых определятся только
температурой и давлением, плотность твердых тел зависит также от строения молекул и поэтому изменяется в зависимости от структуры кристалла и степени кристалличности.
Втом случае, когда твердые частицы являются аморфными или частично
аморфными, их плотность зависит от способа получения и обработки.
Поэтому, в отличие от жидкостей, плотности двух химически эквивалентных
твердых веществ могут быть различными, и это отличие отражает соответствующие различия в строении твердых тел.
Плотность частиц – важная физическая характеристика фармацевтических субстанций.
Плотность твердой частицы может принимать различные значения в зависимости от метода, используемого для измерения ее объема. Следует различать три уровня выражения плотности:
-кристаллическая плотность, которая используется только для твердой фракции материала; кристаллическую плотность также называют истинной
плотностью;
-плотность частиц, которая характеризует также объем пор внутри твердых
частиц;
-объемная плотность, которая включает объемы пустот между частицами, образованные в слое порошка; объемную плотность называют также кажущейся плотностью (насыпной массой).
Кристаллическая плотность - это средняя масса единицы объема вещества, не считая всех пустот, которые не являются неотъемлемой характеристикой кристаллической
решетки вещества. Это свойство, присущее веществу и, поэтому ее величина не должна
зависеть от способа определения. Кристаллическая плотность может быть определена как расчетным путем, так и путем простого измерения.
А. Расчетную кристаллическую плотность получают исходя из
кристаллографических данных (размер и состав элементарной ячейки) идеального кристалла, например, при анализе дифракции рентгеновских лучей, и молекулярной массы вещества.
В. Измеренная кристаллическая плотность - это отношение массы к объему,
определенных для монокристалла вещества.
Плотность частицы вещества
Плотность частицы вещества учитывает как кристаллическую плотность, так и пористость частицы вещества (закрытые и открытые поры). Таким образом, плотность
частицы зависит от величины установленного объема, что в свою очередь зависит от метода измерения. Плотность частицы вещества может быть установлена следующими
двумя методами.
А. Пикнометрическая плотность определяется путем измерения объема, занимаемого известной массой порошкообразного вещества, который эквивалентен
объему газа, вытесненного веществом (для его определения используется пикнометр газового вытеснения 2.9.23). Объем вещества, установленный таким способом, включает объем, занимаемый открытыми порами, однако он не включает объем закрытых не
доступных для газа пор. Благодаря высокой диффузионной активности гелия, который является наиболее предпочтительным для определения газом, большинство открытых
пор доступны для газа. Поэтому пикнометрическая плотность тонко измельченного
порошка, в общем, не сильно отличается от значений кристаллической плотности.
В. Плотность, установленная при помощи ртутного измерителя пористости, также
называемая гранулярной плотностью. В этом методе также не учитывается объем, занимаемый закрытыми порами, однако объем открытых пор учитывается только для пор, размер которых превышает некоторый предел. Значение этой предельной величины пор зависит от давления, под которым подается ртуть во время измерения, и при обычных
условиях измерения ртуть не проникает в мельчайшие поры, доступные для гелия. Для одного образца могут быть получены различные значения гранулярной плотности, так как возможно определение плотности для каждого используемого рабочего давления, что соответствует различным значениям предельных величин пор при различных давлениях.
Плотность (насыпная масса) при свободной засыпке и уплотнении
Плотность при свободной засыпке порошка учитывает вклад свободного объема
между частицами порошка. Следовательно, плотность при свободной засыпке зависит как от плотности частиц порошка, так и от их пространственного расположения в слое порошка.
Плотность при свободной засыпке зачастую очень сложно измерить, так как самые незначительные нарушения в слое могут приводить к новому значению плотности.
Таким образом, важным требованием при сообщении значений насыпной плотности
является указание метода ее определения.
А. Значение плотности при свободной засыпке определяется измерением объема известной массы порошка, прошедшего через сито, мерным цилиндром (2.9.15).
В. Плотность при уплотнении достигается путем механического постукивания
мерным цилиндром, содержащим образец порошка. После измерения начального объема
цилиндр подвергается механическому постукиванию, и наблюдение за объемом продолжают до тех пор, пока дальнейшее изменение объема становится
незначительным (2.9.15).
2.2.43. МАСС-СПЕКТРОМЕТРИЯ
Масс-спектрометрия основана на прямом измерении отношения массы к
числу элементарных положительных или отрицательных зарядов ионов (m/z), полученных из анализируемого вещества и находящихся в газовой фазе. Данное отношение выражается в атомных единицах массы (1 а.е.м. = одной двенадцатой массы нуклида 12С) или в дальтонах (1 Да = массе атома водорода).
Ионы, образовавшиеся в ионном источнике, ускоряются и перед попаданием
в детектор разделяются с помощью масс-анализатора. Все эти действия
происходят в камере, в которой насосная система поддерживает вакуум от 10−3 до
10−6 Па.
Результирующий масс-спектр является графиком зависимости относительного количества различных ионов от отношения m/z. Сигнал,
отвечающий иону, представлен несколькими пиками, соответствующими
статистическому распределению различных изотопов этого иона. Такой сигнал называется изотопным профилем, а пик (по крайней мере, для небольших
молекул), представляющий наиболее распространённые изотопы для каждого
атома, - моноизотопным пиком.
Масс-спектрометрический анализ даёт важную качественную (определение
молекулярных масс; информация, касающаяся структуры определяемых фрагментов) и количественную (с использованием внешнего или внутреннего
стандартов) информацию с пределом обнаружения от пикомоля до фемтомоля.
ВВОД ОБРАЗЦА
Первой стадией анализа является ввод образца в прибор без значительного
нарушения вакуума. В широко распространённом методе, называемом прямым вводом жидкости, образец помещается на конец цилиндрического штока (в
кварцевом тигле, на проволоке или на металлической поверхности). Этот шток через вакуумный шлюз, в котором поддерживается первичный промежуточный вакуум между атмосферным давлением и вторичным вакуумом прибора, вводится в
спектрометр.
Другие системы ввода позволяют проанализировать компоненты смеси, так как они разделяются с помощью соответствующего прибора, соединённого с масс-
спектрометром.
Газовая хроматография/масс-спектрометрия. При использовании
подходящих колонок (капиллярных или полукапиллярных) возможно непосредственное введение конца колонки в ионный источник прибора без
применения сепаратора.
Жидкостная хроматография/масс-спектрометрия. Такая комбинация особенно полезна для анализа полярных соединений, которые являются недостаточно летучими либо слишком термолабильными, для того чтобы их можно
было проанализировать методом газовой хроматографии в сочетании с масс-
спектрометрией. Данный метод осложняется трудностью получения ионов в газовой фазе из жидкой фазы, что требует применения специальных интерфейсов, таких
как:
-прямой жидкостный ввод: подвижная фаза распыляется и растворитель испаряется перед ионным источником прибора,
-интерфейс с пучком частиц: подвижная фаза, скорость которой может достигать 0,6 мл/мин, распыляется в десольватационной камере, в результате в ионный источник прибора попадают лишь анализируемые вещества в нейтральной форме; данный способ может быть использован для соединений с относительно низкой
полярностью и молекулярными массами меньше 1000 Да,
-интерфейс с движущейся полосой: подвижная фаза, скорость которой может достигать 1 мл/мин, наносится на поверхность движущейся полосы; после
выпаривания растворителя анализируемые вещества переносятся к ионному
источнику прибора, где подвергаются ионизации; данный способ плохо подходит
для очень полярных или термолабильных соединений.
Другие виды интерфейсов (электрораспыление, термораспыление,
химическая ионизация при атмосферном давлении) могут рассматриваться как самостоятельные методы ионизации и описаны в разделе, посвящённом способам
ионизации.
Сверхкритическая флюидная хроматография/масс-спектрометрия.
Подвижная фаза, обычно состоящая из находящегося в сверхкритическом состоянии диоксида углерода, переходит в газообразное состояние после
прохождения через нагретую заслонку, находящуюся между колонкой и ионным
источником.
Капиллярный электрофорез/масс-спектрометрия. Элюент вводится в
ионный источник, в некоторых случаях после добавления дополнительного растворителя, для того чтобы достичь скорости потока порядка нескольких миллилитров в минуту. Ограничениями данного метода являются малые
количества вводимого образца и необходимость использовать летучие буферные
растворы.
СПОСОБЫ ИОНИЗАЦИИ
Электронный удар. Образец, находящийся в газообразном состоянии, ионизируется потоком электронов, энергия которых (обычно 70 эВ) больше энергии ионизации образца. При этом кроме молекулярного иона M+ образуются осколочные ионы характерные для данной молекулярной структуры. Главным ограничением данного способа является необходимость испарения образца, что
делает невозможным его применение для полярных, термолабильных или высокомолекулярных соединений. Ионизация электронным ударом может быть
использована в газовой хроматографии, соединённой с масс-спектрометрией, а в некоторых случаях и в жидкостной хроматографии.
Химическая ионизация. Этот тип ионизации предполагает использование
реагентного газа, такого как метан, аммиак, монооксид азота, диоксид азота или
кислород. Спектр характеризуется ионами (M + H)+ или (M − H)- типов или ионамиаддуктами, образованными из аналита и используемого газа. Осколочные ионы образуются реже, чем при ионизации электронным ударом. Для термолабильных
веществ используется разновидность данного метода ионизации, при которой
образец, находящийся на проволоке, очень быстро испаряется вследствие
эффекта Джоуля-Томсона (десорбционная химическая ионизация).
Бомбардировка быстрыми атомами (FAB) или ионизация бомбардировкой быстрыми ионами (жидкостная масс-спектрометрия вторичных ионов – LSIMS). Образец, растворённый в вязкой матрице, например в глицерине, наносится на металлическую поверхность и ионизируется потоком нейтральных атомов, например, аргона или ксенона или обладающими большой кинетической энергией ионами цезия. Образуются ионы (M + H)+ или (M − H)– типов либо ионы-аддукты, сформированные матрицей или образцом. Данный тип ионизации хорошо подходит для полярных, термолабильных соединений, имеющих
молекулярную массу до 10000 Да. При добавлении к подвижной фазе 1 – 2%
глицерина он может быть использован для жидкостной хроматографии, однако скорость подвижной фазы должна быть очень низкой (несколько микролитров в
минуту). При нанесении тонкого слоя матрицы на поверхность хроматографических
пластинок такой способ ионизации может быть использован и в тонкослойной хроматографии.
Полевая десорбция или полевая ионизация. Образец испаряется около
вольфрамовой проволоки, покрытой микроиглами (полевая ионизация) или помещается на эту проволоку (полевая десорбция). Сильное электрическое поле
(напряжение около 10 кВ), возникающее между проволокой и противоэлектродом, ионизирует образец. При данных способах ионизации образуются
преимущественно молекулярные ионы M+ и (M + H)+ ионы. Эти способы используются для малополярных и/или термолабильных соединений.
Лазерная десорбция – ионизация с помощью матрицы (MALDI). Образец
смешанный с подходящей матрицей и помещённый на металлическую подложку,
ионизируется импульсным лазерным излучением с длиной волны от УФдо ИКдиапазона (продолжительность импульсов может составлять от пикосекунды до нескольких наносекунд). Данный способ ионизации играет важную роль при анализе
соединений с очень большой молекулярной массой (более чем 100 000 Да), но ограничен использованием времяпролётного анализатора (см. ниже).
Электрораспылительная ионизация. Данный способ ионизации проводится
при атмосферном давлении. Образец, находящийся в растворе, вводится в
источник через капилляр, конец которого имеет потенциал порядка 5 кВ. Для
облегчения распыления может использоваться газ. Испарение молекул растворителя из образующихся микрокапель приводит к образованию в газовой
фазе однозарядных или многозарядных ионов. Скорости подвижной фазы при данном виде ионизации могут изменяться от нескольких микролитров в минуту до 1 мл/мин. Такая ионизация подходит для полярных соединений, а также для исследования биомолекул с молекулярными массами до 100 000 Да. Она может
сочетаться с жидкостной хроматографией или капиллярным электрофорезом.
Химическая ионизация при атмосферном давлении (APCI). Ионизация
проводится при атмосферном давлении под действием электрода, имеющего потенциал несколько киловольт и помещённого на пути подвижной фазы, которая
распыляется как вследствие тепловых эффектов, так и благодаря использованию потока азота. Образующиеся ионы являются однозарядными и относятся к (M + H)+
типу в случае положительного заряда и к (M − H)– типу в случае отрицательного. Возможность использования высоких скоростей подвижной фазы (до 2 мл/мин) делает этот способ ионизации идеальным для сочетания с жидкостной хроматографией.
Термораспылительная ионизация. Образец, находящийся в подвижной фазе, состоящей из воды, органических модификаторов и содержащей летучий электролит (обычно ацетат аммония) вводится в распыленной форме после
прохождения через металлический капилляр, температура которого
контролируется. Допускаются скорости подвижной фазы порядка 1 мл/мин – 2 мл/мин. Ионы электролита ионизируют анализируемое соединение. Такой процесс ионизации может быть заменён или усилен электрическим разрядом величиной около 800 вольт, особенно при использовании только органического растворителя. Данный способ ионизации может быть использован в методе жидкостной хроматографии-масс-спектрометрии.
АНАЛИЗАТОРЫ
Различия в работе анализаторов зависят, главным образом, от двух
параметров:
-диапазон, в котором могут быть измерены отношения m/z (массовый диапазон),
-разрешение, характеризуемое возможностью разделить два иона одинаковой
интенсивности с отношениями m/z, отличающимися на M и перекрывающиеся при
определённом процентном превышении базовой линии; например, разрешение (M/ M) равное 1000 с 10% превышением базовой линии позволяет провести
разделение отношений m/z 1000 и 1001 с интенсивностями, на 10% превышающими базовую линию. В некоторых случаях (времяпролётные анализаторы,
квадрупольные анализаторы, ионные ловушки) разрешение может быть определено как отношение между молекулярной массой и шириной пика на
половине высоты (50% превышение базовой линии).
Магнитные и электростатические анализаторы. Ионы, образовавшиеся в ионном источнике, ускоряются потенциалом V и сосредотачиваются, в зависимости от устройства прибора, между магнитным (магнитное поле B) или электростатическим анализаторами (электростатическое поле E). В соответствии с
законом Лапласа они перемещаются по траектории с радиусом r:

m / z = B 2r 2
2V
Для накопления и измерения различных ионов, образовавшихся в источнике, могут быть использованы два типа сканирования: изменение B при постоянной величине V или изменение V при постоянной величине B. За магнитным анализатором обычно следует электрический сектор, который действует как фильтр
кинетической энергии и позволяет заметно увеличить разрешение прибора. Максимальное разрешение такого прибора (двойной сектор) изменяется от 10000
до 150000 и в большинстве случаев позволяет достаточно точно оценить величину отношений m/z для определения элементного состава соответствующих ионов. Для
однозарядных ионов массовый диапазон равен от 2000 до 15000 Да. Некоторые ионы могут разрушаться спонтанно (метастабильные переходы) или при
столкновении с газом (диссоциация, активированная столкновением (CAD)) в областях между ионным источником и детектором, где отсутствует поле. Исследование этих процессов разрушения очень полезно для определения структуры и характеристики определённого соединения, входящего в состав смеси,
и делает возможным использование тандемной масс-спектрометрии. В зависимости от места, в котором протекают процессы разрешения, известно много методик, которые могут быть использованы для этого:
-режим дочернего иона (определение состава ионов, образовавшихся при
разрушении определённого родительского иона): B/E = константа, MIKES (Massanalysed Ion Kinetic Energy Spectroscopy),
-режим родительского иона (определение всех ионов, образовавшихся при разрушении иона с определённым отношением m/z): B2/E = константа,
-режим нейтральных частиц (определение всех ионов, которые теряют идентичные фрагменты):
B/E(1 − E/E0)1/2 = константа, где E0 базовый потенциал электрического сектора.
Квадрупольные анализаторы. Анализатор состоит из четырёх
параллельных металлических стержней, имеющих цилиндрическое или
гиперболическое поперечное сечение. Они расположены симметрично по
отношению к траектории движения ионов; пары противоположных стержней присоединены к электрическому источнику. Потенциалы двух пар стержней противоположны. Они состоят из постоянного и изменяющегося компонентов. Ионы, образовавшиеся в ионном источнике, пропускаются и разделяются вследствие изменения потенциалов, приложенных к стержням, причём отношение постоянного потенциала к переменному должно оставаться неизменным. Квадрупольные анализаторы обычно имеют массовый диапазон от 1 атомной единицы массы до
2000 атомных единиц массы., хотя в некоторых случаях он может достигать 4000 атомных единиц массы. Несмотря на то, что такие анализаторы имеют меньшее
разрешение, чем магнитные, они позволяют получать моноизотопный профиль однозарядных ионов, полученных во всём массовом диапазоне. Для получения спектров можно использовать три квадруполя, соединённых друг с другом, Q1, Q2, Q3 (Q2 служит ионизационной камерой, и в действительности не является
анализатором; чаще всего в качестве ионизирующего газа применяют аргон).
Наиболее часто используемыми режимами сканирования являются:
- режим дочернего иона: Q1 выбирает m/z ион, фрагменты которого, полученные
при ионизации в квадруполе Q2, анализируются квадруполем Q3,
-режим родительского иона: Q3 пропускает ионы только с определённым
соотношением m/z, в то время как Q1 сканирует определённый массовый диапазон. Детектируются только ионы, при разрушении которых образуется ион, отбираемый
квадруполем Q3.
-режим нейтральных частиц: Q1 и Q3 сканируют определённый массовый
диапазон, но в интервале, соответствующем фрагментам, которые теряет определённое вещество или группа веществ.
Для получения спектров возможно сочетание квадрупольных анализаторов с магнитными или электростатическими, такие приборы называют гибридными масс-
спектрометрами.
Ионные ловушки. Данные анализаторы имеют такой же принцип работы, что
и квадрупольные, но в них используются трёхмерные электрические поля.
Анализаторы такого типа позволяют получать спектры ионов нескольких генераций
(MSn).
Циклотронно-резонансные масс-анализаторы. Ионы, образовавшиеся в ячейке и подвергнутые действию интенсивного магнитного поля, движутся по круговым траекториям с частотами, которые могут быть непосредственно связаны с величинами отношений m/z для этих ионов при помощи преобразования Фурье.
Данное явление называется ионно-циклотронным резонансом. Анализаторы такого
типа состоят из сверхпроводящих магнитов и обладают очень высокой
разрешающей способностью (до 1000000 и выше), а также позволяют получать MSn спектры. Их недостатком является необходимость использования очень глубокого
вакуума (порядка 10−7 Па).
Времяпролётные анализаторы. Ионы, образовавшиеся в ионном источнике, ускоряются потенциалом величиной от 10 кВ до 20 кВ. Они проходят через анализатор, состоящий из бесполевой трубы длиной от 25 см до 1,5 м, обычно называемой пролётной трубой.
Время (t), за которое ион достигает детектора, пропорционально квадратному корню из отношения m/z. Теоретически массовый диапазон для такого анализатора
бесконечен. Практически он определяется методом ионизации или десорбции.
Времяпролётные анализаторы используются, главным образом, для высокомолекулярных соединений (вплоть до нескольких сотен тысяч дальтон).
Данный способ ионизации обладает очень высокой чувствительностью (достаточно
несколько пикомолей вещества). Точность измерений и разрешение таких приборов могут быть значительно улучшены при использовании электростатического зеркала (рефлектрона).
ПОЛУЧЕНИЕ СИГНАЛА
Имеются три возможных режима получения сигнала.
Режим полного спектра. Записывается полный сигнал, полученный для выбранного массового диапазона. Спектр представляет собой зависимость относительной интенсивности различных ионов от величины m/z. Получаемые
результаты являются, главным образом, качественными. Для более быстрой
идентификации возможно использование библиотек спектров сравнения.
Фрагментометрический режим (селективное сканирование ионов).
Получаемый сигнал ограничен одним (сканирование одного иона, single-ion monitoring (SIM)) или несколькими (множественное ионное сканирование, multipleion monitoring (MIM)) ионами, характерными для анализируемого вещества. В этом