

Билет 18. Кинетич. закон действующих масс
.docxБилет 18. Химическая кинетика
Химическая кинетика, как и термодинамика, является теоретической основой химической технологии, тепловой энергетики и металлургии.
Принципиальная возможность, направление и глубина протекания химической реакции, например, реакции горения, определяются законами термодинамики, которая анализирует, прежде всего, начальное и конечное состояние. Однако только кинетика выясняет реальную возможность и время достижения конечного состояния, в том числе путем анализа промежуточных состояний.
Например, исходная смесь H2 и O2, несмотря на чрезвычайную выгодность реакции H2 + O2 = H2O, rG(298) = -229 кДж/моль, может существовать очень долго и мгновенно, с взрывом протекает при ее инициировании искрой.
Термодинамический анализ реакции получения HJ позволил рассчитать выход продукта при 718 К, который при эквимолярном соотношении реагентов составил 77,2 % (см. пример 1 в разделе 3.9).
|
|
Рис. 3.2. Изменение концентрации H2 , I2 при синтезе (а) HI и его термической диссоциации (б) во времени при Р=1 атм и температуре 718 К. |
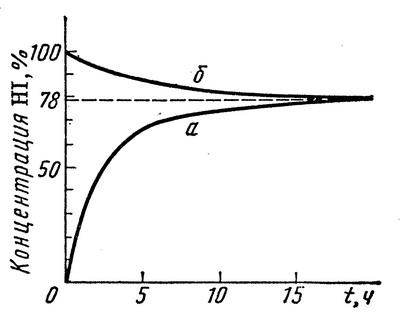
Специально поставленные Боденштейном кинетические измерения показали, что конечное равновесное состояние в этих условиях достигается за время 20 ч. При этом процесс взаимодействия водорода с йодом представляется совокупностью промежуточных элементарных реакций с их различным долевым участием, зависящим от температуры и соотношения реагентов.
H2 = H˙ + H˙, J2 = J˙ + J˙, H˙ + J˙ = HJ,
H˙ + J2 = HJ + J˙, J˙ + H2 = HJ + H˙
Кинетическое исследование взаимодействия H2 и O2 и ряда других процессов горения позволили Н.Н. Семенову разработать теорию цепных реакций, получить за это звание академика, а впоследствии и Нобелевского лауреата.
При нормальных условиях реакция образования воды из H2 и O2 не происходит из-за высокой энергии активации (см. далее). Инициирование реакции зарождает цепь созданием активных радикальных частиц H, OH, O:
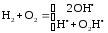
В результате элементарных реакций, составляющих звено цепи (ЗЦ) разветвленной цепной реакции, наряду с продуктом реакции образуются новые радикалы.
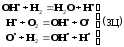
Баланс между ростом числа радикалов и их гибелью на окружающих стенках, инертных частицах определяет скорость процесса, которая при малых потерях радикалов носит экспоненциальный, взрывной характер.
Можно утверждать, что ядерная физика позаимствовала химические представления, например, в части цепной реакции деления ядер, условием возникновения которой является наличие размножающихся нейтронов.
Скорость химической
реакции по номенклатурным правилам
IUPAC
определяется как скорость возрастания
химической координаты реакции .
Напомним, что для реакции общего вида
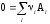 (i <
0 для реагентов), координата реакции
имеет вид: =
(ni–
ni0)/i
.
(i <
0 для реагентов), координата реакции
имеет вид: =
(ni–
ni0)/i
.
Таким образом, скорость гомогенной (объемной) реакции:
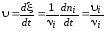 , (3.55)
, (3.55)
где υi – скорость изменения количества i‑го компонента, моль/с.
Более важна связанная с концентрацией ci скорость реакции r, отнесенная к единице объема V. При учете ni = ciV
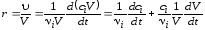 .
(3.56)
.
(3.56)
При постоянном объеме (V = const)
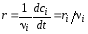 ,
,
 , (3.57)
, (3.57)
где компонентная скорость изменения концентрации ri > 0 для продуктов и ri < 0 для исходных веществ.
Для элементарной
реакции
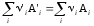 выражение
выражение
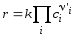 (3.58)
(3.58)
определяет основной закон кинетики или кинетический закон действующих масс (КЗДМ):
Скорость элементарной реакции равна произведению концентраций реагентов в степенях их стехиометрических коэффициентов.
Коэффициенты 'i в кинетическом ЗДМ – только целые числа 1, 2, 3. Коэффициент пропорциональности k называется константой скорости.
Впервые КЗДМ сформулирован Гульдбергом и Вааге в 1887 г.
Для двусторонней обратимой реакции
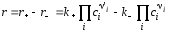 , (3.59)
, (3.59)
где k+, k− - константы скорости прямой и обратной реакций.
При достижении
равновесия (r =
0)
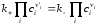 .
Таким образом, для простой двусторонней
реакции
.
Таким образом, для простой двусторонней
реакции
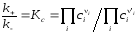 (3.60)
(3.60)
отношение констант скоростей прямой и обратной реакций оказывается равным константе равновесия. Это позволяет по k+, рассчитывать k− и наоборот.
В области далекой от равновесия коэффициенты, даже для элементарных реакций, i и 'i не тождественны стехиометрическим коэффициентам. На практике такое различие, в большинстве случаев, объясняется не элементарностью и скрытой многостадийностью большинства химических реакций.
По этой же причине n = 'i, называемая порядком реакции или ее молекулярностью, не совпадает с величиной, найденной из обработки кинетических кривых.
Объединение формул (3.57) и (3.58) дает дифференциальное уравнение, в данном случае односторонней реакции
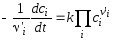 . (3.61)
. (3.61)
Приведем примеры решения или интегрирования этого уравнения для отыскания зависимостей концентраций реакционных компонентов от времени (т.н. кинетических кривых). Отметим, что во втором и третьем из рассмотренных простейших примеров представлены выражения для констант скоростей, из которых при необходимости могут быть получены явные зависимости с(t). Дополнительным индексом “0” отмечены начальные концентрации реагентов.
|
(1) |
A продукты |
|
|
(2) |
A + B продукты |
|
|
(3) |
A + B + C + ... продукты (c0A=c0B=..., cA=cB=...) |
|
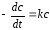 ,
,
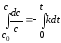 ,
ln c
–
ln c0
= – kt,
c
= c0e–kt
,
ln c
–
ln c0
= – kt,
c
= c0e–kt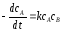 ,
,
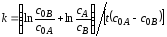
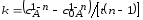
Более сложные задачи химической кинетики, включающие обратимые реакции и тем более системы из нескольких последовательно и параллельно протекающих реакций, не имеют аналитических решений и требуют применения численных методов.