

161
The reaction of TXT with two equivalents of substituted amines was carried out with the addition of NaOH, preventing acidification of the reaction mixture to give the disubstituted product 6-chloro-N2,N4-diphenyl-1,3,5-triazine-2,4-diamine 1.21 (Scheme 1.9).
Scheme 1.9
At the next stage, the authors carried out the synthesis of target hybrid compounds based on the reaction between compound 1.22, acetophenone, and ethyl 3-oxobutanoate using Bi(NO3)3 (Scheme 1.10).
Scheme 1.10
Cytotoxic activity of ethyl 1-(4,6-bis(phenylamino)-1,3,5-triazin-2-yl)-6-methyl-4- phenyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate 1.23 was evaluated in vitro with the cell lines: HeLa, MCF-7, HL-60 (human promyelocytic leukaemia cell), HepG2 (human hepatocellular carcinoma cell line) and MCF-12A (ductal-derived epithelial cell line human mammary gland) by the MTT method. Cisplatin was used as a comparator. The results showed that compound 1.23 exhibited moderate activity against cell lines HeLa (IC50 = 96.6 ± 0.5 μM), MCF-7 (IC50 = 87.4 ± 0.8 μM), HL-60 (IC50 = 83 .7 ± 0.3 μM), however, it does not show activity against the MCF-12A cell line, which indicates the selectivity of the cytotoxic effect of the resulting 1,3,5-triazine derivative.
Continuing to consider options for the synthesis of biologically active 1,3,5- triazines, we would like to highlight a study on the synthesis of 2,4-diamino-6-aryl-1,3,5- triazines, which were obtained using solid-phase synthesis [22]. The different amination reactivity of the monoaryl-substituted triazines results in the resin-bound amine selectively displacing chlorine in the crude reaction mixture to give triazine 1.24. The third chlorine

162
atom is then replaced by an amine in DMF at high temperature (Scheme 1.11). Resin cleavage was carried out by treatment with 50 % trifluoroacetic acid in dichloromethane. Thus, 2,4-diamino-6-aryl-1,3,5-triazines 1.25–1.27.
Scheme 1.11
Most of the obtained compounds demonstrated antiproliferative activity against cell lines of myelogenous leukaemia (K562), human prostate adenocarcinoma (PC-3 and HO8910). In particular, compound 1.26 exhibited marked inhibitory activity with IC50 values of 1.01, 2.23 and 1.06 μM against K562, PC-3 and HO8910 cell lines, respectively.
The synthesis of β-lactams, to which the dimorpholinotriazine fragment is attached, turned out to be successful [23]. The starting compound 4,4'-(6-chloro-1,3,5-triazine-2,4- diyl)dimorpholine (1.28) was synthesised from TXT in acetone and treated with a mixture of triethylamine and morpholine at −10 °C to give the adduct 1.28. The subsequent reaction of 1.28 with 4-nitroaniline in the presence of potassium carbonate and DMF led to the formation of trisubstituted triazine 1.29, in which the nitro group underwent reduction to form compound 1.30 (4,6-dimorpholino-N-(4-nitrophenyl)-1,3,5-triazine-2-amine; Scheme 1.12).
Рекомендовано к изучению сайтом МедУнивер - https://meduniver.com/

163
Scheme 1.12
Then Schiff bases 1.31 obtained from 1.30 reacted with substituted acetic acids in the presence of triethylamine and tosyl chloride in various molar ratios to give cis-β- lactams 1.32–1.33 in 75–90 % yields (Scheme 1.13).
Scheme 1.13
The effect of the obtained β-lactam-triazines on the HEK293, SW-1116, HepG2, and MCF-7 cell lines was assessed using the MTT test [23]. Notably, none of the described compounds showed inhibitory activity against non-tumour HEK293 cells. Against HepG2 and MCF-7 cell lines, compounds 1.32 and 1.33 showed inhibitory activity with IC50 values > 100 μM.
Another paper [24] described the synthesis of 2-N,4-N-bis[2-(2-{2-[4-(1,2-dicarba- closo-dodecaboran(12)-1-ylmethyl)-1H-1,2,3-triazol- 1-yl]ethoxy}ethoxy)ethyl]-6-N-(2- {2-[2-(2-nitro-1H-imidazol-1-yl)ethoxy]ethoxy}ethyl)-1,3,5-triazine-2,4,6-triamine, which was obtained from o-carborane. A fragment containing 2-nitroimidazole was synthesised according to Scheme 1.14 by nucleophilic substitution of mesylate with 2- nitroimidazole in DMF with Cs2CO3 as base at 110 °C. After treatment and purification, compound 1.34 was isolated in 72 % yield. Next, the azido group of compound 1.34 was reduced to an amine by the Staudinger reaction with triphenylphosphine in a two-phase system of Et2O and HCl. An aromatic nucleophilic substitution was then carried out by reaction with cyanuric chloride in the presence of diisopropylethylamine base (DIPEA; Scheme 1.14). Sequential introduction of substituents into the triazine framework was
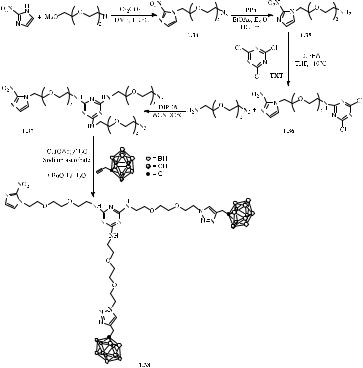
164
carried out in two stages under temperature control to ensure successive substitutions of chlorine atoms. To prevent double or triple substitution of the first chlorine atom in cyanuric chloride, successive reactions were carried out using an excess of TXT. The substitution of the remaining two chlorine atoms was carried out at room temperature. After the slow addition of DIPEA, the reaction mixture was stirred at 80 °C. Flash chromatography (dichloromethane (DCM) with 0–5 % MeOH solvent gradient) gave product 1.36 as a thick yellow oil (71 % yield). The last step in the preparation of compound
1.37 was associated with the use of Huisgen’s azide-alkyne cycloaddition reaction catalysed by copper (I); t-BuOH / H2O (9:1) was used as a solvent. The reaction was stopped after 16 h, and after purification by flash chromatography (DCM with 0–5 % MeOH), compound 1.38 was obtained in 65 % yield.
The authors then tested the effect of compound 1.38 on the viability of tumour cell lines derived from patients with multiple myeloma (MM) using the MTT assay.
Scheme 1.14
Рекомендовано к изучению сайтом МедУнивер - https://meduniver.com/
165
Compound 1.38 showed no toxicity to primary human fibroblasts, and for tumour cell lines of glioblastoma (SF-539) and myeloid leukaemia (HL-60(TB)) GI50 values were
1.18, 2.58 μM, respectively; and for the cell line of lung adenocarcinoma EKVX, myelogenous leukaemia K562, minimally invasive lung adenocarcinoma NCI-H322M
IC50 was more than 100 µM.
1.3.Preparation of hybrid polynitrogenous heterocyclic derivatives of 1,3,5-
triazine
Recently, many works have been devoted to the synthesis and study of the biological activity of nitrogen-containing heterocycles. This fact is primarily since the introduction of heterocycles of this series (tetrazol, triazole, etc.) into an organic substrate molecule often leads not only to an increase in efficiency, but also to a prolongation of the drug action [25–30]. In this case, as a rule, there is no increase in acute toxicity. Tetrazoles are successfully used as components for obtaining medical materials, kits for biochemical studies, and diagnostic test systems[31]. In addition, it is worth noting that most vitamins, nucleic acids, enzymes, coenzymes, hormones and alkaloids contain nitrogen-containing heterocycles.
Considering synthetic approaches to the introduction of heterocycles into the triazine framework, it is also worth mentioning the substitution reactions of chlorine in TXT. Thus, in the article [32], it is reported that chlorine in cyanuric chloride can be replaced by a heterocyclic amine in a DCM medium. This uses K2CO3 to neutralise HCl to prevent salt formation of the product. To replace the chlorine atom in TXT at room temperature, we took the heterocyclic amine 4-arylthiazol-2-amine and its nitro derivative. As a result of the reaction, N-(4,6-dichloro-1,3,5-triazin-2-yl)-4-phenylthiazol-2-amine (1.39), N-(4,6-dichloro-1, 3,5-triazin-2-yl)-4-(4-nitrophenyl)thiazol-2-amine (1.40) in 64.8 % and 61.9 % yields, respectively (Scheme 1.15).

166
Scheme 1.15
Moreno et al. [33] reported the synthesis of some new derivatives of chalcone-1,3,5- triazine-2-pyrazoline hybrids (Scheme 1.16). Amino-1,3,5-triazine derivative 1.41 was obtained in 83 % yield from TXT and 4-aminoacetophenone in acetone with successive introduction of Na2CO3 at a reduced temperature to obtain monosubstituted triazine 1.42
1-[4-[[4,6-bis(2-hydroxyethylamino)-1,3,5-triazin-2-yl]amino]phenyl]ethanone
Scheme 1.16
At the next stage, compound 1.42 was treated with ethanolamine in dioxane under microwave synthesis for 5 min to give compound 1.25, which was then subjected to Claisen–Schmidt condensation with 4-(trifluoromethyl)benzaldehyde 1.43 to give the corresponding 1,3,5-triazine chalcone (E)-1-(4-((4,6-bis((2-hydroxyethyl)amino)-1,3,5- triazin-2-yl)amino)phenyl)-3-(4-(trifluoromethyl)phenyl) prop-2-en-1-one 1.44 in 85 % yield (Scheme 1.17).
Scheme 1.17
Next, 1,2-dinucleophilic cyclocondensation reactions were carried out of (E)-1-(4- ((4,6-bis((2-hydroxyethyl)amino)-1,3,5-triazin-2-yl)amino)phenyl)-3-(4-
Рекомендовано к изучению сайтом МедУнивер - https://meduniver.com/

167
(trifluoromethyl)phenyl)prop-2-en-1-one 1.44 with (3,5-dichlorophenyl)hydrazine 1.45 in ethanol (Scheme 1.18).
Scheme 1.18
Due to the rather difficult substitution of the third chlorine atom in TCT, the authors of the study [34] used tetrahydrofuran (THF) to obtain N2,N4-dicyclohexyl-N6-(4- arylthiazol-2-yl)-1,3,5-triazin-2, 4,6-triamines 1.48, 1.49 in 23–28 % yields (Scheme 1.19). The product was isolated by extraction using an acidified water : dichloromethane (2:1) system to remove excess amine or any salts formed.
Scheme 1.19
In addition to cyclohexylamine, the authors used an ammonia solution to obtain diamino derivatives N2,N4-diamino-N6-(4-arylthiazol-2-yl)-1,3,5-triazine-2,4,6- triamines1.50, 1.51 (Scheme 1.20).
Scheme 1.20
When predicting biological activity, the authors of the study evaluated the affinity of compounds 1.50 and 1.51 for the enzyme dihydrofolate reductase, which catalyses the synthesis of tetrahydrofolate, which is a cofactor in DNA synthesis. Thus, compounds 1.50 and 1.51 can cause cell cycle inhibition. After predicting dihydrofolate reductase inhibitory

168
activity, the most promising compounds were tested against six different tumour cell lines. The resulting compounds 1.50 and 1.51 were studied for cytotoxic activity in vitro. The tested compounds showed the highest activity against human gastric adenocarcinoma (AGS) and human colon carcinoma (HCT-116) cell lines with IC50 of 6.05 and 13.56 μM, respectively. Continuing the study, the authors established the possibility of carrying out the reaction of TXT with 4-amino-4H-1,2,4-triazole-3-thiol [34]. A series of 4-((4,6- dichloro-1,3,5-triazin-2-yl)amino)-5-R-4H-1,2,4-triazole-3-thiols obtained in methylene chloride in the presence of K2CO3 The authors isolated 1.52–1.54 in 77–79 % yields. Substituents at the 4th and 6th positions of the triazine were introduced by successive reactions with ammonia and cyclohexylamine 1.55 and 1.56 in THF at reflux until two chlorine atoms were completely replaced (Scheme 1.21). The different nature of the nucleophilic agent (both aqueous ammonia and cyclohexylamine) and the substituent at the carbon atom of the triazole ring did not significantly affect the yield in this reaction (>60 %).
Scheme 1.21
To study the mechanism of action of compound 1.54, the authors performed computer simulation of its interaction with human dihydrofolate reductase by molecular docking using the FlexX module in LeadIT 2.1.8 (software package BioSolveIT GmbH, 2014). Confirming the data of biological activity in vitro, compounds containing free amino groups in the triazine ring showed the presence of non-covalent interactions in the
Рекомендовано к изучению сайтом МедУнивер - https://meduniver.com/

169
active site of the enzyme (Fig. 1.3). Two amino groups in the triazine ring of the cytostatic imitate two amino groups in the pteridine ring of methotrexate, which are involved in the formation of hydrogen bonds with the amino acid residue Glu 30.
Fig. 1.3. Scheme of non-covalent interactions of compound 1.54 in the active site of human dihydrofolate reductase.
Next, the synthesised derivatives 1.52–1.56 were tested for antineoplastic activity against a line of non-small cell lung carcinoma (A549). Methotrexate was used as a reference substance. Compound 1.55 appeared to be the most active of the study group, and three compounds (1.53, 1.54 and 1.56) showed activity greater than methotrexate, while compound (1.52) was equivalent to it in terms of antineoplastic activity (IC50 = 28–
50 μM). It was shown that the presence of a free amino group in the triazine ring, as in compound 1.54, enhanced the cytotoxic activity.
In 1996, at the Federal State Budgetary Institution ‘National Research Centre of Oncology named after N. N. Petrov’ of the Ministry of Health of Russia, the substance [5- [[4,6-bis(aziridin-1-yl)-1,3,5-triazin-2-yl]-amino]-2,2-dimethyl-1,3-dioxan-5-yl]- methanol (1.57) with antitumour activity, which belongs to the group of alkylating agents of the ethyleneimine class. The advantages of this compound are its amphiphilicity, which allows the drug to be administered in oily and aqueous solutions, as well as the presence of a pronounced contact antitumour effect, and its local and systemic toxic effect is much less than that of drugs based on platinum coordination compounds [35].
This substance showed antitumour activity in in vitro experiments on models of glioblastoma cell lines [36]. In the works [37,38] a comparative analysis of the antitumour activity of [5-[[4,6-bis(aziridin-1-yl)-1,3,5-triazin-2-yl]-amino]-2,2 dimethyl-1,3-dioxan- 5-yl]-methanol, cisplatin and their combinations in the model of ascitic ovarian tumour in female Wistar rats. The antitumour activity of the tested drugs was assessed by the increase in the life span of the animals. It was shown that compound 1.57 and cisplatin showed
170
comparable antitumour activity, increasing the lifespan of animals by 63.1 % and 48.1 %, respectively. At the same time, a synergistic antitumour effect of the tested compounds was noted with lower toxicity and better tolerability compared to cisplatin [39]. Similar results were obtained when studying the antitumour effect of the combination of gemcitabine and [5-[[4,6-bis(aziridin-1-yl)-1,3,5-triazin-2-yl]-amino]-2,2-dimethyl-1,3- dioxan-5-yl]-methanol (1.57) in mouse models with Ehrlich’s ascitic tumour. A higher antitumour activity was noted when using a combination of drugs against the background of no increase in toxicity, which was expressed in a significantly greater increase in the time of onset of ascites and an increase in median survival [40]. In experiments to study the increase in antitumour activity using regional hyperthermic intraperitoneal chemotherapy (HIPEC) in combination with cytostatics, it was shown that compound 1.57 statistically significantly increases the average survival of animals compared to cisplatin [41].
Efficacy of [5-[[4,6-bis(aziridin-1-yl)-1,3,5-triazin-2-yl]-amino]-2,2-dimethyl-1,3- dioxan-5-yl]-methanol 1.57 has been shown in fragmented clinical trials conducted in patients with common forms of malignant neoplasms of various localisations: breast cancer, non-small cell lung cancer, small cell lung cancer, undifferentiated lung cancer, gastric cancer, skin melanoma, kidney cancer, colon cancer colon, cervical cancer, liver cancer, osteogenic sarcoma, soft tissue sarcomas, lymphogranulomatosis, non-Hodgkin’s lymphomas and ovarian cancer [42]. The most pronounced antitumour effect of compound
1.57 was observed in the treatment of advanced forms of ovarian and breast cancer, including in patients with ascites and previously treated with other alkylating agents [43]. Additionally, clinical trials of [5-[[4,6-bis(aziridin-1-yl)-1,3,5-triazin-2-yl]-amino]-2,2- dimethyl-1,3-dioxane-5-yl]-methanol 1.57 as an antitumour agent for hepatic artery chemoembolisation in patients with primary liver cancer and liver metastases [44], colorectal cancer and in inoperable and advanced forms of kidney cancer [45]. The effectiveness of chemoembolisation using compound 1.57 was shown in the form of achieving an objective antitumour response and increasing patient survival [46,47]. Results of a collaborative clinical study of [5-[[4,6-bis(aziridin-1-yl)-1,3,5-triazin-2-yl]-amino]- 2,2-dimethyl-1,3-dioxan-5-yl]-methanol 1.57 in phase II give grounds to conclude that, being used in single doses of 15 mg intravenously by bolus or intraperitoneally at intervals of 72-96 h up to a total dose of 90–120 mg, the drug is very effective for chemotherapy of
Рекомендовано к изучению сайтом МедУнивер - https://meduniver.com/