

ТеорИмба
.pdfЭлектрохимические методы анализа
Несмотря на повсеместное распространение современных высокоинформативных методов анализа органических веществ, электрохимические методы анализа находят широкое применение для количественного определения различных веществ, в том числе и биологически активных.
Электрохимические методы анализа (ЭХМА) основаны на применении электрохимических процессов, проводимых в гальванической ячейке. Такая ячейка есть электрохимическая система, состоящая из взаимодействующих между собой электролитов и электродов. Между компонентами такой электрохимической системы протекают электрохимические реакции на границе раздела фаз.
В состав гальванической ячейки обычно входят два электрода – индикаторный и электрод сравнения, однако иногда требуется использование дополнительного третьего электрода, называемого вспомогательным. Индикаторный электрод реагирует на возбуждения или изменения состава раствора. Использование электрода сравнения необходимо для создания замкнутой цепи и обеспечения неизменного значения потенциала индикаторного электрода. Вспомогательный же электрод, иногда называемый противоэлектродом, вместе с рабочим электродом включен в цепь, через которую проходит электрический ток.
ЭХМА могут быть классифицированы в зависимости от протекающих на электродах процессов:
1.Методы, не связанные с электродной реакцией (кондуктометрия);
2.Методы, основанные на протекании электродной реакции, в ходе которой ток через границу фаз не протекает (потенциометрия);
3.Методы, основанные на протекании электродной реакции между электродом и приэлектродной частью раствора, в ходе которой ток протекает через границу раздела фаз (кулонометрия, вольтамперометрия и др.).
1

Потенциометрия
Основой данного метода является зависимость равновесного электродного потенциала от концентрации (а точнее, от активности) ионов в растворе, в который погружен электрод. При потенциометрии измеряют электродвижущую силу (ЭДС) гальванических элементов, составленных из электродов, которые погружены в анализируемый раствор. Чаще всего применяют двухэлектродную схему, при этом оба электрода могут быть погружены как в один раствор (в этом случае говорят об «элементе без переноса»), так и в два различных по составу раствора, соединенных солевым мостиком («элемент с переносом»).
Если на электроде устанавливается обратимое равновесие с участием потенциалопределяющих ионов, то возникающий на нем потенциал, определяется по уравнению Нернста:
E = E0 ± nR××FT ln a
где E0 – стандартное значение потенциала, R – универсальная газовая постоянная, T – температура, F – константа Фарадея, n – заряд потенциалопределяющего иона, а – его активность.
Из существующего множества разнообразных электродов, в потенциометрическом методе анализа применяют два класса индикаторных электродов:
1.Электронообменные электроды – электроды, на межфазных границах которых протекают электрохимические процессы с участием электронов. Такие электроды чаще всего применяются в потенциометрии с использованием окислительно-восстановительных реакций, при этом их изготавливают обычно из инертных металлов, таких как платина и золото. Металлические индикаторные электроды представляют собой тонкую пластину, скрученную проволоку или металлизированное стекло. При помещении такого электрода в анализируемый раствор равновесие устанавливается обычно очень быстро.
2.Ионоселективные электроды – электроды, на межфазных границах которых протекают ионообменные процессы и реакции комплексообразования. Ионоселективные электроды (ИСЭ) позволяют селективно определять
2
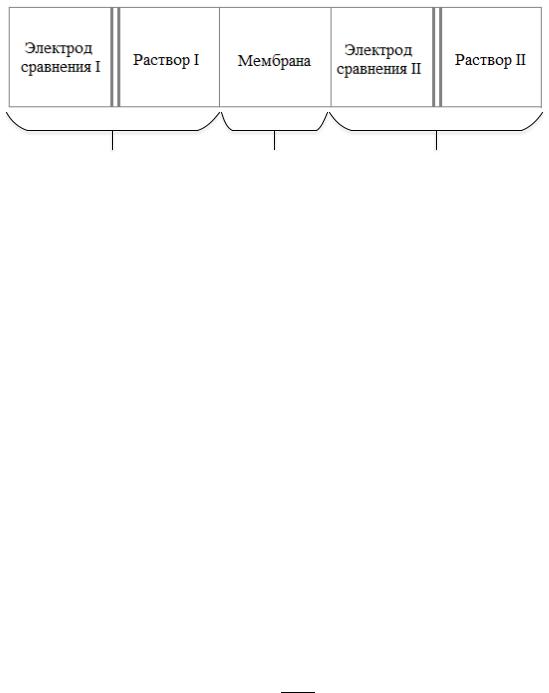
активности одних ионов в растворе в присутствии других. Данные электроды представляют собой гальванические полуэлементы, состоящие из ионселективной мембраны, внутреннего раствора и внутреннего электрода сравнения. Вторым полуэлементом является внешний электрод сравнения, погруженный в стандартный электролит.
При потенциометрии с использованием ионоселективных электродов измеряют ЭДС следующей ячейки (рисунок 1).
E1 ΔφM E2
Рис. 1.
E = E2 + DjM - E1
Значения E1 и E2 являются табличными или могут быть рассчитаны, ΔφM – мембранный потенциал, определяемый как сумма разности граничных потенциалов двух разделов фаз и диффузионного потенциала (разности потенциалов внутри мембраны).
Измеряя мембранный потенциал, можно определить активность ионов в анализируемом растворе. В том случае, когда мембрана электрода с обеих сторон контактирует с растворами одного и того же иона (мешающие ионы отсутствуют), мембранный потенциал выражается уравнением, с виду похожим на уравнение Нернста:
DjM |
= |
R ×T |
ln |
aM 1 |
|
n × F |
aM 2 |
||||
|
|
|
где аМ1 и аМ2 – активности иона М в растворах 1 и 2.
Потенциал ионоселективного электрода описывается следующим выражением:
EИСЭ = const + nR××FT ln aM 1
3
Если же в анализируемом растворе присутствует мешающий ион Х с активностью ах, то потенциал ИСЭ выражается через уравнение Никольского:
|
R ×T |
é |
|
n |
ù |
|
EИСЭ = const + |
|
lnêaM 1 |
+ kM , X × aX 1 Z x ú |
|||
n × F |
||||||
|
ë |
û |
||||
где kM,X – коэффициент селективности ИСЭ по определяемому иону М в присутствии мешающего иона Х, ZХ – заряд мешающего иона, аХ1 – активность мешающего иона.
Коэффициент селективности позволяет давать количественную оценку влияния мешающих ионов (Х) на результаты измерения активности определяемых ионов (М) с помощью ИСЭ.
Помимо вышеописанного коэффициента, важными характеристиками работоспособности ИСЭ являются предел обнаружения и время отклика. Предел обнаружения показывает, какое минимальное количество вещества (иона) может быть определено данным ИСЭ с заданной достоверностью. Для большинства ИСЭ предел обнаружения колеблется в районе 10-5 – 10-7 моль/л. Время отклика определяется как время, в течение которого потенциал ИСЭ достигает 90% величины общего изменения от E1 до Е2 и составляет от 10-15 секунд (для концентрированных раствором) и до нескольких минут (для разбавленных растворов).
|
ИСЭ классифицируются в зависимости от природы активного материала |
мембраны и могут быть: |
|
а) |
стеклянными; |
б) |
с твердой мембраной (гомоили гетерогенной); |
в) |
с жидкой мембраной на основе ионообменников; |
г) |
газовыми. |
Потенциометрическое титрование
Одним из вариантов реализации метода потенциометрии является потенциометрическое титрование. Данный метод основан на фиксировании точки эквивалентности по резкому изменению потенциала индикаторного электрода,
вызванного |
изменением |
активности |
того |
или |
иного |
компонента. |
Потенциометрическое титрование может |
быть |
основано |
на реакциях |
|||
4
нейтрализации, комплексообразования или окислительно-восстановительных процессах.
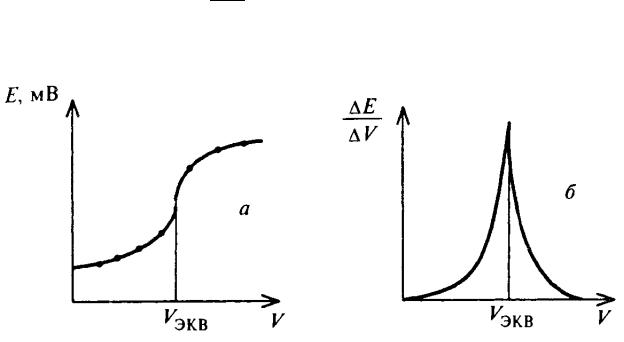
Во всех случаях в процессе потенциометрического титрования регистрируют ЭДС электрохимической ячейки после введения очередной порции титранта, при этом по мере приближения к точке эквивалентности необходимо уменьшать вносимую порцию титранта для более точного определения. Точку эквивалентности чаще всего находят графически по кривой титрования, построенной в координатах «потенциал электрода – объем титранта» (рисунок 2а). Более наглядно точку эквивалентности можно зафиксировать, построив кривую
титрования в координатах DE (рисунок 2б), так как в таком случае она будет
DVт
иметь вид острого пика.
Рис. 2. Кривые потенциометрического титрования, построенные в разных координатах.
В случае, когда требуется повышенная точность определения точки эквивалентности, кривую титрования можно построить в координатах второй
производной потенциала по объему |
D2 E |
(рисунок 3). В данном случае точка |
|||||
DVт |
2 |
||||||
|
|
|
|
|
|||
эквивалентности будет соответствовать значению производной |
D2 E |
= 0. |
|||||
DVт |
2 |
||||||
|
|
|
|
|
|||
5
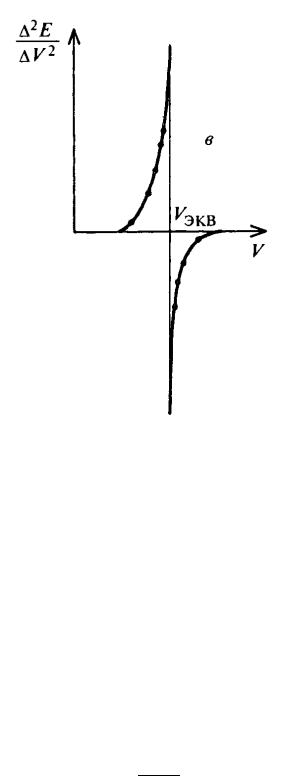
Рис. 3.
На практике для измерения ЭДС используют потенциометры. Такие приборы также называют pH-метрами. Для проведения анализов с применением ИСЭ используются иономеры - pH-метры с небольшими модификациями, чьи шкалы откалиброваны как в единицах pH, так и в мВ.
Кулонометрия
Данный метод основан на измерении количества электричества, израсходованного на электрохимические реакции, результирующие электроокисление или электровосстановление определяемого вещества. В основе количественного определения лежит закон Фарадея:
g = M ×Q n × F
где g – масса превращенного вещества, г; М – его молярная масса, г/моль; Q
– количество электричества, Кл; n – число электронов, принимающих участие в электрохимической реакции; F – константа Фарадея.
Различают прямую кулонометрию и кулонометрическое титрование. Для реализации анализа по методу прямой кулонометрии требуется, чтобы
6

определяемое вещество было электроактивным, то есть на электроде должна протекать одна из следующих реакций:
A + ne ® B (электровосстановление)
или
A - ne ® B (электроокисление)
Кулонометрическое титрование же применимо и для электронеактивных веществ, так как в этом случае в анализируемый раствор вводят вспомогательный реагент (обозначим его как D), который вступает в электрохимическую реакцию с образованием продукта (P). Продукт Р, в свою очередь, количественно взаимодействует с определяемым соединением А. При таких условиях на катоде происходят следующие взаимодействия:
A + ne ® B
D+ me ® P
Ав растворе происходит химическая реакция:
mA + nP ® mB + nD
То есть, измеряя общее количество электричества, затраченное на протекающие реакции, можно рассчитать массу определяемого соединения А. Здесь стоит отметить, что важным условием для реализации метода кулонометрического титрования является отсутствие побочных электрохимических реакций. Иными словами, все расходуемое количество электричества должно затрачиваться на превращение определяемого соединения А.
Кулонометрия может быть реализована в двух вариантах. С контролируемым потенциалом, когда измеряется количество электричества, затраченное на электрохимическое превращение определяемого вещества, а потенциал рабочего электрода в процессе анализа поддерживается постоянным. И с контролируемым током, когда анализ проводят с фиксированным предварительно подобранным значением силы тока электролиза, а потенциал рабочего электрода в процессе анализа изменяется.
Для кулонометрического титрования в качестве электролизера применяют ячейку, изображенную на рисунке 4, состоящую из двух или трех изолированных камер.
7
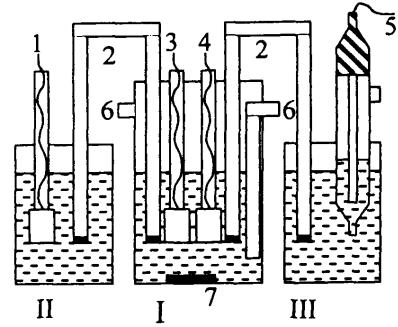
Рис. 4. Ячейка для кулонометрического титрования. 1 – вспомогательный электрод, 2 – электролитические ключи, 3 – генераторный электрод, 4 – индикаторный электрод, 5 – электрод сравнения, 6 – патрубки для входа и выхода инертного газа, 7 – якорь магнитной мешалки.
Современные приборы для кулонометрии соединяют в себе все необходимые узлы и работают под управлением программного обеспечения с графическим интерфейсом. Такие условия позволяют реализовывать любые варианты как прямой кулонометрии, так и кулонометрического титрования.
Кулонометрия позволяет определять от 1 мкг до 100 мг вещества с относительной погрешностью, не превышающей 0.3%. Данный метод характеризуется рядом достоинств, например, высокой точностью и воспроизводимостью измерений.
Кондуктометрия
В основе данного метода лежит зависимость между электропроводностью раствора и концентрацией ионов в этом растворе. Электропроводность, в свою очередь, является результатом диссоциации молекул растворенного вещества и миграции ионов под действием приложенной разности потенциалов. Различают удельную электропроводность æ – электропроводность 1 см3 раствора, находящегося между электродами площадью 1 см2 на расстоянии 1 см друг от друга, и эквивалентную электропроводность λ – электропроводность раствора,
8
содержащего 1 моль-экв электролита, измеренная при расстоянии 1 см между электродами.
æ = a ×С × F ×(z+u+ + z-u- ) |
é |
1 |
= |
1 ù |
|||||
ê |
|
|
|
ú |
|||||
Ом ×см |
|
|
|||||||
|
|
|
|
ë |
|
См û |
|||
l = |
1000 |
× æ |
é |
См ×см2 |
ù |
|
|||
|
|
ê |
|
|
|
ú |
|
||
|
С |
|
|
|
|||||
|
|
|
|
|
|||||
|
|
|
ë |
моль - экв û |
|||||
где α – степень диссоциации электролита, С – концентрация электролита, моль-экв/см3, F – константа Фарадея, u+, u-, z+, z- - скорость миграции (м/с) и заряд катионов и анионов при напряженности электрического поля 1 В/см.
В кондуктометрии аналитическим сигналом является электропроводность раствора λ, учитывающая вклад всех присутствующих в растворе ионов:
l = åCi ×li × Zi
Как уже говорилось ранее, анализ, основанный на измерении электропроводности раствора, может быть реализован в виде прямой кондуктометрии и методом кондуктометрического титрования. Прямую кондуктометрию применяют редко из-за неселективности аналитического сигнала. Большее распространение нашёл метод кондуктометрического титрования, основанный на проведении химической реакции, результатом которой является резкое изменение электропроводности раствора. Для этого метода анализа используют реакции почти всех типов, наибольшее применения находят кислотноосновные взаимодействия, комплексообразование, осаждение и окислительновосстановительные реакции.
Для рассмотрения процесса кондуктометрического титрования в общем случае примем сильный электролит АВ за определяемое вещество, а сильный электролит CD за титрант. При этом при титровании образуется малодиссоциированное (или слаборастворимое в данном растворителе) вещество AD.
A+ + B - + C + + D- ® AD + C + + B-
Впроцессе титрования происходит замена ионов А+ в растворе на С+. Так как электропроводность раствора является функцией его состава, а подвижность ионов зависит только от их природы, то по мере титрования до точки
эквивалентности на ход кривой титрования будет влиять соотношение
9

подвижности ионов A+ и С+. При этом возможны три вида кривых (рисунок 5).
Рисунок 5. Возможные варианты вида кривых кондуктометрического титрования.
Одной из наиболее распространенных реакций, применяемых в кондуктометрическом титровании, является кислотно-основное взаимодействие. В данном случае вид кривых титрования будет зависеть от силы взятых кислоты и основания, а также от их концентраций. Кривые титрования кислот различной силы представлены на рисунке 6.
Рис. 6. Кривые кондуктометрического титрования: а) сильной кислоты сильным основанием; б) слабой кислоты сильным основанием; в) слабой кислоты
10