

Механизм пластификации
При растворении в полимере жидкости макромолекулы оказываются окруженными молекулами этой жидкости. Это ведет к понижению взаимодействий между макромолекулами. Кроме того, молекулы низкомолекулярной жидкости являются более подвижными и легче обмениваются местами, чем макромолекулы полимера. Снижение межмакромолекулярного взаимодействия и наличие в системе подвижного низкомолекулярного компонента ведет к повышению молекулярной подвижности всей системы. Это вызывает изменение всего комплекса свойств полимера: изменяются его прочностные, деформационные, температурные, реологические электрические свойства. С молекулярной точки зрения под пластификацией полимеров понимается увеличение подвижности структурных элементов полимера при введении в него специально подобранных жидкостей – пластификаторов, не взаимодействующих химически с полимером. Из механизма пластификации следует, что макромолекулы полимера должны быть разделены молекулами пластификатора. Выполнение этого условия предусматривает обязательную растворимость пластификатора в полимере. Кроме того, молекулы пластификатора должны обладать значительно более высокой подвижностью, чем макромолекулы полимера.
Как уже отмечалось, пластификатор вводят для изменения деформационных, прочностных, электрических, теплофизических и других свойств полимера. Предсказание изменения свойств полимера при пластификации очень важно при научном подходе к вопросу созданияполимерных композитов. Поэтому важно наличие теории, предсказывающей эти изменения.
По одному из предположений полярные группы полимера сольватируются полярными группами пластификатора. Будучи экранированными полярные группы макромолекул не взаимодействуют между собой, что снижает Тс. Из этого следует, что понижение температуры стеклования ΔТс должно быть пропорционально числу молей введенного пластификатора n2:
ΔТс = kм · n2 ,
где k – коэффициент пропорциональности зависящий от природы компонентов.
Это уравнение называют правилом молярных концентраций. Правило действует только в ограниченной области составов и только для полярных полимеров и пластификаторов.
При пластификации неполярных полимеров низкомолекулярными жидкостями предложено другое правило – правило объемных концентраций:
ΔТс = kф · φ2 ,
где k - коэффициент пропорциональности зависящий от природы компонентов, φ2 – объемная доля пластификатора.
По мнению авторов, в этом случае основную роль играет не взаимодействие полимер – пластификатор, а конформационные превращения цепей полимера в растворе. Если в растворах объем, занятый растворителями, один и тот же, то число конформаций, которое могут принять цепи, должно быть одинаковым для различных растворителей. Следовательно, понижение Тс должно быть пропорционально объемной доле пластификатора.
Это правило не справедливо даже для многих неполярных полимеров и пластификаторов.
Оба рассмотренные подходы очень ограничены. Они не учитывают ни растворимость, ни молекулярную массу, ни гибкость пластификатора, которые оказывают большое влияние на изменение свойств полимеров при пластификации.
32. Высокоэластическое состояние полимеров. Полимеры, находясь в высокоэластическом состоянии, способны к большим (4-5-кратным) обратимым деформациям.
Высокоэластическое состояние специфично для полимеров: низкомолекулярные материалы таким свойством не обладают. Температурный интервал высокоэластического состояния на термомеханической кривой находится в пределах Тт-Тс. По мере увеличения температуры от Тс до Тт доля свободного объема возрастает в соответствии с уравнением (3.13).
Для полимеров, находящихся в высокоэластическом состоянии, сохраняется ближний порядок во взаимном расположении сегментов макромолекул, но подвижность их существенно выше, нежели в стеклообразном состоянии: время релаксации сокращается на 5-6 десятичных порядков. Модуль упругости полимерных тел, находящихся в высокоэластическом состоянии, снижается до 0,1-0,3 Мпа. Существенно изменяется и сжимаемость полимера. Если в стеклообразном состоянии она для различных волокнообразующих полимеров заключена в пределах (1÷5)10-12 Па-1, то в результате расстекловывания полимерного субстрата сжимаемость возрастает до (3÷6)10-10 Па-1.
Наиболее вероятному состоянию полимерного тела соответствует максимальная энтропия:
![]()
Это состояние объясняется наиболее энергетически выгодной конформацией свободносочлененной цепи.
Обратимая деформация пространственной сетки полимерного субстрата, построенной из таких статистических клубков, приводит к изменению конфигурационной энтропии ΔSк. Вместе с тем деформируемость такой сетки характеризуется модулем высокоэластичности Евэ:
![]()
где ρп - плотность полимера при температуре Ti; k - константа Больцмана; Мс - молекулярная масса сегмента.
Следовательно, определяя Евэ, можно оценить размер сегмента полимерной цепи в высокоэластическом состоянии. Величина eвэвозрастает при повышении Т.
При анализе процессов деформирования полимеров в высокоэластическом состоянии подвижность кинетических элементов структуры (сегментов) принимается аналогичной подвижности частиц идеальных газов. Это допущение оказывается справедливым для деформаций не более 50%. Большие деформации, характерные для полимеров в высокоэластическом состоянии, реализуются за счет не только εвэ, но и εу и εп (см. рис. 3.7). Эти деформации обусловливают изменение не только ΔSк, но и энтальпии полимера ΔH.
При больших деформациях наблюдаются также уменьшение объема полимерных тел, уплотнение их структуры и выделение некоторого количества тепла, повышающего температуру образца на 1,5-2 град.
Температурная зависимость механических свойств аморфных полимеров выше Тс может быть описана так называемой функцией приведения aT. Эта величина представляет собой отношение времени релаксации при некоторой температуре Т ко времени релаксации при температуре Т > Т1 ≥ Тс, т.е.

Величина aT обусловлена температурной зависимостью гибкости макромолекул. Вместо времен релаксации τT и τTc могут быть выбраны другие временны́е характеристики, зависящие от релаксационных свойств полимеров: вязкость, напряжение, деформация, коэффициент диффузии. Например,
![]()
где η и ρ - вязкость и плотность полимерной системы при температуре Т, а η1 и ρ1 - при температуре приведения Т1 = Tс + (50 ± 4).
Сопоставляя (3.13), (3.17) и (3.18), получаем
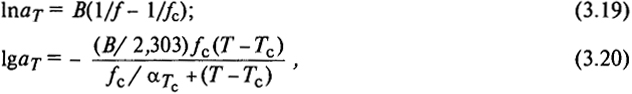
где αTc - коэффициент теплового расширения при Тс.
Очевидно, в общем виде можно записать

где А и С - постоянные, зависящие от доли свободного объема fc.
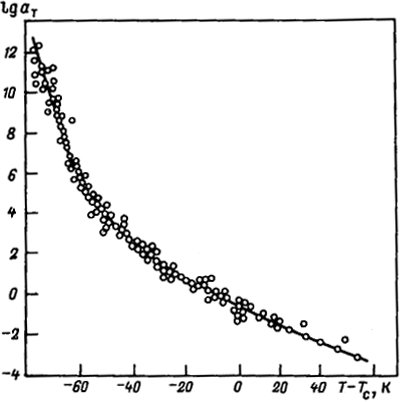 Рис.
3.9. Зависимость lgaT от (T - Tс)
для различных полимеров
Рис.
3.9. Зависимость lgaT от (T - Tс)
для различных полимеров
Соотношение (3.21) известно как формула Вильямса - Ланделла - Ферри (ВЛФ).
На рис. 3.9 представлена зависимость lgaT от Т - Тс для различных систем, свидетельствующая об их универсальности. Константы, входящие в эту формулу, определяют температурные характеристики релаксационных свойств, в том числе и ηэф. Они универсальны по отношению ко многим волокнообразующим полимерам.
Если в качестве температуры приведения выбраны Тс или температура менее (Тс + 50), то А = -17,44; С = 51,6. Если же в качестве температуры приведения используется Т1 = (Тс + 50), что целесообразно для многих эластомеров, то А = -8,86; С = 101,6.
В высокоэластическом состоянии основную долю составляет εвэ, хотя вклад εу и εп также довольно существен. Повышение температуры уменьшает εу и увеличивает εвэ и εп.