

1.3.4.Кинетика ферментативных реакций.
Браун А, и Анри В. впервые высказали мысль о том, что в основе ферментативной реакции лежит обратимое взаимодействие S и Е с образованием комплекса, который позже распадается с образованием продуктов и регенерацией исходного фермента.
Ферментативную реакцию в простом случае одностороннего превращения одного субстрата можно выразить общим уравнением:
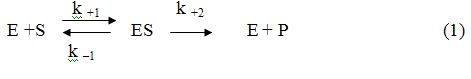
В соответствии с этой схемой [S] обратимо реагирует с [E] с образованием ФСК [ES]. Этот комплекс называют комплексом Михаэлиса.
Допустим, что ФСК все время находится в равновесии с исходными веществами. Другими словами равновесие на первой стадии устанавливается быстро и не нарушается высвобождением фермента от ES в направление k +2

Скорость
ферментативных превращений субстрата
равна скорости образования продукта
(2-я стадия), если [S] >> [E]0, что бывает
чаще, где [E]0 – концентрация фермента в
начальный момент и равен
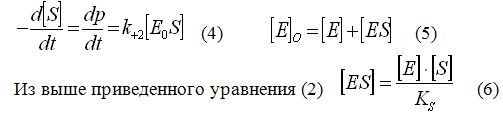
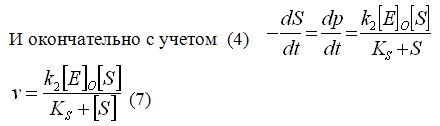 Это
и есть уравнение Михаэлиса-Мэнтен.
Это
и есть уравнение Михаэлиса-Мэнтен.
KS – субстратная константа = 1/KP (KP- константа равновесия).
Однако в этом уравнении отсутствует константа образования продукта k+2. Холдейн и Бригс вывели уравнение более полно описывающее зависимость скорости от концентрации субстрата.
Исходя из уравнения ферментативной реакции мы можем написать уравнение скоростей образования и распада ФСК.

При достаточно быстром протекании стадии образование ФСК и его превращения в продукт может быть реализовано состояние, когда концентрация [ЕS] меняется во времени медленнее, чем концентрация [S] и [Е]. Это случай возможен при [E]0 << [S].
Экспериментально доказано, что в реакциях при оптимальных условиях и при [S] >> [E]0 быстро наступает стационарное течение процесса, при котором распад [ES] по направлению k-1 и k+2 уравновешивается его образованием в направлении k+1. Тогда для стационарного состояния можно написать
k-1 [ES]+k+2 [ES] = k+1 [E][S]
или (k -1 +k +2) [ES] = k +1 [E] [S] (11)
Обозначим общую концентрацию фермента через [E]0, тогда
[E]0 = [E]+[ES] и отсюда [E]=[E]0-[ES](12).
Подставим [E] из уравнения 12 в уравнение 11 и получим:
(k –1 + k +2) [ES] = k +1 ([E]0 – [ES]) [S] (13). Отсюда найдем [ES].
![]() (14)
(14)
разделим числитель и знаменатель на k +1 получим выражение:
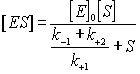 (15)
(15)
Обозначив
![]() (15)
(15)
Получим
![]() (16)
(16)
Скорость может быть выражена
![]() (17)
(17)
Подставим значение [ES] из уравнения 16 в уравнение 17 и получим уравнение Холдейна-Бригс (ХБ), более известного как уравнение Михаэлиса-Ментен (ММ).
Получим
![]() (18)
(18)
Рассмотрим внимательно выражение:
![]() KM – константа Михаэлиса, одна из
важнейших констант в кинетике
ферментативных реакций.
KM – константа Михаэлиса, одна из
важнейших констант в кинетике
ферментативных реакций.
Если k–1 >> k+2, т.е. если обратный распад ФСК на исходные вещества происходит гораздо быстрее его превращение в продукт, то можно пренебречь k+2 и уравнение ХБ. прейдет в уравнение ММ. Это условие совпадает с предположением о существовании равновесия на первой стадии в ходе всего процесса. Т.е. принцип стационарности позволяет получить более общую форму уравнения М.М., уравнение Х.Б.
Уравнение Х.Б. в целом хорошо описывает
экспериментальные данные по кинетике
ферментативных реакций. Чаще всего оно
принимается в дифференциальной форме,
связывающий начальную скорость
превращения субстрата с его начальной
концентрацией при заданном количестве
внесенного фермента:
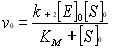 (19)
(19)
Это объясняется тем, что в ходе реакции могут появиться некоторые дополнительные эффекты – торможение продуктами, инактивация ферментов и т.д., которые искажают ход временной зависимости по сравнению с уравнениями М.М., Х.Б.
Взаимосвязь начальной скорости реакции [v]0 и начальной концентрации субстрата [s]0 по уравнению Х.Б. представляет собой функцию имеющую свои характерные особенности (Рис.1.15).
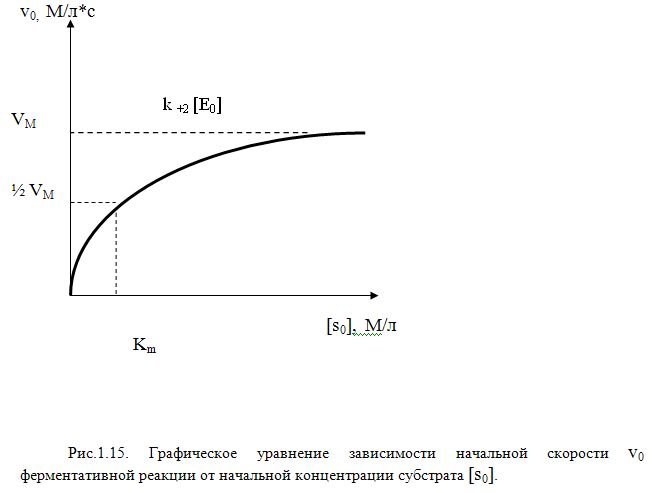
При достаточно малых концентрациях
субстрата, когда KM >> [s0] уравнение
переходит в линейную форму
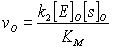 (20) , а при больших [s]0, если [s]0
>> KM кривая стремится к предельному
значению k+2[E]0 , которую
обозначают через Vm. Тогда Vm
= k+2[E]0 (21). Если V0=1/2
VM, то [s]0=KM. В силу
изложенного уравнения Х.Б. часто
представляют в форме, удобной для
практической обработки экспериментальных
данных:
(20) , а при больших [s]0, если [s]0
>> KM кривая стремится к предельному
значению k+2[E]0 , которую
обозначают через Vm. Тогда Vm
= k+2[E]0 (21). Если V0=1/2
VM, то [s]0=KM. В силу
изложенного уравнения Х.Б. часто
представляют в форме, удобной для
практической обработки экспериментальных
данных:
![]() (21)
(21)
Константы этого уравнения характеризуют активность фермента и его сродство к данному субстрату, поэтому целью кинетического исследования является прежде всего нахождение значений VM и КM . наиболее простое такое исследование выполняется в виде серии экспериментов при постоянной концентрации фермента [E]0 и изменяющихся начальных концентрациях субстрата [s]0. В ходе каждого опыта изучается лишь начальный участок кинетической кривой для определения начальной скорости реакции V0 имея набор значений V0 при известных [s]0 легко найти кинетические константы.
Для этой цели удобны уравнения и графики
Лайнуивера – Берка (Рис.1.16). Общим
свойством дробных рациональных функций,
имеющих в числители произведения, а в
знаменателе сумму некоторых величин
является их способностью переходить в
линейную форму при обращении левой и
правой части равенства:
![]() (22)
(22)
при 1/s0=0 имеем
![]() т.е. прямая пересекает ось ординат в
точке 1/VM, а при 1/v0=0 1/s0=-
1/KM . Таким образом, при продолжение
прямой в отрицательную область до
пересечения с осью абсцисс она отсекает
на оси отрезок =- 1/КM
т.е. прямая пересекает ось ординат в
точке 1/VM, а при 1/v0=0 1/s0=-
1/KM . Таким образом, при продолжение
прямой в отрицательную область до
пересечения с осью абсцисс она отсекает
на оси отрезок =- 1/КM
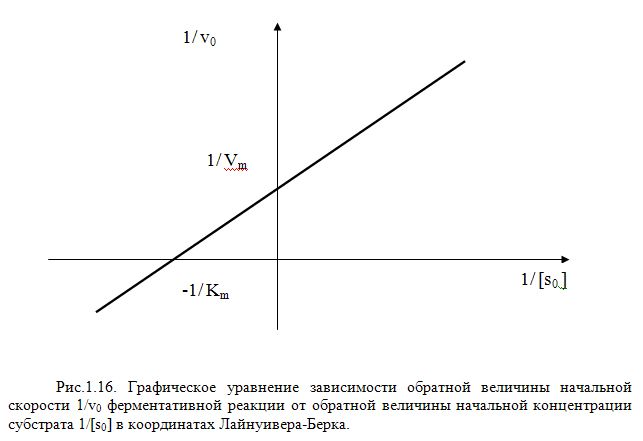
Недостатком координат Лайнуивера-Берка (ЛБ). является экстраполяция прямой влево до пересечения с осями ординат, что приводит к ошибкам.
Одно из удобных решений исключающих
недостаток графиков Л.Б. заключается в
использовании координат Эди-Хофсти
![]() (Рис.1.17) и описываемых уравнением
Эди-Хофсти
(Рис.1.17) и описываемых уравнением
Эди-Хофсти
![]() (24)
(24)
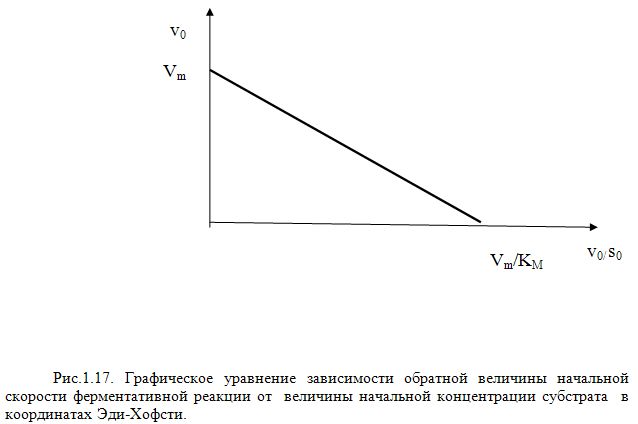
В координатах с отрицательным углом наклона отсекается на осях ординат отрезок, равный VM, а на оси абсцисс отрезок равный VM/KM.
Недостаток такого метода обработка является возможность возрастания ошибок при делении, 2-х величин V0 и s0 каждая из которого измерена с ошибкой.
Определение VM=k2[E]0
позволяет по известной мольной
концентрации катализатора найти k +2
– константу скорости распада ES. Константа
Михаэлиса определяет стационарную
концентрацию ФСК
![]() (25). Однако с помощью КМ в общем
случае нельзя найти ни отдельные
элементарные константы k -1 и k+1
ни их отношение – субстратную
константу КS . для определения К1
нужно изучать кинетику нестационарной
реакции на самых первых стадиях, что
для быстрых каталитических реакций
представляет большие экспериментальные
трудности и часто связано с диффузионными
ограничениями. Поэтому обычно величину
КМ рассматривают как некоторую
эффективную величину, представляющую
совокупность элементарных констант.
Естественно, что при k +2 << k -1
стационарная концентрация комплекса
E-S мало отличается от равновесной, а КМ
равно субстратной константа КS
константе диссоциации комплекса ES:
КМ=КS . Для многих ферментативных
реакций раздельное определение КS
и KM показало, что эти величины
отличаются мало, исключение составляет
каталаза, пероксидаза. Как обычно KM
лежит в пределах 1-10-8м/л, но чаще
встречаются значения 10-4 M/л.
(25). Однако с помощью КМ в общем
случае нельзя найти ни отдельные
элементарные константы k -1 и k+1
ни их отношение – субстратную
константу КS . для определения К1
нужно изучать кинетику нестационарной
реакции на самых первых стадиях, что
для быстрых каталитических реакций
представляет большие экспериментальные
трудности и часто связано с диффузионными
ограничениями. Поэтому обычно величину
КМ рассматривают как некоторую
эффективную величину, представляющую
совокупность элементарных констант.
Естественно, что при k +2 << k -1
стационарная концентрация комплекса
E-S мало отличается от равновесной, а КМ
равно субстратной константа КS
константе диссоциации комплекса ES:
КМ=КS . Для многих ферментативных
реакций раздельное определение КS
и KM показало, что эти величины
отличаются мало, исключение составляет
каталаза, пероксидаза. Как обычно KM
лежит в пределах 1-10-8м/л, но чаще
встречаются значения 10-4 M/л.
При уверенности отсутствия посторонних
факторов и располагая математическим
выражением для скорости ферментативной
реакции, (уравнение Х.Б. М.М.) мы можем
определить изменение концентрации
реагентов во времени путем интегрирования
уравнения методом разделения переменных:
![]()
![]()
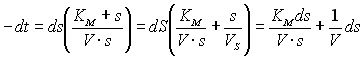
![]() (26)
(26)
интегрирование в пределах от 0 до t и
соответственно от S0 до S приводит к
уравнению Уоелкера-Шмидта (Рис.1.18).
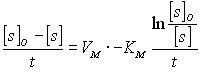 (27)
(27)
Надо иметь виду, что на практике это уравнение проще использовать для расчета времени t, необходимого для достижения определенных концентраций субстрата, а не наоборот.