

1NhCl при различии концентрации раствора нитрата серебра (б)
По данному способу возможно титрование смеси ионов. При этом необходимо помнить, что дифференциальное титрование этих ионов возможно только в том случае, если ПР образующихся малорастворимых соединений отличаются друг от друга не менее чем на три порядка. Если при потенциометрическом титровании способом осаждения могут встречаться следующие ошибки: явления адсорбции, изменение растворимости осадка в присутствии посторонних электролитов и т.п.
Переведение в малодиссоциированные комплексы. Сущность данного способа заключается в следующем: при добавлении титранта к исследуемому pacтвоpу количество ионов, концентрацию которых нужно определить, уменьшается, т.к. они связываются в малодиссоциированный комплекс. В связи с этим будет изменяться потенциал индикаторного электрода и, следовательно, ЭДС гальванического элемента.
Рассмотрим пример титрования цианид-ионов раствором нитрата серебра. Индикаторным электродом гальванического элемента служит серебряная проволока, электродом сравнения - каломельный полуэлемент. Титрантом является раствор нитрата серебра.
Ионы серебра с ионами цианида образуют комплекс [Ag(CN)2], причем процесс комплексообразования обратим:
Ag+ + 2CN- [Ag(CN-)2]-
Константа нестойкости этого комплекса равна:
 .
(18)
.
(18)
Воспользовавшись этой формулой, можно рассчитать концентрацию ионов серебра, а по формуле Нернста (5) - потенциал серебряного электрода. В процессе титрования потенциал последнего и, следовательно, ЭДС гальванического элемента будут возрастать (рис.16,а), и в точке эквивалентности 1 обнаружится резкий скачок потенциала. При дальнейшем титровании вслед за скачком потенциала, соответствующим

Рис.16. Кривые потенциомётрического титрований по способу комплексообразования: цианид-ионов раствором нитрата серебра (а); ионов кальция раствором ЭДТА (б) при различных рН окончанию процесса комплексообразования, возникает почти горизонтальный участок.
Это объясняется тем, что дальнейшее добавление нитрата серебра ведет к образованию в растворе малодиссоциированного осадка:
[Ag(CN-)2]- + Ag 2AgCN
Таким образом, концентрация ионов серебра в растворе, от которой зависит величина потенциала индикаторного электрода, определяется растворимостью образующегося осадка. До тех пор, пока весь комплекс не превратится в осадок, потенциал системы остается практически постоянным.
Скачок потенциала в точке эквивалентности тем больше, чем меньше константа диссоциации комплекса и чем больше начальная концентрация титруемого раствора.
При использовании большинства органических комплексообразующих лигандов процесс титрования сильно зависит от рН раствора, так как при различных рН образуются комплексы с различной константой нестойкости и различного состава. На рис.16,б приведены кривые потенциометрического титрования ионов Са2+ раствором динатриевой соли этилендиаминтетра-уксусной кислоты (ЭДТА) при различных рН. Очевидно, что при рН=8 точка эквивалентности будет определена менее правильно, нежели при рН=9 или рН=12.
Окислительное титрирование. При протекании окислительно-восстановительной реакции в растворе имеются ионы в окисленной и восстановленной форме. Если подобрать индикаторный электрод, обратимый относительно этих ионов, то по мере течения реакции концентрация ионов, находящихся в окисленной и восстановленной форме, будет изменяться. Тогда, в соответствии с формулой Нернста, будет меняться величина электродного потенциала и, следовательно, ЭДС гальванического элемента.
В качестве примера окислительно-восстановительного титрования рассмотрим процесс титрования хлорида железа (III) раствором хлорида олова (II)
2Fe3+ + Sn2+ 2Fe2+ + Sn4+
Стандартные потенциалы электродных процессов равны:
2Fe3+
+ 2e
2Fe2+
![]()
Sn2+
+ 2e
Sn4+
![]()
Так
как
![]() ,
то ион Fe3+
будет
окислять ион Sn2+.
Составим гальванический элемент, в
котором индикаторный электродом служит
платина 2, электродом сравнения -
каломельный полуэлемент 3, и заполним
его хлоридом железа(III)
(рис.17). Титрантом является раствор
хлорида олова I,
перемешивание осуществляется магнитной
мешалкой 4. Потенциал платинового
индикаторного электрода в растворе
хлорида железа (III)
описывается уравнением:
,
то ион Fe3+
будет
окислять ион Sn2+.
Составим гальванический элемент, в
котором индикаторный электродом служит
платина 2, электродом сравнения -
каломельный полуэлемент 3, и заполним
его хлоридом железа(III)
(рис.17). Титрантом является раствор
хлорида олова I,
перемешивание осуществляется магнитной
мешалкой 4. Потенциал платинового
индикаторного электрода в растворе
хлорида железа (III)
описывается уравнением:
![]() .
(19)
.
(19)
Тогда ЭДС гальванического элемента можно записать в виде:
 .
(20)
.
(20)

Рис. 17. Установка для окислительно-восстановительного потенциометрического титрования
Естественно,
что изменение концентраций ионов Fe3+
и Fe2+
в процессе титрования повлечет за собой
изменение величины![]() и, следовательно, ЭДС гальванического
элемента, которое зафиксируется
измерительным прибором.
и, следовательно, ЭДС гальванического
элемента, которое зафиксируется
измерительным прибором.
В
процессе титрования раствора хлорида
железа(III)
раствором хлорида олова (II)
(рас.18) электродный потенциал
![]() и, следовательно, ЭДС гальванического
элемента вначале незначительно
уменьшаются, в точке эквивалентности
I
происходит резкий скачок потенциала,
а затем он начинает изменяться на
незначительную величину. В результате
получается характерная кривая
потенциометрического титрования.
и, следовательно, ЭДС гальванического
элемента вначале незначительно
уменьшаются, в точке эквивалентности
I
происходит резкий скачок потенциала,
а затем он начинает изменяться на
незначительную величину. В результате
получается характерная кривая
потенциометрического титрования.
Скачок потенциала в точке эквивалентности тем больше, чем больше разность стандартных окислительно-восстановительных потенциалов и концентрация титруемого раствора и титранта.

Рис.18. Потенциометрическая кривая титрования раствора железа (III) раствором хлорида олова (II)
Кислотно-основное титрование. Титрование сильных и слабых кислот сильными и слабыми основаниями (и наоборот) связано с изменением концентрации ионов водорода Н+ в растворе. Поэтому индикаторным электродом гальванического элемента в этом случае наиболее часто служит стеклянный электрод 1 (рис.19). Электродом сравнения обычно служит каломельный полуэлемент 2 (или хлорсеребряный).
Поскольку потенциал стеклянного электрода обусловлен обменом ионов щелочных металлов, находящихся в стекле, с ионами водорода из раствора, то изменение концентрации последних в исследуемом растворе при титровании повлечет за собой изменение электродного потенциала и ЭДС элемента.

Рис.19. Установка для кислотно-основного потенциометрического титрования
При титровании раствора сильной кислоты раствором сильной щелочи (или наоборот) происходит уменьшение концентрации ионов H+ например:
HCl + NaOH = H2O + NaCl
или
H+ + OH- = H2O
Это ведет к уменьшению потенциала индикаторного электрода и, соответственно, к уменьшению ЭДС. В результате получается типичная кривая потенциометрического титрования с резким скачком потенциала в эквивалентной точке (рис.20, кривая 1).
Таким образом, потенциал индикаторного электрода при кислотно-основном титровании будет зависеть от рН раствора. Действительно, если в качестве индикаторного электрода взять стеклянный электрод, то в этом случае
![]() (21)
(21)
Так
как стандартный потенциал водородного
электрода
![]() принят за нуль, то
принят за нуль, то
![]()
Поскольку
![]() окончательно
имеем:
окончательно
имеем:
![]() (22)
(22)
Сложнее обстоит дело при титровании слабых кислот и оснований, особенно в случае, когда титрант тоже является либо слабым основанием, либо слабой кислотой.

Рис.20. Кривая потенциометрического титрования кислот разной силы сильным основанием
Это объясняется тем, что слабые кислоты или основания диссоциируют не полностью, поэтому концентрация ионов Н+ в растворе заметно меньше, чем при диссоциации сильных кислот или оснований. Кроме того, в процессе титрования слабых кислот или оснований в растворе будет получаться буферная смесь остатков, например, слабой кислоты и ее соли, образующейся в процессе нейтрализации. Все это ведет к тому, что при титровании слабых кислот или оснований получается потенциометрическая кривая с небольшим скачком потенциала в точке эквивалентности (рис.20, кривая 2). В связи с этим очень слабые кислоты и очень разбавленные растворы титровать вообще нельзя, т.к. на кривой титрования не будет скачка потенциала и, следовательно, не будет обнаружена эквивалентная точка (рис.20, кривая 4). Поэтому при титровании слабых кислот и оснований нужно учитывать их константу диссоциации. Чем больше константа диссоциации, тем больше степень диссоциации слабой кислоты и, следовательно, выше концентрация ионов H+ в растворе. В результате величина скачка потенциала на кривой титрования больше и можно более точно определить точку эквивалентности (см. рис.20). Скачок потенциала при кислотно-основном титровании тем больше, чем больше концентрация титруемой кислоты или щелочи и чем больше их степень диссоциации. Минимальные концентрации удовлетворительно титруемых кислот и оснований следующие:
константа диссоциации;
кислоты - 10-7 до 5·10-7 до10-6 до10-5;
концентрация, г-экв/л - не титруется 1 0,1 0,001.
Поскольку ход кривой потенциометрического титрования зависит от константы диссоциации, то можно титровать смесь кислот, отличающихся значениями Kg (рис.21, кривая 1).

Рис.21. Кривые титрования смеси сильной (1) и слабой (2) кислоты
В этом случае на кривой титрования получается два скачка потенциала: сначала оттитровывается сильная кислота (эквивалентная точка V'экв) а затем слабая (V''экв). Для того, чтобы при титровании смеси кислот получались четкие кривые титрования, необходимо, чтобы значения Кg этих кислот отличались не менее чем на четыре порядка:
![]() .
(23)
.
(23)
В
противном случае на кривой
потенциометрического титрования может
не получиться четко выраженных скачков
потенциала (рис.21. кривая 2), Необходимо
отметить, что отсутствие ярко выраженного
скачка потенциала у кривой титрования
в координатах Е; В-V,
мл еще не означает, что эквивалентную
точку определить нельзя. Необходимо
построить дифференциальную кривую
титрования в координатах
![]() мл,
что для ряда случаев позволит получить
значения эквивалентной точки.
мл,
что для ряда случаев позволит получить
значения эквивалентной точки.
2.1.2.3. Титрование в неводных, растворителях. В потенциометрическом титровании довольно широко применяются также неводные растворители. По способности отдавать или принимать протоны неводные растворители можно разделить на четыре группы:
- растворители, проявляющие только кислотные свойства, например, ледяная уксусная кислота;
- растворители, проявляющие только основные свойства, например, жидкий аммиак;
- амфипротонные растворители, проявляющие как кислотные, так и основные свойства, например, этанол;
- апротоннке растворители, не проявляющие ни кислотных, ни основных свойств, например, бензол и четыреххлористый углерод.
Применение неводных растворителей дает возможность проводить потенциометрическое титрование таких веществ, титрование которых в водном растворе осуществляется с трудом или вообще невозможно, (например, веществ, не растворимых в воде или образующих стойкие эмульсии и суспензии). Хороший результат можно получить и при титровании смесей кислот или оснований, которые не титруются раздельно в водном растворе. Так, при титровании смеси хлористоводородной и монохлоруксусной кислот в водном растворе гидроксидом натрия скачки потенциалов, соответствующие эквивалентным точкам, определить трудно (рис.22, кривая 1).

Рис.22. Кривые потенциометрического титрования в различных растворителях: 1- вода; 2- ацетон
Если в качестве растворителя взять ацетон, то получается кривая с четко выраженными скачками потенциала, по которым можно точно определить эквивалентные точки для обеих кислот (V´экв и V"экв).
Как известно, при производстве сульфатной целлюлозы основным отходом производства является черный сульфатный щелок. Из него в дальнейшем выделяют сульфатное мыло, из которого получают талловое масло - сырье для получения талловой канифоли и синтетических моющих средств. Таловое масло в основном (92%) состоит из жирных (олеиновой, линоленовой, стеариновой и т.д.) и смоляных (абиетиновой) кислот, которые не растворимы в воде. Определение их содержания проводят методом экстракции. Однако этот метод является весьма длительным (анализ длится почти двое суток) и многооперационным. В связи с этим явный интерес представляет потенциометрическое титрование жирных и смоляных кислот в неводных растворителях - этаноле или бутаноле - гидроксидом натрия. При этом получаются типичные кривые потенциометрического титрования (индикаторным электродом является стеклянный электрод, электродом сравнения - хлорсеребряный). Описанный метод обладает достаточной точностью, а время проведения опыта составляет 10-15 минут.
2.1.2.4. Способы нахождения конечной точки титрования. Основной задачей при потенциометрическом титровании является обнаружение скачка потенциала, отвечающего конечной точке титрования, и нахождение эквивалентной точки. Для этого применяют два способа - расчетный и графический.
Расчетный способ. Кривые титрования можно построить на основании расчетных величин потенциала в зависимости от концентрации раствора, а потенциал в точке эквивалентности рассчитать на основании типа реакции. Этот случай рассматривать не будем, т.к. в практических работах используется графический способ нахождения конечной точки титрования.
Графический способ. Строится кривая потенциометрического титрования в координатах «ЭДС гальванического элемента (или потенциал индикаторного электрода) - объем прибавленного титранта». На рис.23,а приведена интегральная кривая потенциометрического титрования. Для нахождения конечной точки титрования проводят две параллельные касательные к пологим нижней и верхней ветвям кривой. Третью касательную проводят к восходящей (или нисходящей) части кривой до пересечения с двумя первыми касательными) (см. рис. 23,а). Полученный отрезок прямой m n делят пополам и получают конечную точку титрования А (эквивалентную точку). Опуская из точки перпендикуляр к оси абсцисс, получают эквивалентный объем титранта Vэкв.
Более
точным способом нахождения конечной
точки титрования А является построение
дифференциальной кривой потенциометрического
титрования в координатах
![]() мл (рис.23,б), где
мл (рис.23,б), где![]() - разность между вторым и первым значением
ЭДС. между третьим и вторым и т.д. В этом
случае максимум на кривой соответствует
конечной точке титрования. При титровании
смеси кислот различной силы на кривой
титрования появится несколько максимумов.
- разность между вторым и первым значением
ЭДС. между третьим и вторым и т.д. В этом
случае максимум на кривой соответствует
конечной точке титрования. При титровании
смеси кислот различной силы на кривой
титрования появится несколько максимумов.

Рис.23. Различные графические способы нахождения конечной точки при потенциометрическом титровании
В ряде
случаев, когда конечную точку титрования
надо зафиксировать наиболее точно, ее
находят по кривой зависимости
![]() ,
мл (рис.23,в). Для этого соединяют концы
обеих ветвей кривой, которые находятся
с разных сторон оси абсцисс (прямаяdg).
Точка пересечения прямой dg
с
осью абсцисс дает объем титранта,
соответствующий конечной точке
титрования.
,
мл (рис.23,в). Для этого соединяют концы
обеих ветвей кривой, которые находятся
с разных сторон оси абсцисс (прямаяdg).
Точка пересечения прямой dg
с
осью абсцисс дает объем титранта,
соответствующий конечной точке
титрования.
Ранее
отмечалось, что при кислотно-основном
титровании потенциал индикаторного
электрода зависит от рН исследуемого
раствора (уравнение II).
Поэтому кривые окислительно-восстановительного
титрования часто строят в координатах
![]() ,
мл;
,
мл;![]() ,
мл;
,
мл;![]() ,
мл.
,
мл.
2.1.3.Аппаратура. В потенциометрии различают два вида измерительных приборов:
- потенциометры, работающие по компенсационной схеме;
- высокоомные вольтметры. Рассмотрим принцип компенсационного метода. Компенсационный метод определения ЭДС, иногда называемый методом Поггендорфа, заключается в следующем (рис.24). При замыкании ключа 1 от источника постоянного тока 2 на реохорд 4 подается некоторое напряжение, регулируемое переменным сопротивлением 3. На реохорде 4 напряжение равномерно повышается от нуля до значения ЭДС источника тока (например, аккумулятора). Нормальный элемент Вестона 6, ЭДС которого постоянна и равна 1,0186 В при 25оС, через ключ 8 и гальванометр 10 присоединен одним концом к движку реохорда II, а другим - к началу реохорда. Параллельно нормальному элементу Вестона подключена электролитическая ячейка 7, состоящая из индикаторного электрода и электрода сравнения, ЭДС которой нужно определить.

Рис.24. Схема установки для измерения ЭДС гальванических элементов компенсационным способом
Сомкнув ключи 1 и 8, мы навстречу ЭДС аккумулятора 2 включаем ЭДС нормального элемента Вестона и, передвигая подвижной контакт реохорда 4, добиваемся компенсация, т.е. ЭДС аккумулятора, скомпенсированная сопротивлением реохорда 4, становится равной ЭДС нормального элемента Вестона (стрелка гальванометра в этот момент должна стоять на нулевой отметке). В этот момент ЭДС реохорда равна ЭДС нормального элемента Вестона:
![]() .
(24)
.
(24)
Известно, что
![]() ,
(25)
,
(25)
где
ЕАК - ЭДС аккумулятора;
l - отрезок реохорда от положения движка;
L - общая длина реохорда.
Тогда, учитывая равенство электродвижущих сил, имеем
![]() .
(26)
.
(26)
Таким образом, мы узнали цену деления реохорда. Далее, отключим контакт 8 и замкнем контакт 9. В этом случае навстречу проградуированной ЭДС источника тока включена ЭДС гальванического элемента 9, которую нужно определить. Точно так же, передвигая контакт 11, добиваемся момента компенсации (стрелка гальванометра должна стоять на нулевой отметке). Поскольку цена деления реохорда известна, определяем ЭДС гальванического элемента:
![]() .
(27)
.
(27)
Принципиальная схема потенциометра, работающего по компенсационной схеме, представлена на рис.25. в настоящее время промышленностью выпускаются высокоомные потенциометры типа ППТВ-1 .Р-300, Р-307 и другие.

Рис.25. Схема типового потенциометра:
А. - аккумулятор;
R1, R2, R3, - набор сопротивлений;
н.э.- нормальный элемент Вестона;
х - исследуемый гальванический элемент;
Г – гальванометр.
Устройства,
измеряющие ЭДC
по некомпеасационной схеме, называются
высокоомными вольтметрами. Это, например,
рH-метры
(ионометры), шкала которых проградуирована
в милливольтах (мВ) и рХ. Таким образом,
эти приборы могут работать как
милливольтметры и рХ-метры, Другими
словами, при потенциометрическом
титровании можно строить
кривые
в координатах Е,В - V,
мл;
![]() ,
мл; или рХ -V,
мл; или
,
мл; или рХ -V,
мл; или
![]() ,
мл (естественно, что, если проводится
кислотно-основное титрование, то кривые
строятся в координатахpH-V,
мл); или
,
мл (естественно, что, если проводится
кислотно-основное титрование, то кривые
строятся в координатахpH-V,
мл); или
![]() мл.
мл.
В настоящее время промышленностью выпускаются рН-метры рH-121, рН-340, рН-673, ЭВ-74 и другие.
Кроме того, выпускаются вольтметры прямого отсчета, снабженные электронным усилителем для получения сигнала, который регистрируется с помощью цифрового счетчика (Щ-1413; Щ-4313; И-120). Вольтметры прямого отсчета выпускаются также с измерительным устройством, шкала которого проградуирована в единицах рН и милливольтax.
Приборы типа высокоомного вольтметра дают менее высокую точность, нежели приборы, работающие по компенсационной схеме. Однако рН-метры проще в эксплуатации и незаменимы в работе с автоматическими титраторами.
Из электродов сравнения наиболее широко применяется хлорсеребряный электрод, поскольку он, в отличие от каломельного электрода сравнения, не имеет в своем составе ртути. В настоящее время выпускаются хлорсеребряные электроды сравнения ЭВЛ-1М3, ЭВЛ-1M1 и т.п.
Применение в потенциометрии того или иного индикаторного электрода зависит от ряда условий:
- применяемого титрования;
- определяемого элемента;
- наличия примесей в растворе;
- концентрации раствора.
Обычно стараются применить наиболее стойкий и чувствительный в данных условиях электрод.
Металлические индикаторные электроды изготовляют из плоской металлической пластинки, скрученной проволоки и т.п. Отечественная промышленность выпускает тонкослойный платиновый электрод ЭТПЛ-1, а также проволочный платиновый электрод ЭПЛ-1. Все более широко отечественной промышленностью выпускаются ионоселективные электроды:
-стеклянные электроды для определения рН: ЭСП-11Г-05; ЭСЛ-41Г-04; ЭСЛ-63-07;
- стеклянные электроды для измерения активности ионов Na+ (ЭС A-51-07) и ионов К+ (ЭСЛ-91-07);
- пленочные пластифицированные электроды ЭМ-С1O4-О1; ЭМ-NO3-O1.
В заключение следует отметить, что метод потенциометрии незаменим в тех случаях, когда те или иные измерения необходимо проводить в условиях отсутствия электрического тока (приборы не к чему подключать). Действительно, имея небольшой переносной потенциометр компенсационного типа, портативный аккумулятор, элемент Вестона и набор электродов, можно проводить анализы в походах, экспедициях и т.п.
2.2. Вольтамперометрия. Только что рассмотренный метод потенциометрического анализа основан на электродных реакциях в отсутствие внешнего тока (ток во внешней цепи появляется за счет работы гальванического элемента). В противоположность этому вольтамперометрия основана на электродных реакциях, протекающих за счет приложенного извне постоянного электрического тока.
2.2.1. Явления на электродах электрохимической ячейки при прохождении постоянного электрического тока. Ранее отмечалось, что при пропускании тока через электрохимическую ячейку перенос электричества осуществляется:
- по металлическим проводам и электродам - за счет направленного перемещения электронов;
- в растворе электролита - вследствие миграции ионов. При этом положительно заряженные ионы (катионы) движутся к отрицательно заряженному электроду (катоду), а отрицательно заряженные ионы (анионы) перемещаются к положительно заряженному электроду (аноду);
- через межфазную границу «электрод-электролит» - за счет электродных окислительно-восстановительных реакций.
Напомним кратко, какие электродные процессы происходят в электрохимической ячейке, заполненной водным раствором ZnCl2, в который опущены цинковый и платиновый электрода, при прохождении постоянного электричества тока (рис. 26). В растворе электролита ионы цинка и хлора движутся хаотично. Однако если мы подключим цинковый электрод к отрицательному полюсу источника тока, а платиновый электрод - к положительному полюсу, то катионы начнут перемещаться к катоду, а анионы к аноду. Если, мы приложим к электродам ячейки необходимую разность потенциалов, на катоде начнется выделение металлического цинка:
Zn2+ + 2e Zn0
на нерастворимом аноде будет выделяться хлор:
2Cl- - 2e Cl2
В зависимости от природы электролита и материала электродов на последних не обязательно выделяются продукты растворенной соли. Например, при электролизе раствора Nа2SO4 на катоде будет выделяться водород:
2H2O + 2e = H2 + 2OH-
а на аноде кислород;
2H2O - 4e = O2 + 4H+
Это происходит потому, что для разряда каждого вида иона на электроде должен быть достигнут потенциал его выделения. В последнем случае потенциалы выделения ионов Na+ и S042- не достигнуты, поэтому они на электродах не выделяется.

Рис.26. Электрохимическая ячейка
Могут быть и другие случаи. Например, при электролизе FeCl3 на катоде получается двухвалентное железо Fe3++e Fez+, которое на аноде вновь может окисляться до трехвалентного состояния:
Fe2+ - e Fe3+
Однако всегда на катоде электроны переходят от электрода к ионам, а на аноде - от ионов к электроду. Для прохождения постоянного тока через ячейку (т.е. чтобы на электродах происходили электродные реакции) на ее электроды нужно подавать необходимое напряжение, определяемое по формуле
![]() ,
(28)
,
(28)
где
Е - напряжение на клеммах ячейки, В;
φa - электродный потенциал анода, В;
φк - электродный потенциал катода, В;
iR падение напряжения в электролите между электродами, В.
Практически же получается, что к электродам ячейки необходимо приложить большее напряжение, нежели рассчитанное по (28), т.к. в действительности происходит поляризация электродов.
Рассмотрим это на примере. Возьмем электрохимическую ячейку, заполненную электролитом, и на ее электроды подадим напряжение от источника постоянного тока (рис.27).

Рис.27. Вольтамперные кривые
Вначале при увеличении напряжения на клеммах электрохимической ячейки сила тока может быть практически равна нулю. Это происходит потому, что не достигнуты потенциалы выделения данных веществ, следовательно, они не выделяются на электродах за счет окислительно-восстановительных электродных реакций (электрическая цепь разомкнута, т.к. ток не проходит через межфазную границу «электрод-электролит»). Но как только при дальнейшем увеличении напряжения будут достигнуты потенциалы выделения веществ (см. рис. 27. точку a), на электродах начнут происходить электродные реакции и, согласно закону Ома, дальнейшее увеличение напряжения повлечет за собой возрастание силы тока. Если бы сопротивление электролита в ячейке равнялось нулю, то с повышением напряжения сила тока возрастала бы по линии 1. Поскольку же раствор электролита обладает определенным сопротивлением, вследствие чего в нем происходит падение напряжения (iR), сила тока с повышением напряжения на клеммах ячейка, в соответствии с уравнением (28), должна бы повышаться по кривой 2 (см. рис.27). В действительности, снимая подобную зависимость, мы получаем кривую 3. Таким образом, реальный ход i-V кривой будет отличаться от расчетного на величину ΔV. Это приращение напряжения получило название поляризации, или перенапряжения. Поскольку напряжение на клеммах электрохимической ячейки складывается, помимо падения напряжения в электролите, из электродных потенциалов катода и анода, это означает, что электродные потенциалы выделения веществ на катоде и аноде несколько превышают их равновесные потенциалы. При этом потенциал катода смещается в область отрицательных значений, потенциал анода - в область положительных значений. Например, по расчету потенциал выделения вещества на катоде равен 0,5 В, на анода 0,8 В. Их алгебраическая сумма Е=0,8-(-0,5)=1,3 В. В действительности из-за поляризационных явлений электродный потенциал катода смещен в область отрицательных значений на 0,2 В и равен -0,7 В; электродный потенциал анода сместился в область положительных значений на 0,1 В и стал равен 0,9 В. Тогда их алгебраическая сумма будет равна:
![]() ,
т.е. > Е на 0,3 В.
,
т.е. > Е на 0,3 В.
Различают концентрационную поляризацию (или перенапряжение диффузии), химическую поляризацию (перенапряжение реакции), кристаллизационную поляризацию (перенапряжение кристаллизации). Следует отметить, что последний вид поляризации - перенапряжение кристаллизации открыт свердловским ученым, профессором Уральского политехнического института О. Н. Есиным. Как известно, любая электрохимическая реакция многостадийна. Например, реакция выделения ионов меди состоит из трех стадий.
1. Подход ионов меди из глубины электролита к поверхности катода.
2. Собственно акт электрохимического превращения: Сu2+ + 2е →Сu0.
3. Внедрение атома меда в кристаллическую решетку катода.
В случае выделения газов стадий реакции может быть больше. Например, при выделении водорода различают пять стадий.
1. Подход ионов водорода к катоду.
2. Собственно электрохимическое превращение: Н+ + е →Н
3. Образование молекулы водорода: Н + Н → Н2
4. Объединение молекул в пузырьки газа.
5. Отрыв пузырьков от поверхности катода.
Известно, что скорость сложной многостадийной химической реакции определяется скоростью ее наиболее замедленной стадии. Подобным образом определяется, какой вид поляризации присущ данному электрохимическому процессу.
Если наиболее замедленной стадией электрохимической реакции является диффузия ионов из глубины электролита к поверхности катода, мы имеем дело с концентрационной поляризацией или, как ее еще называют, перенапряжением диффузии.
В случае если наиболее замедленной стадией электрохимической реакции является собственно электрохимическое превращение (а в случае выделения, например, газов - также стадия рекомбинации или образования пузырьков газа), мы имеем дело с химической поляризацией (или перенапряжением реакции).
Наконец, если наиболее замедленной стадией является вхождение разрядившихся ионов в кристаллическую, решетку металла, мы имеем дело с кристаллизационной поляризацией или перенапряжением кристаллизации.
Несмотря на различные виды поляризации, необходимо подчеркнуть, что во всех случаях их реальная сущность заключается в приращении потенциала катода при смещении его в область отрицательных значений, равно как и в смещении потенциала анода в область положительных значений. В конечном итоге происходит увеличение напряжения, приложенного к клеммам электрохимической ячейки. Таким образом, при прохождении через электрохимическую ячейку постоянного тока уравнение (28) следует записать в следующем виде:
![]() ,
(29)
,
(29)
где ΔV - дополнительное напряжение на клеммах ячейки вследствие поляризационных явлений на катоде и аноде.
![]() ,
(30)
,
(30)
где
ηа - поляризация (перенапряжение) анода;
ηK - поляризация (перенапряжение) катода.
2.2.2. Теоретические основы метода. Вольтамперометрический метод анализа основан на использовании явления поляризации микроэлектрода, получении и обработке вольтамперных (поляризационных) кривых, отражающих зависимость силы тока от приложенного напряжения. Таким образом, аналитическим сигналом для количественного анализа в данном методе является сила тока, которая зависит от концентрации раствора электролита.
В вольтамперометрии используют два электрода: рабочий поляризуемый электрод с малой поверхностью и неполяриэуемый электрод сравнения. Если, в качестве рабочего электрода выбран электрод с постоянно обновляющейся поверхностью (например, ртутный капающий электрод), то метод анализа называют полярографическим. В случае применения в качестве рабочего электрода таких металлов, как Pt, Fе и т.п., мы имеем дело собственно с вольтамперометрией.
При прохождении тока через электролитическую ячейку напряжение, приложенное к ее клеммам, согласно формуле 29, равно:
![]() .
(31)
.
(31)
Если определяемое вещество способно восстанавливаться при прохождении тока через его раствор в ячейке, то рабочий электрод с малой поверхностью делают катодом; если вещество окисляется - малый электрод делают анодом.
Предположим, что рабочим электродом с малой поверхностью является катод. Сделаем следующее. Добавим к исследуемому раствору «фон» - раствор сильного электролита, хорошо проводящего ток, с концентрацией в 100-1000 раз большей, чем раствор исследуемого вещества. Движение ионов к катоду или аноду происходит под действием двух сил:
- силы диффузии, обусловленной наличием разности концентраций ионов вещества в прикатодном слое и в глубине электролита;
- электролитической силы (движение ионов под действием этой силы называется миграцией).
Ионы фонового электролита являются электрохимически индифферентными, так как не принимают участия в электродных реакциях. Катионы фона (или анионы, в зависимости от того, какой знак имеет рабочий микроэлектрод), двигаясь к катоду, не могут при данном потенциале на нем разряжаться и остаются у поверхности электрода, образуя двойной электрический слой. Электрическое поле катода экранируется этими ионами, и поэтому катионы анализируемого вещества не притягиваются данным полем и двигаются только за счет силы диффузии.
Кроме того, введение фона увеличивает электропроводность электролита, и сопротивление его в ячейке становится весьма небольшим (R ~ 1 кОм). А так как сила тока в вольтамперометрии не превышает 10-5 А, то падением напряжения в электролите iR можно пренебречь. В результате уравнение (23) принимает вид:
![]() .
(32)
.
(32)
Далее, поверхность вспомогательного электрода (в нашем случае - анода) сделаем в сотни раз больше, чем поверхность рабочего микроэлектрода. Поскольку сила тока в вольтамперометрии мала, а поверхность анода велика, последний не будет поляризоваться при изменении силы тока, φa будет оставаться постоянным. Аналогичный вывод можно сделать и тогда, когда в качестве вспомогательного электрода используется каломельный электрод сравнения |Hg2Cl2, КСlнас|Нg. Действительно, каломельный электрод изготовлен на основе металлической ртути и твердого хлорида ртути Hg2Cl2, находящегося в равновесии с водным раствором хлорида калия (см. рис.8). Если такой электрод является анодом, то процесс на нем заключается в том, что атомы ртути отдают электроны, причем ионы ртути образуют с ионами хлора, находящимися в растворе, осадок каломели:
2Нg+2Сl- = Нg2Cl2+2е
Потенциал ртутного анода определяется концентрацией ионов ртути в растворе:
![]() .
(33)
.
(33)
Концентрации Нg22+ в свою очередь определяются концентрацией ионов Cl-:
 .
(34)
.
(34)
Поскольку плотность тока на аноде мала, то концентрация ионов Сl- вблизи анода изменяется настолько мало, что практически не влияет на концентрацию ионов Hg22+. Вследствие этого анод не поляризуется и его потенциал практически не меняется.
Итак, потенциал
анода
![]() в уравнении (32) остается практически
постоянным, следовательно, не влияет
на величину
в уравнении (32) остается практически
постоянным, следовательно, не влияет
на величину![]() .
Тогда уравнение (16) принимает вид
.
Тогда уравнение (16) принимает вид
![]() .
(35)
.
(35)
Таким образом, величина напряжения, приложенного к электролитической ячейке, определяется изменением потенциала катода, который мы избрали в качестве рабочего микроэлектрода. Ясно, что если рабочий микроэлектрод является анодом, то
![]() .
(36)
.
(36)
На рис.28,а приведена полярографическая установка, электролитическая ячейка которой имеет капающий катод 1 (рабочий микроэлектрод); вспомогательным электродом 2 (анодом), имеющим большую поверхность, является ртуть, налитая на дно ячейки, которая через платиновую проволоку соединяется с полярографом 4. В этом случае поляризоваться будет только рабочий микрокатод (капля ртути, втекающая из капилляра) и, естественно, только на нем возможен электрохимический процесс восстановления. Ячейка имеет трубки 3 для пропускания азота, чтобы удалить из ячейки растворенный кислород. На ртутном капающем катоде происходит процесс:
Men++ne Me (амальгама)
Образовавшийся свободный металл диффундирует в глубь капли ртути. Затем капля амальгамы падает, и процесс начинается на новой капле ртути.

Рис.28. Полярографические установки
На рис.28,б показана полярографическая установка, у ячейки которой вспомогательным электродом служит каломельный электрод сравнения 2, соединенный с исследуемым раствором, либо электролитическим ключом 5, либо стеклянной пористой пластинкой. Ячейки, представленные на рис.28, а, б являются простейшими.
На рис.28,в показана полярографическая установка с твердыми электродами (1 и 2). Если мы будем непрерывно увеличивать напряжение, подводимое к электродам ячейки, то будет возрастать потенциал рабочего микроэлектрода. Измерив силу тока, проходящего через ячейку, и построив зависимость силы тока i от потенциала V, получим кривую, называемую полярографической волной (рис.29).
При малых значениях
потенциала ионы анализируемого вещества
не разряжаются на микроэлектроде, т.к.
потенциал их разряда еще не достигнут.
Небольшой ток на участке 1 обусловлен
разрядом более положительных, чем ионы
анализируемого вещества, примесей и
называется остаточным током. При
дальнейшем увеличении напряжения на
клеммах ячейки достигается потенциал
разряда исследуемых ионов
![]() ,
которые начинают разряжаться на рабочем
микроэлектроде. В результате этого ток
резко возрастает (участокII).
Это так называемый фарадеевсний ток.
Он увеличивается до некоторого предельного
значения, и с дальнейшим ростом потенциала
его значение практически остается
постоянным (участок III).
Возникает предельный ток диффузии
(iпред.диф).
,
которые начинают разряжаться на рабочем
микроэлектроде. В результате этого ток
резко возрастает (участокII).
Это так называемый фарадеевсний ток.
Он увеличивается до некоторого предельного
значения, и с дальнейшим ростом потенциала
его значение практически остается
постоянным (участок III).
Возникает предельный ток диффузии
(iпред.диф).

Рис.29. Полярографическая волна
Постоянство iпред.диф обусловлено тем, что в данной области потенциалов число ионов анализируемого вещества, продиффундировавших из глубины раствора электролита к поверхности рабочего микроэлектрода, равно их количеству, разрядившемуся на нем. Таким образом, скорость диффузии в этих условиях контролирует скорость электрохимического процесса в целом. Следовательно, величина предельного тока диффузии будет пропорциональна концентрации анализируемых ионов, вступивших в электрохимическую реакцию на рабочем микроэлектроде.
Полярографическая
волна (или полярограмма) содержит ценную
аналитическую информацию. Для этого на
полярограмме необходимо определить
величину потенциала полуволны
![]() и значениеiпред.диф.
и значениеiпред.диф.
Потенциалом
полуволны
![]() называется потенциал середины
полярографической волны (точкаm).
Потенциал полуволны не зависит от
концентрации анализируемых ионов, а
зависит только от их природы. Таким
образом, величина φ1/2
служит качественной характеристикой
полярографически активного вещества,
поскольку каждый ион имеет свой постоянный
потенциал полуволны. В справочниках
имеются значения потенциалов полуволны.
Поэтому, определив из подпрограммы
величину
называется потенциал середины
полярографической волны (точкаm).
Потенциал полуволны не зависит от
концентрации анализируемых ионов, а
зависит только от их природы. Таким
образом, величина φ1/2
служит качественной характеристикой
полярографически активного вещества,
поскольку каждый ион имеет свой постоянный
потенциал полуволны. В справочниках
имеются значения потенциалов полуволны.
Поэтому, определив из подпрограммы
величину
![]() ,
по табличным данным можно найти, ионы
какого вещества участвуют в электрохимической
реакции на рабочем микроэлектроде.
,
по табличным данным можно найти, ионы
какого вещества участвуют в электрохимической
реакции на рабочем микроэлектроде.
Зависимость предельного тока диффузии при использовании ртутного капающего электрода выражается уравнением Ильковича:
![]() ,
(37)
,
(37)
где
n - заряд иона, разряжающегося на микроэлектроде;
D - коэффициент диффузии, см2с-1;
m - масса ртути, вытекающая из капилляра за секунду;
τ - время жизни ртутной капли, с;
С - объемная концентрация исследуемых ионов, моль-л-1.
Для определенного иона и капилляра с постоянной характеристикой все величины правой части уравнения (37) постоянны, тогда:
![]() .
(38)
.
(38)
Следовательно, определив из полярограммы величину iпред.диф, рассчитывают концентрацию анализируемого вещества.
Для твердого микроэлектрода уравнение зависимости iпред.диф от концентрации исследуемых ионов имеет вид
![]() ,
(39)
,
(39)
где
S - площадь микроэлектрода, см2;
δ - толщина диффузионного слоя, см;
F - число Фарадея, Кл.
Таким образом, по величине потенциала полуволны можно провести качественный анализ исследуемого раствора, а по значению предельного тока диффузии - количественный анализ (определить концентрацию раствора).
Если раствор электролита содержит ионы нескольких веществ, способных, например, восстанавливаться на рабочем микрокатоде, полярограмма может иметь несколько волн (рис.30). Определив из полярограммы величину φ´1/2 φ´´1/2, φ´´´1/2, по таблицам идентифицируем вещества, которые присутствуют в исследуемом растворе, а по найденным значениям iпред.диф находим их концентрацию. Чтобы полярографические волны не сливались, необходимо, чтобы потенциалы разряда определяемых ионов отличались не менее чем на 100-150 мВ. Полярографические измерения на ртутном капающем электроде могут быть использованы для определения концентрации веществ от 0,2 до -1,9 В. Однако работа с таким электродом требует тщательного соблюдения всех мер, предусмотренных правилами техники безопасности при работе со ртутью.

Рис.30. Полярограмма раствора, содержащего несколько ионов, разряжающихся на микроэлектроде
Твердые электроды более удобны и безопасны, однако область их использования пока ограничена.
Остановимся еще на одном важном явлении, которое встречается при полярографии - возникновении максимумов на полярографических кривых.
В ряде случаев вместо нормального хода полярограммы получается кривая с максимумом. Это происходит вследствие того, что в некотором интервале потенциалов возникает ток, превышающий предельный ток диффузии (рис.31).
В этом случае определение величины iпред.диф (иногда говорят «определение высоты полярографической волны») становится невозможным.
Максимумы первого рода появляются при отсутствии поверхностно-активных веществ (ПАВ) на фоне слабоконцентрированных электролитов и имеют форму пика (см. рис.31,а).

Рис.31. Максимумы на полярографических кривых:
а- максимум первого рода; б- максимум второго рода
Это объясняется движением поверхности ртутной капли, вызывающей дополнительное перемешивание раствора, что увеличивает доставку ионов к рабочему микроэлеитроду и, следовательно, увеличивает силу диффузионного тока.
Максимумы второго рода появляются в концентрированных растворах при работе с быстро капающим ртутным электродом и имеют более сглаженную форму (см. рис.31,б). Они объясняются движением внутри самой ртутной капли, вызываемым вытеканием ртути из капилляра. В результате этого процесса происходит перемешивание раствора, вследствие чего увеличивается диффузионный ток.
Естественно, что для проведения полярографического анализа от максимумов нужно избавляться. Максимумы первого и второго рода могут быть сведены на нет добавкой ПАВ, тормозящих движение поверхности ртути (рис.32). В качестве таких ПАВ применяют желатин, агар-агар, столярный клей и т.п.
2.2.3. Методы вольтамперометрического анализа. Основной задачей вольтамперометрического анализа является качественный и количественный анализ исследуемого раствора. Если качественный анализ по потенциалу полуволны затруднений не вызывает, то для, проведения количественного анализа по величине iпред.диф существует ряд методов.

Рис.32. Полярографические кривые:
1- с максимумом второго рода; 2 - та же кривая с добавкой ПАВ
Метод расчета. Заключается в том, что с помощью полученной полярограммы измеряют величину iпред.диф а также определяют массу ртути m и время образования капли τ (значения коэффициента диффузии D берут из таблиц). Затем по уравнению Ильковича (37) определяют концентрацию анализируемого вещества:
![]() .
(40)
.
(40)
Этот метод интересен с теоретической точки зрения, но применяется редко.
Метод калибровочных кривых. Готовят ряд эталонных растворов определяемого вещества, концентрация которых точно известна. Строят калибровочный график (рис.33) в координатах «h – С», где h - высота вольтамперометрической кривой, равная величине iпред.диф; С - концентрация, моль·л-1. Затем снимают вольтамперную кривую исследуемого раствора и, определив ее высоту h, находят по калибровочному графику концентрацию анализируемого раствора Сх.
Метод стандарта. Готовят один эталонный раствор, снимают его вольтамперную кривую и такую же кривую для анализируемого раствора. Зная концентрацию стандартного раствора (С ст) и определив высоту стандартного (hст) и анализируемого (hx) растворов, можно рассчитать концентрацию определяемого вещества по формуле
![]() .
(41)
.
(41)

Рис.33. Калибровочный график
Метод добавок. вначале снимают вольтамперную кривую анализируемого раствора, затем в ячейку прибавляют по каплям стандартный раствор, определяемого иона с точно известной концентрацией с таким расчетом, чтобы высота волны возросла вдвое. Снимает вольтамперную кривую полученного раствора и делают расчет по формулам
![]() ,
,
где
Сх - концентрация анализируемого иона, мг/л;
hx - высота волны анализируемого иона,мм;
Сст - концентрация добавляемого иона, мг/л;
hCT - высота волны стандартного раствора, мм;
hобщ - суммарная высота определяемого вещества в анализируемом и прибавляемом растворах, мм.
Концентрацию определяемого иона, введенного в ячейку в виде стандартного раствора, вычисляют по формуле:
![]() ,
(42)
,
(42)
где
Сст - первоначальная концентрация стандартного раствора, мг/л;
VCT - количество стандартного раствора, добавленного в ячейку, мл;
Vx - объем анализируемого раствора в ячейке, мл.
2.2.4. Новые направления в вольтамперометрии. Чувствительность классического метода вольтампероматрии во многих случаях бывает явно недостаточной.
Новые отрасли науки и техники предъявляют особые требования, например, к чистоте материалов и, соответственно, к методам их контроля. В связи с этим возникла необходимость разработки методов, позволяющих определять примеси при содержаний их в анализируемом веществе до 10-5–10-7 %. Так появились другие разновидности вольтамперометрического анализа, которым свойственна повышенная селективность, большая чувствительность, лучшая воспроизводимость. Кратко рассмотрим важнейшие из них.
Вольтамперометрия с линейной разверткой потенциала. Сущность данного способа анализа заключается в том, что напряжение на микроэлектрод и вспомогательный электрод подается со скоростью изменения до 100 В/с, а получаемая вольтамперная кривая регистрируется на осциллографе. Это дает возможность изучать процессы, мгновенно протекающие на микроэлектроде (до 10-7 с), а также повышает чувствительность метода, поскольку imax на осциллограммах в несколько раз больше iпред.диф получаемого на полярограммах.
Значительно большие, чем в классическом методе вольтамперометрии, скорости изменения поляризующего напряжения приводят к изменению формы вольтамперной кривой (рис,34,а): вместо полярографической волны наблюдается кривая с четко выраженным максимумом (пиком). Потенциал максимума φmax используется для качественного анализа вещества в растворе, т.е. он характеризует природу вещества. Максимальная высота h пика, соответствующая iпред.диф пропорциональна концентрации вещества, окисляющегося или восстанавливающегося на микроэлектроде, и используется для количественного анализа. Определив высоту пика, концентрацию вещества в растворе рассчитывают по формуле
![]() ,
(43)
,
(43)
где V- скорость изменения поляризующего напряжения.
Переменнотоковая вольтампорометрия. В классическом способе вольтамперометрии на электроды ячейки подается постоянный ток с изменяющимся во времени напряжением. В данном же способе анализа на электроды наряду с постоянным напряжением, медленно изменяющимся во времени, накладывается переменное напряжение небольшой амплитуды (до 50 мВ). Это дает возможность уменьшить почти до нуля конденсаторный ток, который сильно искажает форму вольтамперных.

Рис.34. Вольтамперные кривые:
А - с линейной разверткой напряжения; б - переменнотоковая;
в - инверсионная кривых при малых концентрациях вещества в растворе.
Вследствие чего увеличивается чувствительность и разрешающая способность данного способа анализа.
Так же, как и в вольтамперометрии с линейной разверткой, вольтамперная переменнотоковая кривая имеет форму кривой с максимумом (рис.34,б) и содержит такую же аналитическую информацию: потенциал максимума характеризует природу вещества, а максимальная высота пропорциональна его концентрации.
Инверсионная вольтамперометрия. Определяемое вещество с концентрацией 10-7-10-9 м/л некоторое время подвергают электролизу в небольшом объеме стационарного ртутного электрода или на поверхности твердого электрода при потенциале, несколько более отрицательном, чем потенциал полуволны определяемого иона. Определяемое вещество, например, в случае полярографии, при этом концентрируется в ртути в виде амальгамы. Последняя затем анодно растворяется при потенциале, непрерывно изменяющемся от значения, при котором проводилось катодное выделение элемента на ртути, до более положительных потенциалов. В результате кривая, анодного тока имеет вид характерного зубца (рис.34, в), глубина l которого соответствует iпред.диф и пропорциональна концентрации определяемого иона в растворе при условии постоянства других факторов. Значение φmin характеризует природу вещества.
2.2.5. Аппаратура. Для проведения вольтамперометрического анализа необходимы электролитическая ячейка и прибор-полярограф. Электроли-тическая ячейка состоит из стеклянного сосуда емкостью от 1 до 50 мл с погруженными в него рабочим минроэлектродом и вспомогательным электродом. Микроэлектродом могут служить либо ртутная капля, либо такие металлы, как Рt, Au, Ag и т.п., а также графит специальной обработки. В качестве вспомогательного электрода используют слой ртути с большой поверхностью либо электроды сравнения (каломельный, хлорсеребряный и т.п.).
Полярограф представляет собой прибор, поляризующий электроды ячейки постоянно изменяющимся напряжением и в тоже время регистрирующий изменение силы тока в ячейке от подаваемого напряжения.
В ходе
анализа вольтамперная кривая записывается
пером на диаграммной бумажной ленте,
которая перемещается синхронно с
подаваемым напряжением. В результате
получается вольтамперная кривая в
координатах «i
– φ». Если скорость изменения напряжения,
подаваемого на ячейку, велика (до
нескольких десятков вольт в секунду),
самопишущие регистраторы в силу их
инерционности использовать нельзя.
Тогда вольтамперная кривая регистрируется
на экране осциллографа, и определение
iпред.диф
и
![]() производят по осциллограммам.
производят по осциллограммам.
В настоящее время выпускают различные марки полярографов: ППТ-1, ПУ-1, IP-7, I П-7в, ОН-102, OH-105 и др.
В заключение следует отметить, что помимо анализа растворов, содержащих ионы неорганических веществ, вольтамперометрия дает неплохие результаты при анализе многих органических веществ. К последним относятся соединения, содержащие карбонильные группы, двойные углерод-углеродные связи, связи углерод-галоген, азот-кислород, диазогруппы. Соединения такого типа характерны для процессов и продуктов бумажной, гидролизной и лесохимической промышленности, а также для производства древесных плит и пластиков. Поскольку многие органические соединения плохо растворяются в воде, в качестве растворителей часто используют органические жидкости.
2.3. Амперометрическое титрование. При проведении амперометрического титрования необходимо соблюдать условия вольтамперометрического анализа:
- один из электродов ячейки должен иметь очень малую поверхность (индикаторный микроэлектрод), поверхность другого электрода должна быть велика (вспомогательный электрод). В этом случае поляризуется только индикаторный микроэлектрод, потенциал вспомогательного электрода остается практически постоянным;
- в исследуемый раствор необходимо вводить фоновый электролит (в результате чего падением напряжения в электролите можно пренебречь);
- ионы определяемого вещества должны восстанавливаться или окисляться на индикаторном микроэлектроде.
Дополнительным
условием для проведения амперометрического
титрования является то, что оно проводится
при потенциале предельного тока диффузии.
Изменение последнего в процессе
титрования раствора является аналитическим
сигналом в этом методе анализа.
Действительно, в соответствии с уравнением
Ильковича (37)
![]() величина предельного тока диффузии
пропорциональна концентрации вещества
в растворе. Таким образом, изменение
концентрации определяемого иона повлечет
за собой изменение величины iпред.диф
(рис.35,а).
величина предельного тока диффузии
пропорциональна концентрации вещества
в растворе. Таким образом, изменение
концентрации определяемого иона повлечет
за собой изменение величины iпред.диф
(рис.35,а).

Рис.35. Вольтамперные кривые при различной концентрации
вещества в растворе (а) и кривая амперометрического титрования (б); С1>С2>С3>С4
Если по оси ординат отложить значения iпред.диф, а по оси абсцисс - объем добавляемого титранта, то в результате получим кривую амперометрического титрования (рис.35,б).
Кривая амперометрического титрования состоит из двух прямолинейных участков, пересечение которых соответствует точке эквивалентности.
В зависимости от характера реакции, протекающей между определяемым веществом и титрантом, а также от того, какое вещество окисляется или восстанавливается на индикаторном микроэлектроде при потенциале титрования, различают несколько типов кривых амперометрического титрования.
Первый тип кривой (рис.36,а) наблюдается, когда исследуемое вещество электрохимически активно, т.е. при заданном потенциале окисляется или восстанавливается на индикаторном микроэлектроде. Титрант в этом случае индифферентен (не участвует в электродных реакциях).

Рис.36. Типы кривых амперометрического титрования
Кривые первого типа получаются при титровании ионов Ag+, Рb2+, Bi3+ или каких-либо других ионов, способных восстанавливаться на индикаторном микрокатоде, каким-либо осадителем.
Например, раствор AgN03 титруется раствором КI при потенциале предельного тока диффузии разряда ионов Ag+ на катоде Ag+ + е → Ag. В результате реакции осаждения Ag+ + I- → AgI концентрации ионов Ag+ в исследуемом растворе будет уменьшаться. Следовательно, в соответствии c уравнением Илъковича (37), для данного случая по мере добавления титранта будет уменьшаться значение предельного тока диффузии.
![]()
В точке эквивалентности предельный ток диффузии достигает минимального значения и далее остается постоянным.
Второй тип кривых наблюдается тогда, когда ионы исследуемого вещества при заданном потенциале электрохимически неактивны (не участвуют в электродной реакции), а титранта электрохимически активен. Например, раствор, содержащий ионы Ва2+, титруется хроматом:
Ba2+ + Cr2-4 = BaCrO4
До точки эквивалентности (рис.36,б) диффузионный ток иона CrO4 невелик (соответствует остаточному току полярографической волны), т.к. концентрация ионов CrO42- в растворе мала. По мере добавления титранта концентрация ионов СrО42- достигает такой величины, что становится возможным их разряд на рабочем микроэлектроде:
CrO2-4 + 3e +8H+ Cr3+ + 4H2O
Дальнейшее
добавление титранта в соответствии с
уравнением Ильковича
![]() повлечет
за собой увеличение предельного тока
диффузии. Третий тип кривых наблюдается,
когда оба компонента - ионы исследуемого
вещества и титрант - принимают участие
в электрохимических реакциях. Например,
раствор, содержащий ионы Рb2+,
титруется хроматом:
повлечет
за собой увеличение предельного тока
диффузии. Третий тип кривых наблюдается,
когда оба компонента - ионы исследуемого
вещества и титрант - принимают участие
в электрохимических реакциях. Например,
раствор, содержащий ионы Рb2+,
титруется хроматом:
Pb2+ + CrO2-4 PbCrO4
Вначале
при добавлении титранта снижение
![]() обусловливается уменьшением концентрации
ионов Pb2+,
связывающихся в осадок. В точке
эквивалентности ток имеет минимальное
значение. При дальнейшем добавлении
титранта в растворе увеличивается
концентрация ионов СrO42-,
что
влечет за собой возрастание
обусловливается уменьшением концентрации
ионов Pb2+,
связывающихся в осадок. В точке
эквивалентности ток имеет минимальное
значение. При дальнейшем добавлении
титранта в растворе увеличивается
концентрация ионов СrO42-,
что
влечет за собой возрастание
![]() (рис.36, в).
(рис.36, в).
В разобранных примерах использовался один индикаторный микроэлектрод. Если определяемый ион восстанавливается, микроэлектрод должен быть катодом, если окисляется - анодом.
Однако возможно амперометрическое титрование с двумя индикаторными электродами. В этом случае в анализируемый раствор помещают два одинаковых электрода, между которыми поддерживается небольшая разность потенциалов (10-50 мВ). Наличие тока в ячейке связано с электрохимическими процессами на двух электродах. При титровании по этому методу часто отпадает необходимость в построении кривой титрования, т.к. точка эквивалентности может быть определена по резкому прекращению или появлению тока.
В процессе амперометрического титрования происходит увеличение объема раствора. Чтобы не вводить поправку на изменение объема, концентрация раствора титранта должна быть в 10-20 раз выше концентрации исследуемого вещества.
2.3.1. Аппаратура. Установка для амперометрического титрования компактна и собирается из доступных и недорогих приборов (рис.37).

Рис.37. Амперометрическая установка с одним индикаторным микроэлектродом
В нее входят: источник постоянного тока 1 (аккумулятор, сухой элемент), потенциометр, или магазин переменного сопротивления 2 (примерно на 1 кОм), вольтметр постоянного тока 3, микроамперметр постоянного тока 4 чувствительностью 10-6-10-9 А, микробюретка 5, электролитическая ячейка с индикаторным микроэлектродом 6 и вспомогательным электродом 7, магнитная мешалка 8.
Установив потенциометром 2 на вольтметре 3 значение потенциала предельного тока диффузии определяемого иона, в ячейку из микробюретки 5 добавляют титрант и следят за показаниями микроамперметра 4. Строят график в координатах «iпред.диф -V мл» и определяют эквивалентный объем титранта, пошедшего на титрование. Рассчитывают концентрацию исследуемого вещества в анализируемом растворе по формуле:
![]() ,
(44)
,
(44)
где
С - концентрация исследуемого вещества, г/л;
N - нормальность стандартного раствора титранта, экв/л;
Vэ - объем стандартного раствора титранта, пошедшего на титрование, мл;
Э -эквивалент исследуемого вещества;
Vn - объем пробы исследуемого вещества, мл.
В качестве индикаторного микроэлектрода применяют ртутный капающий или твердый электрод (Pt, Au, Ti, W, графит и т.д.). В качестве вспомогательного электрода используют слой ртути на дне ячейки или металл (последний должен быть нерастворим); можно использовать также каломельный или хлорсеребряный электроды сравнения.
При титровании с двумя индикаторными электродами используют два одинаковых поляризуемых электрода (например, платиновых) площадью 1-2 см2 каждый.
Аналитические возможности метода амперометрического титрования широки. Этим методом можно определять практически все элементы периодической системы и большое число органических соединений, используя реакции осаждения, комплексообразования, окисления - восстановления и кислотно-основного взаимодействия. Нижний предел определяемых концентраций 10-6 м/л. Метод прост и не требует сложной аппаратуры.
2.4. Кулонометрия. Кулонометрия объединяет методы анализа, основанные на измерении количества электричества, затраченного на электрохимическую реакцию определяемого иона. Аналитическим сигналом в данном методе анализа является сила тока, проходящего через электролитическую ячейку в процессе электролиза.
2.4.1. Электролиз. Процесс электролиза проводят в электрохимической ванне, являющейся системой, в которой за счет приложенного извне постоянного электрического тока происходят химические превращения веществ на электродах (см.рис.1). Таким образом, электролизом называются химические превращения веществ под действием электрического тока. Для процесса электролиза необходимо, чтобы на электродах ванны были достигнуты потенциалы разряда (выделения) ионов, находящихся в растворе электролита. Тогда на катоде ванны пойдут процессы восстановления, например:
Men+ +nе → Me0,
на аноде - процессы окисления:
Ме0 –nе→ Men+
Основные законы электролиза установлены Фарадеем:
- количество вещества, выделившееся на электродах при электролизе, пропорционально количеству электричества, прошедшему через раствор электролита;
- при прохождении через раствор электролита одного и того же количества электричества на электродах выделяется одно и тоже количество эквивалентов вещества. Эти законы выражаются формулой:
![]() ,
(45)
,
(45)
где
m - масса вещества, выделившегося на электродах, г;
i -сила тока, прошедшего через электролит, А;
Т - время, с;
Э - г-эктз вещества;
F - число, или постоянная, Фарадея, 96496 Кл.
Законы Фарадея являются одними из наиболее точных законов природа. Согласно им, при прохождении через растворы электролитов количества электричества, равного одному Фарадею, на электродах должен выделиться 1 г-экв вещества: 1 г водорода; 8 г кислорода; 63,5 г меди; 107,8 г серебра и т.д.
Однако в практической деятельности часто встречаются видимые отклонения от законов Фарадея. Например, при электролизе ZnCl2 на катоде выделяется цинк: Zn2++2e→Zn°
При прохождении через раствор количества электричества, равного одному Фарадею, на катоде должен выделиться 1 г-экв цинка (32,7 г). Однако в действительности цинка на катоде выделяется меньше. Это обусловлено тем, что на катоде, помимо выделения цинка, может идти выделение водорода из молекул воды: 2Н2О + 2е → H2f + 2OH- или разряд растворенного в воде кислорода: O2 + 2H2O + 4e → 4OH.
Поскольку на осуществление этих реакций тратится какое-то количество электричества, на процесс выделения цинка его остается меньше чем один Фарадей. В результате цинка на катоде выделяется меньше одного грамм-эквивалента.
Подобные отклонения от законов Фарадея обусловлены совместным разрядом ионов. Они могут возникнуть и тогда, когда продукты электролиза, выделившиеся на электродах, частично растворяются; могут быть и другие случаи.
Обычно в процессах электролиза используется величина, называемая «выход по току», которая рассчитывается по формуле:
![]() ,
(46)
,
(46)
где
А - выход по току, %;
mпракт - масса вещества, выделившегося на электроде в процессе электролиза, г;
mтеорет - масса вещества, которая должна выделиться на электроде теоретически в соответствии с формулой (45), г.
Если отклонений от закона Фарадея нет, масса вещества, выделившегося на электроде, равна массе вещества, рассчитанной по формуле (45), и выход по току составит 100%. Если в процессе электролиза существуют видимые отклонения от законов Фарадея, то mпрокт будет меньше mтеорет и выход по току будет меньше 100%.
Из законов Фарадея следует, что количество электричества, прошедшее через раствор электролита, может быть при известных условиях определено по массе продуктов электролиза, выделившихся на электроде при прохождении тока. Прошедшее количество электричества измеряют с помощью кулонометров.
Кулонометр представляет собой электрохимическую ванну, в которой протекает хорошо известная электрохимическая реакция со 100%-м выходом по току. Принцип действия кулонометра основан на том, что его включают в электрическую цепь последовательно с электрохимической ванной, в которую залит анализируемый раствор. Таким образом, за некоторый промежуток времени через анализируемый раствор в ванне и кулонометр пройдет одно и то же количество электричества. Поскольку в кулонометре проходит известная электрохимическая реакция (например, Сu2+ +2е → Сu0), то измерение количества электричества сводится к определению массы вещества, выделившегося на катоде кулонометра.
Действительно, зная массу выделившейся на катоде меди и ее грамм-эквивалент, по формуле (45) можно рассчитать количество электричества, прошедшего через анализируемый раствор:
![]() .
(47)
.
(47)
В зависимости от способа измерения различают электрогравиме-трические, газовые, титрационные и электронные кулонометры. В электрогравиметрическах кулонометрах определяют массу, например, металлической меди, выделившейся при электролизе сульфата меди. В газовых кулонометрах определяется объем газа, выделившегося в результате электрохимического процесса, и по этому объему рассчитывают количество электричества. Электронный кулонометр непосредственно отсчитывает число кулонов.
2.4.2. Теоретические основы метода. Кулонометрия основана на законах Фарадея, дающих связь между количеством электричества, прошедшего через раствор электролита, и массой вещества, выделившегося на электроде в результате электрохимической реакции. Если известны количество электричества и природа вещества, можно определить весовое содержание, или концентрацию определяемого вещества в растворе. При этом необходимо, чтобы все расходуемое количество электричества затрачивалось на электрохимическую реакцию определяемого вещества, т.е. чтобы процесс протекал со 100%-м выходом по току.
В зависимости от происходящих в растворе электрохимических процессов различают прямую кулонометрию и косвенную (кулонометрическое титрование).
В свою очередь, прямая кулонометрия подразделяется на потенциостатическую кулонометрию и амперостатическую кулонометрию). В первом случае анализ выполняется при поддержании постоянного значения потенциала рабочего электрода, во втором - при постоянной величине тока.
Потенциостатическая кулонометрия. Каждый ион, участвующий в электрохимической реакции, выделится на электроде только в том случае, если достигнет его потенциал разряда. В противном случае электрохимического акта не будет.
На рис.38 представлена вольтамперная кривая для многокомпонентной системы.

Рис.38. Вольтамперная кривая многокомпонентной системы
При значении потенциала электрода φ1 начинается разряд одного из компонентов раствора, потенциал φ2 соответствует началу разряда второго компонента. При потенциале, φ3 начинает разряжаться третий компонент. Таким образом, устанавливая то или иное значение потенциала электрода, можно добиться, чтобы на рабочем электроде выделялся определенный ион. Например, если нам необходимо проанализировать первый компонент, то значение потенциала рабочего электрода не должно превышать φ1. При этом условии на рабочем электроде будет выделяться только первый компонент системы, т.к. потенциалы выделения второго и третьего компонентов не достигнуты. Следует иметь в виду, однако, что если достигнут потенциал φ3, то на рабочем электроде будет выделяться не только третий компонент, но также первый и второй. Это необходимо учитывать при проведении данного вида анализа.
Потенциостатическая кулонометрия основана на измерении количества электричества, прошедшего на электрохимическое восстановление или окисление определяемого вещества при постоянном значении потенциала рабочего электрода. Чтобы процесс шел с максимальной скоростью и 100%-м выходом по току, электролиз следует проводить при потенциале, соответствующем середине площадки предельного тока диффузии определяемого вещества. Для первого компонента этот потенциал соответствует φ1 (см. рис.38).
По мере восстановления или окисления на электродах определяемого иона его концентрация в растворе электролита уменьшается и сила тока в цепи падает. Общее количество электричества, затраченное на полное электрохимическое превращение определяемого иона, выражается площадью, ограниченной кривой в координатах «сила тока – время» (рис.39,а) и рассчитывается по формуле:
![]() .
(48)
.
(48)
До полного электрохимического превращения исследуемого вещества нужно бесконечно большое время, т.к. iτ → 0 при τ → ∞. Поэтому для практических целей величину тока в любой момент времени можно определить по уравнению:
![]() ,
(49)
,
(49)
где
iτ - величина тока к моменту времени t, А;
i0 - ток в момент начала электролиза, А;
К - константа, зависящая от условий электролиза, с-1.
В соответствии с уравнением (49), уменьшение тока идет по экспоненциальной кривой (рис.39,б).
Прологарифмировав уравнение (49), получим:
![]() .
(50)
.
(50)

Рис.39. Зависимости силы тока от времени электролиза:
а- теоретическая -кривая ток-время в потенциостатических условиях;
б- кинетическая кривая в потенциостатических условиях;
в- кинетическая кривая в полулогарифмических координатах
Полученное
выражение представляет собой прямую
линию (рис.39,в), точка пересечения которой
с осью ординат соответствует
![]() ,
а тангенс угла ее наклона - К. Определив
значение К количество электричества
находят по формуле:
,
а тангенс угла ее наклона - К. Определив
значение К количество электричества
находят по формуле:
![]() .
(51)
.
(51)
Рассмотренный расчетный способ определения количества электричества предложен Мак-Невилом и Байкером. Кроме того, количество электричества, необходимое для проведения анализа, может быть также определено с помощью химического или электронного кулонометров.
Содержание определяемого вещества находят по закону Фарадея. Простейшая потенциостатическая схема представлена на рис.40:

Рис.40. Схема установки для потенциостатической кулонометрии:
1- источник постоянного тока; 2- делитель напряжения; 3- вольтметр;
4- миллиамперметр; 5- кулонометр; 6- электрохимическая ячейка с пористой перегородкой, разделяющей катодное и анодное пространства;
7- блок измерения электродного потенциала
Для того, чтобы на электродах поддерживалась постоянная разность потенциалов, сопротивление ячейки 6 должно быть во много раз меньше сопротивления делителя напряжения 2. Для устранения влияния продуктов электролиза, образующихся на вспомогательном электроде, катодное и анодное пространства должны быть разделены пористой перегородкой или электролитическим ключом. Об окончании процесса электролиза можно судить по резному уменьшению тока (практически до нуля), регистрируемого миллиамперметром 4.
Преимуществом потенциостатический кулонометрии является высокая селективность метода т.е. возможность поочередного определения отдельных веществ в их смеси.
Амперостатическая кулонометрия. Основана на определении количества электричества, затраченного на электрохимические превращения определяемого вещества при постоянном значении тока, протекающего через электролитическую ячейку.
Поскольку определение содержащегося в растворе вещества при этом методе проводят при постоянном значении силы тока, то, определив время электролиза, можно рассчитать количество электричества по формуле:
![]() .
(52)
.
(52)
Однако в практике проведения этого вида анализа дело обстоит сложнее. В процессе электролиза по мере уменьшения концентрации анализируемого вещества изменяется потенциал рабочего электрода и появляется возможность разряда другого компонента раствора. Следовательно, будут происходить затраты электричества на побочные электрохимические процессы, что скажется на точности результатов анализа (выход по току для определяемого вещества будет меньше 100%). Чтобы избежать этого, принимаются специальные меры. Например, в исследуемый раствор вводится вспомогательный реагент, который участвует в электрохимической реакции, а продукт этой реакции должен стехиометрически взаимодействовать с определяемым веществом. Простейшая гальваностатическая схема представлена на рис.41.

Рис.41. Схема установки для амперостатической кулонометрии:
1- источник постоянного тока; 2- высокоомное сопротивление;
3- миллиамперметр; 4- кулонометр; 5- электрохимическая ячейка с пористой перегородкой, разделяющей катодное и анодное пространства;
6- рабочий электрод; 7- вспомогательный электрод; 8- индикаторные электроды; 9- индикаторный блок
Для определения момента окончания электролиза применяют как визуальные (с помощью индикаторов), так и инструментальные (потенциометрия, амперометрия, фотометрия и др.) методы. Иногда об окончании процесса можно судить по изменению потенциала электрода.
Кулонометрическое титрование. Если в прямой кулонометрии определяемое вещество подвергается непосредственному электрохимическому превращению, то в кулонометрическом титровании определяемое вещество А может быть неэлектроактивно. В электрохимическую реакцию на рабочем электроде вступает вспомогательный реагент В, добавляемый в ячейку, который, например, восстанавливается до промежуточного реагента С:
В + е → С.
Затем в растворе происходит химическая реакция А + С → АС. Таким образом, кулонометрическое титрование основано на электрохимическом получении промежуточного реагента, стехиометрически вступающего в химическую реакцию с определяемым веществом.
В качестве химической реакции, протекающей между определяемым веществом и промежуточным реагентом, может быть использована любая реакция, применяемая в титриметрии.
Электролиз ведут при постоянной силе тока, а расчет количества электричества, израсходованного на генерацию дополнительного реагента (до промежуточного реагента) и, следовательно, на превращение определяемого вещества, производят по формуле (33). Для того, чтобы изменение концентрации вспомогательного реагента в процессе электролиза было незначительным, его обычно берут в 1000-кратном избытке по отношении в определяемому веществу. При этом обеспечивается стопроцентный выход по току.
Схема установки для кулонометрического титрования такая же, как и для амперостатической кулонометрии (см. рис.41). Окончание, процесса электролиза определяют так же, как в амперостатической кулонометрии.
Кулонометрическое титрование отличается большей чувствительностью, чем все другие известные титриметрические методы.
2.4.3. Аппаратура. Для поддержания постоянного потенциала рабочего электрода в потенциостатической кулонометрии могут быть использованы потенциостаты П-5848 и П-5672М, хроноамперометрическая система CXA-1, 1 или простейшая потенциостатическая схема (см. рис.40). Для поддержания постоянной величины тока в амперостатической. кулонометрии могут быть использованы универсальные источники питания УИП-1 и УМП-2, указанные выше потенциостаты П-5848 и П-6672М, поскольку они могут работать как в потенциостатическом, так и в гальваностатическом режимах, или простейшие гальваностатические схемы (см. рис.41).
При использовании потенциостатов электрохимическая ячейка содержит три электрода: рабочий, вспомогательный и электрод сравнения (каломельный или хлорсеребряный). Потенциал рабочего электрода измеряется относительно электрода сравнения и автоматически поддерживается на заданном уровне электронной схемой прибора.
По сравнению с другими физико-химическими методами кулонометрический анализ обладает рядом преимуществ:
- возможностью определения различных веществ в широком диапазоне концентраций;
- высокой точностью и воспроизводимостью;
- высокой чувствительностью (можно определить 10-10 моля вещества) ;
- возможностью селективного определения веществ в их смеси.
2.5. Электрогравиметрия. Электрогравиметрический анализ основан на использовании электролиза. Суть этого метода состоит в выделении из растворов электролитов металлов (например, меди из раствора сульфата меди), осаждающихся на катоде при прохождении через электрохимическую ячейку постоянного электрического тока. Масса выделившегося на катоде металла соответствует его содержанию в растворе.
Электрогравиметрический анализ можно проводить как при постоянной силе тока, так и при постоянном потенциале рабочего электрода.
Следует иметь в виду, что в первом случав (i = const) в ходе анализа могут возникнуть осложнения, обусловленные следующими обстоятельствами. Как известно, внешнее напряжение, приложенное к электродам электролитической ячейки, описывается уравнением (28):
![]()
В процессе электролиза слагаемые правой части уравнения (28) изменяются во времени - вследствие уменьшения концентрации определяемых ионов в растворе электролита его сопротивление будет возрастать, а ток - уменьшаться. Для того, чтобы поддерживать ток постоянным, необходимо непрерывно увеличивать напряжение. Но при любом изменении Е изменяются анодный (φa) и катодный (φK) потенциалы. В результате может наступить такой момент, когда на рабочем электроде начнет осаждаться другой элемент.
Электролиз при постоянном приложенном извне напряжении (E=const) обеспечивает большую селективность анализа, чем при постоянной силе тока, т.к. величину E можно поддерживать достаточно малой. Однако при этом ток электролиза будет мал, и анализ окажется продолжительным. Данное затруднение можно устранить, если проводить электролиз с контролируемым потенциалом рабочего электрода (φк -const), что обеспечивает не только селективность анализа, но и возможно наибольший в условиях данного анализа ток электролиза. Постоянство потенциала рабочего электрода, поддерживают с помощью потенциостата. Схема установки для электрогравиметрического анализа приведена на рис.42.

Рис.42. Схема установки для электроосаждения металлов:
1- источник постоянного тока; 2- реостат; 3- вольтметр; 4- амперметр;
5- электролизер; 6- катод; 7- анод
Электролизер 5 представляет собой обычный химический стакан, в который опущено 2 электрода: большой катод в виде платиновой сетки 6 и меньшего размера анод в виде платиновой спирали 7. Раствор во время электролиза необходимо перемешивать.
Интересной разновидностью электрогравиметрического метода анализа является метод внутреннего электролиза. Метод назван так потому, что его применение не требует внешнего источника электрической энергии. Электрический ток в этом случае возникает за счет работы гальванического элемента, состоящего из платинового катода и анода, металл которого подбирается таким образом, чтобы при погружении этих электродов в исследуемый раствор возникла разность потенциалов. Если в качестве анода взять, например, металлический цинк, то образуется гальванический элемент
Pt | CuSO4 || ZnSO4 | Zn
C4 C1
На платиновом катоде восстанавливается определяемый металл
(Cu2+ + 2e Cu0),
а на аноде растворяется эквивалентное количество цинка:
(Zn0 – 2e Zn2+).
До установления равновесия токообразующей реакции
Cu2+ Zn0 Cu0 + Zn2+
практически вся медь будет количественно выделена на платиновом катоде.
Метод внутреннего электролиза прост, так как не требует внешнего источника тока, и селективен.
При проведении электрогравиметрического анализа должны соблюдаться определенные требования:
- определяемое вещество должно выделяться на катоде количественно;
- полученный осадок должен быть однородным по составу (содержание соосажденвых примесей должно быть минимальным);
- осадок должен быть мелкозернистым и плотно сцепленным с поверхностью электрода.
2.6. Кондуктометрия. Кондуктометрические методы анализа просты, практически очень удобны, достаточно точны и позволяют решить ряд важных научно-исследовательских и производственных задач, не поддающихся решению классическими химическими методами.
Все ранее рассмотренные методы анализа основаны на протекании электродных реакций либо в отсутствие внешнего тока (потенциометрия), либо под током (вольтамперометрия, кулонометрия, электрогравиметрия). В отличие от них кондуктометрия является методом, в котором электродные электрохимические реакции либо не протекают, либо являются вспомогательными и не учитываются. В связи с этим в кондуктометрии важнейшее значение приобретает одно из свойств растворов электролитов - электропроводность.
В качестве аналитических сигналов в кондуктометрии могут быть использованы:
- изменение сопротивления электролита;
- изменение полного переходного сопротивления границы «электрод – электролит»;
- общее изменение сопротивления электролитической ячейки.
В зависимости от того, какой аналитический сигнал используется при проведении анализа, различают собственно кондуктометрию, низкочастотную кондуктометрию и высокочастотную кондуктометрию.
Кондуктометрический анализ обычно проводят при использовании переменного тока. Поэтому, прежде чем перейти к рассмотрению закономерностей кондуктометрии, остановимся на явлениях, которые возникают в электролитической ячейке при прохождении синусоидального переменного электрического тока; рассмотрим также электропроводность растворов электролитов.
2.6.1. Электропроводность растворов электролитов. Электропроводность растворов является результатом диссоциации растворенного вещества и миграции ионов под действием электрического поля.
Как и все проводники электрического тока, растворы электролитов характеризуются определенным сопротивлением. Величина, обратная сопротивлению раствора, называется электропроводностью среды:
![]() ,
(53)
,
(53)
где
W - электропроводность раствора электролита, Ом-1;
R -сопротивление раствора электролита, Ом.
Известно, что сопротивление раствора электролита прямо пропорционально расстоянию l между погруженными в него электродами и обратно пропорционально их площади S:
![]() ,
(54)
,
(54)
где ρ - удельное сопротивление.
Величина, обратная удельному сопротивлению, называется удельной электропроводностью:
![]() .
(55)
.
(55)
Удельная электропроводность - это электропроводность объема раствора, заключенного между параллельными электродами площадью 1 см2 каждый, находящимися на расстоянии 1 см друг от друга. Размерность удельной электропроводности выражается в Ом-1см-1.
Электропроводность электролитов удобнее относить к числу эквивалентов растворенного вещества. Поэтому введено понятие эквивалентной электропроводности.
Эквивалентной электропроводностью называется электропроводность объема электролита, заключенного между параллельными электродами, расположенными на расстоянии 1 см друг от друга и имеющими такую площадь, чтобы между ними содержался 1 эквивалент вещества. Размерность эквивалентной электропроводности выражается в Ом-1см-1экв-1.
Удельная и эквивалентная электропроводности связаны между собой определенной зависимостью:
![]() ,
(56)
,
(56)
где с - концентрация электролита, экв.
Наконец, различают электропроводность при бесконечно большом разведении λ∞ (иногда ее называют «предельная эквивалентная электропроводность»).
Электропроводность при бесконечном разведении может быть представлена как сумма двух слагаемых, зависящих от природы катиона и аниона, которые входят в состав вещества:
![]() ,
(57)
,
(57)
где lк - подвижность катиона;
lа - подвижность аниона.
Подвижности
ионов представляют собой произведение
абсолютной скорости движения ионов
![]() на
число Фарадея:
на
число Фарадея:
![]()
![]() .
(58)
.
(58)
В бесконечно разбавленных растворах абсолютная скорость движения каждого иона есть величина постоянная (при t= const ). Поскольку число Фарадея также является константой, то подвижность для каждого иона является постоянной величиной (табл.1).
Таблица 1
Подвижности ионов при бесконечном разведении в водных растворах,
Ом-1•cм-1•экв-1 (t =25 0С)
|
Катион |
lк |
Анион |
la |
|
Н+ |
349,8 |
ОН- |
198,3 |
|
NH4+ |
73,6 |
1/2 C2042- |
110,5 |
|
K+ |
73,5 |
I- |
78,8 |
|
1/2 Ва2+ |
63,6 |
Br- |
78,1 |
|
Ag+ |
61,9 |
Сl- |
76,4 |
|
1/2 Са2+ |
59,5 |
NO3- |
71,5 |
|
1/2Cu2+ |
56,6 |
СlO4- |
64,5 |
|
1/2 Fe2+ |
53,5 |
F- |
55,4 |
|
Na+ |
50,1 |
СН3СОО- |
40,9 |
Растворы электролитов характеризуются степенью их диссоциации:
![]() .
(59)
.
(59)
Чем сильнее диссоциирует электролит, тем лучше его электропроводность и, стало быть, меньше его сопротивление. Поскольку принимается, что сильные электролиты диссоциируют нацело (на самом деле это не так), то для них формула (59) записывается в виде:
![]() ,
,
где f - коэффициент электропроводности.
Зависимость электропроводности от концентрации. Электропроводность растворов электролитов зависит от их концентрации. При увеличении концентрации можно было бы ожидать, что удельная электропроводность будет возрастать, т.к. увеличивается число ионов, переносящих ток в растворе. Однако эта зависимость не соблюдается - при достижении определенного максимального значения удельная электропроводность начинает уменьшаться (рис.43).

Рис.43. Зависимость удельной электропроводности от концентрации водных растворов электролитов
Такой характер изменения электропроводности объясняется тем, что при дальнейшем росте концентрации уменьшаются расстояния между ионами, следовательно, понижаются скорости их движения вследствие усиления межзонного взаимодействия. Для сильных электролитов усиливаются релаксационный и электрофоретический эффекты торможения, а для слабых электролитов уменьшается их степень диссоциации. Эквивалентная электропроводность увеличивается с разбавлением (рис.44) и достигает максимального предельного значения при бесконечном разбавлении (с→0). С разбавлением ослабляются силы межзонного взаимодействия и увеличиваются скорости движения ионов. Кроме того, в растворах слабых электролитов по мере уменьшения концентрации растет степень диссоциации.
Зависимость электропроводности от природы электролита, и природы растворителя. Природа электролита оказывает заметное влияние на электропроводность, т.к. различные ионы движутся с разными скоростями. Так, аномально высокой подвижностью в водных растворах обладают ионы водорода и гидроксила (см. табл.1), что объясняется специфическим механизмом их движения в растворе электролита.

Рис.44. Зависимость эквивалентной электропроводности от концентрации водных растворов электролитов
Природа растворителя также оказывает влияние на удельную и эквивалентную электропроводности электролита. Основными свойствами растворителя, влияющими на электропроводность, являются его вязкость и диэлектрическая проницаемость. Это связано с изменением скорости движения ионов, степени диссоциации электролитов. Чем больше вязкость, тем меньше скорость движения ионов. В растворителях с низкими значениями диэлектрической проницаемости наблюдаются процессы ассоциации ионов. Поэтому чем меньше диэлектрическая проницаемость растворителя, тем ниже электропроводность электролитов.
Влияние температуры на электропроводность. Удельная и эквивалентная электропроводности электролитов повышаются с ростом температуры. Это объясняется увеличением скорости движения ионов в связи с понижением вязкости раствора и уменьшением гидратации ионов.
Повышение температуры на, один градус вызывает увеличение электропроводности раствора на 2-2,5%.
2.6.2.
Явления на электродах электролитической
ячейки при прохождении синусоидального
тока.
Ранее было показано, что при пропускании
постоянного тока через электролитическую
ячейку напряжение, приложенное к ее
электродам, складывается из разности
потенциалов поляризованных анода и
катода и падения напряжения в электролите:
![]() .
.
В случае прохождения через ячейку переменного синусоидального тока в уравнении (28) неизменным остается лишь падение напряжения в электролите iR. Слагаемые, связанные с электродными реакциями, претерпевают существенные изменения.
Как известно, при погружений электродов в раствор на межфазной границе «электрод-раствор электролита» возникает двойной электрический слой, представляющий собой реальный физический конденсатор. Его емкостное сопротивление переменному току рассчитывается по формуле:
![]() ,
(60)
,
(60)
где
![]() - частота тока, Гц;
- частота тока, Гц;
С - емкость, Ф.
Если на клеммы электролитической ячейки подать необходимое напряжение переменного тока, то будет иметь место электролиз. Казалось бы, электрическую эквивалентную схему одного из электродов ячейки можно смоделировать в виде конденсатора с током утечки (рис.45).

Рис.45. Упрощенная эквивалентная схема границы «электрод-электролит»
Однако в действительности эквивалентная схема сопротивления границы «электрод-электролит» оказывается значительно сложнее, поскольку мы должны учитывать поляризационные явления на электродах ячейки. Так, в случае концентрационной поляризации при включении тока в приэлектродном слое через очень короткий промежуток времени концентрация разряжающихся ионов резко уменьшится из-за замедленности их доставки диффузией из глубины электролита к поверхности электрода. В результате ток будет уменьшаться, а электродный потенциал расти. Таким образом, между периодически меняющейся силой тока и электродным потенциалом создается сдвиг по фазе - изменение потенциала постоянно отстает во времени от изменения тока. Следовательно, концентрационная поляризация проявляет себя как реактивная составляющая, как емкость в схеме. Смоделировать концентрационную поляризацию можно введением в схему конденсатора СR и сопротивления RR. Однако в отличие от реального физического конденсатора, каковым является, например, емкость двойного электрического слоя, емкость конденсатора CR и сопротивление RR зависят от частоты переменного тока.
Аналогичным образом может быть учтена и химическая поляризация. В результате эквивалентную схему сопротивления границы «электрод-электролит» для одного из электродов ячейки можно смоделировать (рис.46), где Сдв.сл - емкость двойного электрического слоя, не зависящая от частоты переменного тока; Rреакции – активное сопротивление, моделирующее химическую поляризацию, также не зависящее от частоты переменного тока; СR и RR - емкостный и активный компоненты, моделирующие концентрационную поляризацию и зависящие от частоты переменного тока; Cадс - реактивный компонент, моделирующий адсорбционные явления на электродах и зависящий от частоты переменного тока; Rэ - сопротивление электролита, не входящее в полное сопротивление границы «электрод-электролит».

Рис.46. Полная эквивалентная схема сопротивления границы «электрод – электролит»
Полное переходное сопротивление границы «электрод-электролит», называемое импедансом (Zграниц), содержит не только активные сопротивления, но и реактивные, из которых емкость двойного электрического слоя Сдв.сл и Rреакции не зависит от частоты переменного тока, остальные элементы схемы являются частотнозависимыми. Аналогично будет выглядеть схема импеданса для другого электрода ячейки.
Таким образом, полное сопротивление электролитической ячейки (Zячейки), синусоидальному переменному току складывается из сопротивления раствора электролита (Rэ) и импеданса (Zграниц):
![]() .
(61)
.
(61)
Сопротивление электролита Rэ и импеданс Zграниц зависят от природы, состава раствора и частоты переменного тока совершенно по-разному. Следовательно, для методов физико-химического анализа возможно использовать либо измерение Rэ, либо Zграниц, либо, наконец, Zячейки.
Анализ уравнения (61) приводит к следующим результатам. Если в измерениях наличие емкостной составляющей мешает проведению анализа (например, при уравновешивании моста Кольрауша, что будет показано ниже), необходимо повысить частоту до 2000 - 4000 Гц. Но из уравнения (60) следует, что с резким повышением частоты переменного тока ω емкостное сопротивление двойного электрического слоя Rc станет весьма малым и зашунтирует правую половину схемы импеданса (см. рис.46). Следовательно, Zграниц, будет очень малым и на первый план выйдет сопротивление электролита RЭ по изменении которого можно проводить анализ. В результате мы имеем случай прямой кондуктометрии и кондуктометрического титрования.
В других случаях при проведении анализа мы можем оказаться заинтересованными в выделении импеданса, т.е. полного переходного сопротивления границы «электрод-электролит». Для увеличения Zграниц, в соответствии с формулой (60), необходимо снизить частоту переменного тока до 7-50 Гц. Сопротивление раствора можно резко уменьшить добавкой фона, например, крепкой серной кислоты. В результате мы будем иметь случай низкочастотной кондуктометрии.
Наконец, возможен и третий случай - высокочастотная кондуктометрия, основанная на измерении полного сопротивления ячейки (Zячейки) при частоте переменного синусоидального тока 1-ЗОмГц.
2.6.3. Прямая кондуктометрия, Кондуктометрическое титрование. Метод основан на измерении электропроводности (или сопротивления) анализируемого раствора при частоте переменного синусоидального тока 2000-4000 Гц.
2.6.3.1. Теоретические основы метода. Электропроводность зависит от многих факторов, а в частности, от природы вещества и концентрации ионов в растворе. При определенных условиях возможна прямая пропорциональность между этими величинами. Следовательно, измеряя электропроводность, можно количественно определять содержание различных веществ в исследуемом растворе.
Обычно строят график зависимости электропроводности от ряда стандартных растворов с известной концентрацией какого-либо вещества. Затем измеряют электропроводность раствора с неизвестной концентрацией и по графику находят концентрацию.
Следует отметить, что малейшие примеси значительно изменяют электропроводность электролита. Поэтому данный метод находит ограниченное применение в проведении исследовательских работ и в лабораторной практике, хотя в промышленных условиях его применяют довольно широко. Так, прямая кондуктометрия позволяет решать многие практические задачи, например, контролировать качество дистиллированной воды, оценивать загрязненность сточных вод, определять общее содержание солей в минеральной, морской и речной воде.
Кондуктометрическое титрование. Большое распространение в аналитической практике получил метод кондуктометрического титрования, основанный на использовании химической реакции, в результате которой происходит заметное изменение электропроводности раствора. После добавления каждой порции титранта измеряют электропроводность. Полученные данные используют для построения зависимости в координатах «электропроводность - объем добавленного титранта», Эти зависимости представляют собой кривые кондуктометрического титрования. По излому кривых в точке эквивалентности находят объем титранта, вступившего в реакцию.
При кондуктометрическом титровании могут быть использованы химические реакции всех типов: осаждения, комплексообразования, окислительно-восстановительные, кислотно-основного взаимодействия.
Рассмотрим, как изменяется электропроводность в процессе титрования раствора, ВаСl2 раствором Na2S04:
BaCl2 + Na2SO4 BaSO4 + 2NaCl
Запишем реакцию осаждения ВаS04в ионной форме:
Ba2+ + 2Cl- +2Na+ + SO42- BaSO4 + 2Na+ + 2Cl-
В начальном растворе имеются ионы Вa2+ и Cl-, в титранте ионы Na+ и SO42-. По мере добавления титранта ионы Ва2+ связываются с ионами S042- в осадок. Содержание ионов Сl- в растворе остается неизменным. Подвижность ионов Ba2+ равна 63,6; Na+ - 50,1. Следовательно, добавлением титранта более подвижный ион Ва2+ заменяется менее подвижным ионом Na+- электропроводность раствора будет уменьшаться (рис.47,а).
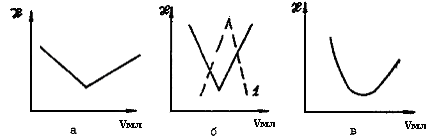
Рис.47. Кривые кондуктометрического титрования
Уменьшение электропроводности будет происходить до тех пор, пока все ионы Ва2+ из раствора не перейдут в осадок. После этого дальнейшее добавление Na2S04 увеличит электропроводность раствора, так как в растворе появляются избыточные ионы Na+ и S042-. Излом кондуктометрической кривой соответствует эквивалентной точке титрования. Зная нормальность титранта и его эквивалентный объем, рассчитывают концентрацию ВaСl2 в исследуемом растворе.
![]() ,
,
откуда
![]() ,
(62)
,
(62)
где
Nx - концентрация исследуемого раствора, экв/л;
Nт - нормальность титранта, экв/л;
Vэ - эквивалентный объем титранта, мл;
Vп - объем пробы исследуемого раствора, взятый для титрования, мл.
Подобного типа кондуктометрические кривые получаются во всех случаях, когда более подвижный ион заменяется при титровании на менее подвижный ион. Так, при нейтрализации сильных кислот сильными основаниями реакция идет согласно уравнению H+ + Cl- + Na+ + OH- H2O + Na+ + Cl-.
По мере добавления титранта более подвижный ион Н+ (lн+=349,8) связывается с ионом ОН- в Н2О, а вместо него появляется малоподвижный ион Na+ (lNa+= 50,1). Уменьшение электропроводности будет продолжаться до тех пор, пока все ионы H+ исследуемого раствора не свяжутся в воду; дальнейшее добавление ОН+ резко увеличит электропроводность раствора (рис.47,б). Кривые титрования кислот средней силы изогнуты и могут давать пологий минимум, не имеющий аналитического значения. Например, кривая титрования монохлоруксусной кислоты (рК=2,75) имеет пологий минимум (рис.47, в).
Еще сложнее обстоит дело при титровании слабых кислот сильными основаниями. Так, формы кондуктометрических кривых при титровании уксусной кислоты сильным основанием зависят от концентрации кислоты (рис.48,а). При титровании 0,1 N раствора СН5СООН раствором NaOH в начале титрования наблюдается минимум электропроводности (рис.48,а, кривая 1).

Рис.48. Кривые кондуктометрического титрования
Однако диссоциация уксусной кислоты быстро подавляется, и на большей часта кондуктометрической кривой, до точки эквивалентности, происходит линейное повышение электропроводности, вызываемое накоплением ионов Nа+ и CH3COO-. По достижении точки эквивалентности дальнейшее добавление титранта вызывает резкое увеличение электропроводности за счет не связывающихся ионов ОН-. С разбавлением уксусной кислоты кривые титрования приобретают форму, характерную для более сильных кислот (см. рис. 48, а) (0,001N CH3COOН-кривая 2, 0,0001N СН3СООН - кривая 3).
Уменьшение электропроводности происходит потому, что образующиеся в растворе ионы СН3СООН- также подавляют диссоциацию СН3СООН. Однако в связи с увеличением степени диссоциации СН3СООН в разбавленных растворах не происходит полного подавления ее диссоциации. Равновесная концентрация водородных ионов в этих условиях остается достаточно высокой, и при их нейтрализации электропроводность понижается.
При титровании очень слабых кислот становится заметным влияние гидролиза образующихся солей, которое выражается в плавном изгибе кондуктометрической кривой вблизи точки эквивалентности (рис. 48, б).
Кондуктометрическое титрование может быть использовано при анализе смесей кислот. Возможность анализа этих смесей зависит от констант диссоциации кислот и их концентрации. Наиболее благоприятные условия создаются при титровании смесей сильных и слабых кислот, т.к. в этом случае сначала нейтрализуется сильная кислота, затем слабая. На рис.48,в представлена кривая кондуктометрического титрования смеси хлористоводородной и борной кислот. Вначале нейтрализуется сильная кислота, и электропроводность понижается до точки эквивалентности. Затем начинает оттитровываться слабая кислота - электропроводность незначительно возрастает до точки эквивалентности, а после нее резко идет вверх. Vэ' и Vэ" на этом графике - эквивалентные объемы титранта, пошедшие на нейтрализацию соответственно сильной и слабой кислот.
Однако не все слабые кислоты (например, с рК≤4) могут быть оттитрованы в таких смесях. Ограничения связаны с возникновением минимума на кривых титрования, сглаживающего первый излом кривой. Это сглаживание становится малозаметным для 0,1 N растворов слабых кислот, когда рК≥4 и для 0,001N растворов, когда рК≥5.
Можно принять следующие границы рК слабых кислот, определяемых в смесях с сильными кислотами:
концентрация кислоты, экв/л 0,1 0,01;
интервал, рК 4-10 5-9.
При кондуктометрическом титровании оснований различной силы кислотами характер изменения электропроводности в общем аналогичен только что рассмотренному титрованию кислот.
Часто
кривые кондуктометрического титрования
строят в координатах «сопротивление
раствора R
- объем прибавленного титранта V,
мл». Поскольку сопротивление раствора
есть величина, обратная его
электропроводности, ход кривых титрования
в этих координатах будет обратен кривым
в
координатах
![]() мл.
Так,
при титровании сильной кислоты сильным
основанием сопротивление раствора R
будет
увеличиваться, т.к. вместо подвижного
иона Н+
появляются
малоподвижные ионы Na+.
После точки эквивалентности дальнейшее
добавление титранта вызовет уменьшение
R
(см. рис. 47,б, кривая 1).
мл.
Так,
при титровании сильной кислоты сильным
основанием сопротивление раствора R
будет
увеличиваться, т.к. вместо подвижного
иона Н+
появляются
малоподвижные ионы Na+.
После точки эквивалентности дальнейшее
добавление титранта вызовет уменьшение
R
(см. рис. 47,б, кривая 1).
2.6.3.2. Измерение сопротивления растворов электролитов мостовыми схемами. Как было указано выше, прямая кондуктометрия и кондуктометрическое титрование основаны на измерении электропроводности растворов электролитов. Для этого должно быть замерено активное сопротивление между электродами, погруженными в анализируемый раствор.
Мостовая схема для измерения электрического сопротивления впервые была разработана Уитстоном. Эта схема была использована Кольраушем применительно к переменному току. В настоящее время четырехплечевой мост (рис.49) получил самое широкое распространение для измерения сопротивления растворов электролитов. Мост состоит из четырех сопротивлений:
rХ - сопротивление анализируемого раствора, залитого в ячейку, в которую опущены два электрода;
RM - магазин сопротивлений;
r1, и r2 - постоянные сопротивления, причем r1=r2.
В диагональ моста включен нуль индикатор n

Рис.49. Четырехплечевой мост
Если питание моста осуществляется постоянным током, мы имеем дело с мостом Уитстона, если мост питается переменным током - с мостом Кольрауша.
В первом случав нуль-индикаторном служит гальванометр постоянного тока, во втором - осциллограф или специальный осциллографический нуль-индикатор.
Включим
мост по схеме Уитстона и вместо
электролитической ячейки с анализируемым
раствором подключим к нему постоянное
сопротивление (например, в 1000 Ом). Из
теории мостов известно, что наивысшая
чувствительность получается тогда,
когда все четыре плеча имеют примерно
одинаковые сопротивления. Поскольку
r1=r2,то,
изменяя значение rМ
магазина сопротивлений, добиваемся
положения, когда стрелка нуль - индикатора
установится на нуле. Мост уравновешен
- точки А и В находятся под одинаковым
напряжением, rм
=rx.
Тогда можно записать баланс моста:
![]() ;
;![]() .
.
Разделим
первое уравнение на второе:
![]() ,
,
откуда
![]() .
(63)
.
(63)
Сделаем следующее - в схему моста Уитcтона вместо постоянного сопротивления подключим электролитическую ячейку с анализируемым раствором rx. Тогда прохождение тока i2 через ячейку вызовет электролиз, и произойдет поляризация электродов. Поскольку явления поляризации смещают потенциал катода в область отрицательных значений, а потенциал анода - в область положительных значений, то через ячейку будет проходить ток i2+iполяризации. Установив магазином сопротивлений rм стрелку нуль-индикатора на нуль, мы уравновесим мост, но баланса моста не получим:
![]() ,
,
![]() .
.
Сократить i1 и i2 уже нельзя, а rX раствора оказывается сложной функцией элементов схемы. Ошибка измерения сопротивления в этом случае сможет составлять сотни процентов, поэтому при измерении сопротивления растворов электролитов мостовыми схемами используют переменный ток, т.е. применяют мост Кольрауша. Использование переменного тока, особенно повышенной частоты (2000 - 4000 Гц), не вызывает поляризации электродов, поэтому, уравновесив мост, мы добиваемся его баланса, и значение сопротивления rм у магазина сопротивления соответствует значению сопротивления раствора электролита между электродами (rХ).
При проведении кондуктометрического титрования используются нерастворимые электроды (лучше платиновые), жестко закрепленные относительно друг друга. Исследуемый раствор перемешивается. Объем раствора в параллельных опытах должен быть одинаков.
Чувствительность моста Кольрауша не очень высока - ошибка в значениях определяемого сопротивления может составлять 10% и более. Это объясняется тем, что при прохождении через ячейку синусоидального переменного тока импеданс (см. рис.46) содержит емкостные сопротивления.
Из теории мостов известно, что мост переменного тока может быть уравновешен только в том случае, если по отдельности будут уравновешены активные и емкостные сопротивления. Поэтому параллельно магазину сопротивлений подсоединяют магазин емкостей (рис.50) и, оперируя ручками обоих магазинов, подбирают на них такие значения, чтобы высота осциллограммы была бы минимальной (у моста Кольрауша нуль-индикаторном служит осциллограф).

Рис.50. Мостовая схема измерения сопротивления ячейки с компенсационной емкостью
Измерения сопротивлений растворов электролитов на таком мосту можно проводить с точностью 0,5-1%, принимая при этом особые меры (тщательная экранировка проводов, симметричность монтажа, правильный подбор точки заземления и др.). Только что приведенная точность замеров означает, что при сопротивлении раствора электролита в 1000 Ом в лучшем случае будет чувствоваться изменение в 10 Ом, при сопротивлении раствора в 100 Ом - 1 Ом. Однако в ряде случаев для анализа растворов нужна более высокая чувствительность моста Кольрауша. Например, при определении сульфат-иона в сульфитном щелоке сопротивление растворов в ячейке составляло 300 Ом,- а кондуктометрическая кривая, т.е. все изменение сопротивления при титровании сульфат-иона, укладывалась в 3 Ом. Следовательно, для получения достаточной точности анализа измерять изменение сопротивления в этом случае необходимо хотя бы с точностью 0,1%, т.е. через 0,3 Ом. Этому требованию отвечает импедансный мост, разработанный профессором С. И. Ремпелем на кафедре физической химии Уральского лесотехнического института. Чувствительность этого моста в 100-1000 раз выше, чем у моста Кольрауша. С помощью разработанного моста было проведено широкое обследование технологических трактов практически всех крупнейших целлюлозно-бумажных комбинатов. Импедансный мост был удачно использован и при определении емкости ионообменных смол.
2.6.3.3. Высокочувствительный импедансный мост для кондуктометрии. Высокая чувствительность импедансного моста (рис.51) достигается за счет применения диодов (1) в левой ветви. При этом через ячейку по-прежнему проходит переменный ток, а уравновешивание моста производится на постоянном токе (точнее, по постоянному полупериодному току). Понятно, что при этом мы измеряем не сопротивление электролита, а полное сопротивление ячейки, состоящее из сопротивления электролита и импеданса (полного переходного сопротивления границы «электрод-электролит»).

Рис.51. Схема импедансного моста высокой чувствительности
При конструировании прибора по указанной схеме и его использовании нужно учитывать следующее:
- диоды должны обладать достаточно малым сопротивлением в проводящем направлении, и это сопротивление должно быть достаточно стабильным во времени и при изменении температуры окружающей среды. Этому требованию в большей мере отвечают кремниевые диоды, например, Д-203, Д-205. Так как сопротивление диодов зависит от силы проходящего по ним тока, питание моста следует стабилизировать;
- желательно работать при соотношении плеч в цепи диодов 1:1 и приблизительном равенстве всех четырех плеч из обычных соображений наибольшей чувствительности моста;
- магазин сопротивлений желательно взять курбелъный. Если измеряемое сопротивление раствора сотни Ом, то магазин должен иметь декады на десятые и сотые доли Ома (например, магазин сопротивлений МСР-6ОМ, класс 0,02Э);
- чувствительность нуль-гальванометра должна быть не менее 0,5 10-6 А;
- питание моста может быть осуществлено от звукового генератора и от сети переменного тока. При этом нужно иметь в виду, что вследствие зависимости импеданса от частоты условия равновесия на высокой и низкой частотах будут различны. Так, ячейка с раствором, имеющая при частоте переменного тока 2500 Гц сопротивление около 100 Ом, при 50 Гц имеет сопротивление порядка 300 Ом.
В заключение приводим примерные значения параметров схемы моста для определения сульфат-иона. При титровании водных растворов сульфат-ионов питание моста осуществлялось от генератора с фиксированной частотой 2000 Гц напряжением 12 В. Сопротивление плеч моста r1 и r2 100 Ом (двухваттные сопротивления MTЛ). При использовании в качестве нуль-инструмента гальванометра с ценой деления 0,5 10-6 А, деление можно было отмечать смещением его стрелки при пользовании на магазине сопротивлений декадой на сотые доли Ома. Другими словами, чувствительность составляла сотые доли Ома. С гальванометром М273/1, который имеет чувствительность 5 10-8 А, чувствительность моста составляет 0,001%.
2.6.3.4, Измерение сопротивления раствора электролита 4-эдектродной ячейкой. В некоторых не особо ответственных случаях кондуктометрическое титрование можно проводить на 4-электродной ячейке (рис.52). Идея метода проста - ток пропускают через одну пару электродов 2, а падение напряжения iR измеряет на каком-либо участке раствора с помощью второй пары электродов 3, не поляризованных током, играющих роль щупов или зондов.

Рис.52, Четырехэлектродная ячейка для кондуктометрического титрования
Теперь несущественно, каким током питать схему - постоянным или переменным. Вопрос решается наличием подходящих источников тока 1 и измерительных приборов 4. При проведении кондуктометрического титрования на 4-электродной ячейке необходимо учесть следующий момент. В ходе титрования сопротивление раствора в ячейке будет меняться, например, увеличиваться. Но тогда, в соответствии с законом Ома, сила тона, проходящего через раствор электролита в ячейке, будет уменьшаться, а произведение iR практически может не изменяться. Для точности анализа необходимо обеспечить постоянство силы тока, проходящего через раствор электролита в ячейке. Осуществить постоянство силы тока можно простым способом. Для этого нужно взять источник напряжения, например, трансформатор на 220 В и из них 216-218 В погасить на сопротивлении 5.
Еще одним необходимым условием в данном методе анализа является то, что напряжение на измерительных электродах нужно обязательно измерять прибором, практически не потребляющим тока (например, высокоомным катодным вольтметром).
Преимуществом методов кондуктометрического титрования является возможность определения концентрации в мутных и окрашенных растворах, концентрации веществ в их смеси, высокая точность, легкое сопряжение с типовыми средствами автоматизации и дистанционного контроля производственными процессами.
2.6.4. Низкочастотное кондуктометрическое титрование. Метод низкочастотного кондуктометрического титрования основан на измерении изменения полного переходного сопротивления границы электрод-электролит (импеданса) в процессе титрования при пропускании через электролит переменного тока низкой частоты - от 7 до 50 Гц. Таким образом, аналитическим сигналом служит изменение импеданса.
В практике весьма часто возникает необходимость определения концентрации хлора в растворах. Например, для приготовления различных растворов часто используют хлорированную воду, для этой же цели используют естественную, речную и артезианскую воду. Содержание в этих водах ионов хлора, естественно, будет различное, что может вызывать нежелательные последствия.
Так, сульфитная варка целлюлозы проводится в импортных биметаллических котлах, внутренняя поверхность которых изготовлена из нержавеющей стали, весьма стойкой в кислотных средах. Однако на Соликамском ЦБК импортные варочные котлы после установки стали быстро выходить из строя. Причина крылась в том, что естественная вода в Соликамске оказалась богата хлоридами. Последние, являясь поверхностно-активными, адсорбируется на поверхности нержавеющей стали и депассивируют ее (разрушают пассивную пленку). В результате неразъемная сталь интенсивно коррозирует, и варочный котел выходит из строя. Вследствие этого предельно допустимая концентрация Сl- в варочной кислоте и сульфитном щелоке не должна превышать 50 кг/л.
Из вышесказанного становится ясным, насколько важен контроль за содержанием Сl- при сульфитной варке.
Обычные методы аргенометрического определения Сl- неприменимы к сульфитным щелокам, турмовой и варочным кислотам и другим технологическим растворам ЦВП, поскольку они мутные и интенсивно окрашенные. Вследствие этого становится невозможным фиксирование эквивалентной точки титрования с помощью цветных индикаторов.
Несколько более точных результатов можно добиться при потенциометрическом титровании, однако при титровании растворов с концентрацией 10-20 мг/л Сl- скачок потенциалов слишком мал и растянут, поэтому эквивалентную точку определить трудно. Кроме того, на ход анализа будут влиять органические вещества, находящиеся в растворе. Органические вещества влияют и на ход кондуктометрического титрования; необходимо также отметить, что кондуктометрическое титрование основано на измерении электропроводности раствора, а изменение последней за счет связывания Cl- (Cl- +Ag+=AgCl) будет настолько незначительным на фоне присутствующих кислот, что не зафиксируется на кривых титрования.
На кафедре физической химии УЛТИ профессором С. И. Ремпелем и М.В.Бураковым разработан новый электрохимический метод анализа растворов сложного состава - низкочастотное кондуктометрическое титрование и сконструирован низкочастотный индикатор кондуктометрического титрования (НИКТ), при помощи которого можно быстро определить, в частности, концентрацию хлоридов в сульфитных щелоках и других растворах ЦБП. Важно отметить, что, в отличие от обычного метода кондуктометрического титрования, при низкочастотном титровании присутствие индифферентных хорошо проводящих электролитов (серной кислоты и др.) не мешает чувствительности определения, не снижает ее, а, наоборот, как будет показано ниже повышает чувствительность определения эквивалентной точки титрования. Таким образом, данный метод имеет повышенную чувствительность и избирательность по сравнению с другими электрометрическими методами анализа. Например, азотнокислым серебром в кислой среде (30-50%Н2S04) можно оттитровать до 0,17 мг/л иона.
Низкочастотное титрование позволяет контролировать конец титрования по величине фарадеевского импеданса электродов при стационарном потенциале или по изменению емкости двойного электрического слоя в ходе титрования. Форма кривых титрования весьма характерна - например, кривая титрования растворов хлоридов азотнокислым серебром имеет в эквивалентной точке резкий излом.
Помимо определения концентрации прибор и сам метод могут быть применены для исследования физико-химических процессов, протекающих в объеме раствора и на электродах. Метод низкочастотного титрования может быть использован для изучения химических реакций и равновесий в растворе, адсорбционных процессов на электродах, определения коэффициентов диффузии и переходных коэффициентов электрохимической реакции.
2.6.4.1. Теоретические основы метода. В основу низкочастотного титрования положено измерение полного переходного сопротивления границы «электрод-электролит», называемого импедансом, при пропускании переменного тока достаточно низкой частоты (7-50 Гц). Сопротивление раствора при этом элиминируется наличием проводящего фона. При соблюдении этих условий в значительном числе случаев оказывается, что изменение импеданса электродов несравненно более резко и четко указывает на конец титрования, чем изменение собственно проводимости раствора (рис.53). Метод низкочастотного титрования особенно выгоден для окислительно-восстановительных реакций и реакций осаждения.

Рис.53. Кривые низкочастотного кондуктометрического титрования:
а- для малых концентраций НСl (1-5 мг/л) в присутствии 50% H2S04;
б- слабоконцентрированного раствора соли Мора на фоне 2NH2S04
Электрохимические явления на границе "электрод-электролит", происходящие в электролитической ячейке, моделируются следующей схемой (рис.54):

Рис, 54. Схема импеданса электродов электролитической ячейки
А - схема, моделирующая электрохимические явления на одном электроде ячейки; данную схему называют импедансом электрода; В - схема, моделирующая электрохимические явления, или импеданс, на другом электроде ячейки; Rэ - сопротивление электролита между электродами ячейки.
Из рис.54 следует, что полное переходное сопротивление границы «электрод-электролит» (или импеданс) для каждого из электродов слагается из:
Сдв. сл - емкости двойного электрического слоя, не зависящей от частоты переменного тока; iRреакции - активного сопротивления, моделирующего химическую поляризацию, обусловленного энергетическими затруднениями самого электрохимического акта разряда, также не зависящего от частоты переменного тока; СR и RR - емкостного и активного компонентов, моделирующих концентрационную поляризацию электродов; Сr и RR зависят от частоты переменного тока:
![]() ,
(64)
,
(64)
![]() ,
,
![]() ,
(65)
,
(65)
где
f - частота переменного тока;
Садс - реактивный компонент, моделирующий адсорбционные явления на электродах и зависящий от частоты переменного тока.
Правая цепочка схемы, моделирующей импеданс электрода, состоящая из Cадс, Rреакции, СR и RС, называется фарадеевской частью импеданса, или фарадеевской проводимостью.
Существуют обратимые и необратимые электрохимические системы. К необратимым электрохимическим системам относятся такие, в которых на электродах ячейки происходят окислительно-восстановительные реакции. Например, на электродах ячейки в растворе НCl при достижении необходимой разности потенциалов начинается процесс электролиза: на катоде идет восстановление ионов Н+:
2H+ + 2e H2
на аноде, в зависимости от величины плотности тока, происходит либо окисление ионов Сl-:
2Cl- - 2e Cl2,
либо окисление молекул воды:
2H2O – 4e O2 + 4H+.
Количество выделившегося газа, в соответствии с законом Фарадея, пропорционально количеству электричества, прошедшего через электролит. Как отмечалось ранее, любой электрохимический процесс сопровождается концентрационной или химической поляризацией; в некоторых процессах имеют место оба вида поляризации.
Согласно рис.54, Rреакции моделирует химическую поляризацию, RR и СR - концентрационную.
Таким образом, в необратимых электрохимических системах изменение импеданса границы «электрод-электролит» при низкой частоте пропускаемого тока определяется изменением фарадеевской части импеданса (точнее, изменением CR и RR, зависящих от частоты). В обратимых электрохимически системах на электродах практически нет окислительно-восстановительных, реакций и, следовательно, процесс электролиза отсутствует. В этом случае через электролит проходит остаточный ток, возникающий вследствие адсорбционных явлений на электродах. Следовательно, фарадеевской частью импеданса можно пренебречь и величина импеданса границы «электрод-электролит» в этом случае будет определяться изменением емкости двойного электрического слоя Сдв.сл (см. рис.54). В методе низкочастотного титрования возможны оба вышеотмеченных случая.
Если при титровании до и после точки эквивалентности возникает необратимая система (например, I/I-; Fe3+/Fe2+), то изменения импеданса при низкой частоте пропускаемого тока определяется изменением фарадеевской части импеданса, зависящей от концентрации.
Если при титровании до и после точки эквивалентности существуют только обратимые системы, то фарадеевской частью импеданса можно пренебречь, все изменения импеданса в этом случае связаны с изменением емкости двойного электрического слоя.
Именно этот случай имеет место при титровании Cl- азотнокислым серебром в присутствии платиновых электродов. При пропускании через такую систему тока порядка 10-20 мкА (при таком токе падение напряжения на платиновых электродах составит величину порядка 5-15 мВ) потенциалы выделения ионов исследуемого раствора не достигаются, т.е. реакций разряда на электродах нет. Если в исследуемый раствор добавить электролит, обладающий хорошей проводимостью (фон), например, серную кислоту, то сопротивлением электролита можно пренебречь. Тогда импеданс электрода будет определяться только изменением емкости двойного электрического слоя Сдв.сл.:
![]() .
(66)
.
(66)
Ионы Сl-, являясь поверхностно-активными, адсорбируются на активных центрах поверхности платиновых электродов, снижая тем самым емкость двойного электрического слоя. В соответствии с уравнением (66) величина 2 возрастает. При титровании раствора, содержащего Cl-, азотнокислым серебром в результате реакции Ag+ + Cl- AgCl сначала будут связываться в осадок ионы Сl-, содержащиеся в электролите (не адсорбированные на электродах). Поскольку это явление не будет сказываться на величине Сдв.сл., то величина Z будет, в принципе, оставаться постоянной. Но как только наступит момент, когда все ионы Cl-, находящиеся в электролите, будут связаны в AgCl. Следующие добавления AgN03 поведут к тому, что в AgCl будут связываться ионы Cl-, адсорбированные на электродах. Десорбция ионов Cl- с поверхности платиновых электродов поведет к резкому возрастанию емкости двойного электрического слоя и, следовательно, к уменьшению Z. Уменьшение Z будет продолжаться до тех пор, пока все ионы Cl- не десорбируются с поверхности платиновых электродов и не свяжутся в AgCl. Момент начала десорбции ионов Cl-, ведущий к уменьшению Z, четко фиксируется на графике п - V, мл, где n - показания прибора в относительных единицах (рис.55):

Рис.55. Кривая низкочастотного титрования при определении в растворе концентрации ионов Cl-
2.6.4.2. Схема прибора НИКТ. Измерение величины импеданса в приборе низкочастотного измерителя конца титрования (НИКТ) осуществляется по методу амперметра-вольтметра (рис.56). При постоянном значении Z катодный вольтметр будет показывать одно и тоже значение. Ясно, что увеличение Сдв.сл, ведущее к уменьшению Z, тут же зафиксируется катодным вольтметром - стрелка прибора отклонится в сторону меньших значений.

Рис.56. Принципиальная схема измерений при низкочастотном титровании: 1- источник переменного тока; 2-переменное сопротивление;
3- ячейка; 4- катодный вольтметр; 5- микроамперметр
Для практического проведения анализов разработано несколько схем прибора для низкочастотного титрования. Одна из простейших схем, рассчитанная на использование тока промышленной частоты, приведена на рис.57. Так как сопротивление диодов зависит от силы проходящего по ним тока, питание прибора следует стабилизировать.

Рис.57. Схема низкочастотного индикатора кондуктометрического титрования (НИКТа), разработанного и изготовленного на кафедре физической и аналитической химии УЛТИ
2.6.4.3. Примеры применения НИКТа. Обработка раствора перед анализом. Для того, чтобы выделить из общего импеданса интересующую нас компоненту - емкость двойного электрического слоя на электродах а свести к минимуму колебания омического сопротивления раствора (Rэ), титрование ведут в сильно кислой среде (50% серной кислоты по весу). Поскольку сульфитные щелока содержат редуцирующие вещества (сахара, альдегиды, муравьиную кислоту и прочее), восстанавливающие азотнокислое сеpебрo до металлического в ходе титрования хлоридов, они должны быть предварительно разрушены.
Это осуществляется обработкой аликвотной части щелока перекисью водорода в щелочной среде. Обработка щелока перекисью водорода в кислой среде не может быть применима, т.к. при ней хлор-ион окисляется в сопряжении с сахарами до свободного хлора и теряется.
Определение концентрации Сl- в водопроводной, речной и артезианской воде. Поскольку в данной работе редуцирующие вещества отсутствуют, специальной обработки исследуемого раствора не проводят. В стакан берут 10-20 мл воды, добавляют 25 мл 23N H2S04, 10 мл дистиллированной воды, после чего раствор охлаждают. Электроды опускают в стакан, включают мешалку и ждут 5-10 минут. Для установления адсорбционного равновесия электроды должны быть полностью покрыты раствором. Затем начинают титрование добавляя
по каплям 0,025 N AgN03 (см.рис.55). По результатам титрования строят кривую показания прибора n - V, мл AgN03. Определяют количество мл AgNo3 в эквивалентной точка и рассчитывают концентрацию Cl-:
![]() ,
(67)
,
(67)
где
N - нормальность раствора;
Vэ - объем азотнокислого серебра, затраченного на титрование пробы до эквивалентной точки, мл;
Vпр - объем пробы, мл;
35,5 - грамм-эквивалент хлор-иона.
После первого титрования проводят второе, добавляя серебро меньшими объемами.
Определение Сl- в сульфитном котловом щелоке. Отбирают пипеткой 10 мл раствора в стаканчик емкостью 50 мл, причем в отобранную аликвотную часть не должны попадать волокна целлюлозы. Прибавляют 1,35 мл 2N- раствора NаOH. Раствор после этого должен реагировать со щелочью на фенолфталеин. Эта дозировка выбрана для нормальных Соликамских щелоков сульфитной варки на смешанном кальциево-натриевом основании, имеющих рН 1,7-1,8. Если щелок более кислый или если изменилось основание, то необходимое количество щелочи должно быть заново установлено построением кривой потенциометрического титрования щелока раствором NаОН. Выбираем количество щелочи, дающее со щелоком рН=11. Если приготовленный раствор щелочи не точно 2N, то ее объем увеличивается или уменьшается пропорционально фактической нормальности. Прибавляют 1 мл перекиси водорода, закрывают стеклом, дают стоять до опадения обильной пены при размешивании. Раствор при этом немного разогревается (до 35-45°). Когда пена опала, добавляют еще 1 мл перекиси водорода, размешивают и осторожно нагревают до кипения, которое ведут 2-3 мин до разложения избытка перекиси и начала кипения раствора крупными пузырями. Снимают стакан с плитки, добавляют еще 1 мл перекиси водорода, после чего повторяют кипячение. Охлаждают стаканчик в воде, добавляют 25 мл 23 N раствора серной кислоты, после чего вновь охлаждают разогревшийся от кислоты раствор с выпавшим коричневым осадком лигносульфоновых кислот. Затем проводят титрование раствора, как указано выше.
Определение содержания Сl- в полукислоте, турмовой варочной кислотах. Поскольку данные кислоты не содержат органических редуцирующих веществ, предварительная щелочная обработка перекисью водорода в их анализе может быть опущена. Однако избыток серной кислоты, имеющийся в этих растворах, частично может восстанавливать азотнокислое серебро и служить причиной неверных результатов. Поэтому главная часть свободного 02 перед титрованием удаляется кипячением раствора, а остатки окисляются перекисью водорода на холоде. В стаканчике для титрования под стеклом 10 мл пробы нагревают до кипения и кипятят 1-2 мин. Турмовую кислоту нужно кипятить очень осторожно («на руках»), т.к. выпадающий моносульфит кальция склонен давать сильные толчки при кипении. Охлаждают в воде, добавляют 25 мл 23NН2S04, 5 мл дистиллированной воды (объем раствора 40 мл), охлаждают, добавляют около 0,5 мл перекиси водорода и титруют, как описано выше.
2.6.5. Высокочастотное кондуктометрическое титрование. Метод основан на измерении полной электропроводности ячейки в процессе титрования при пропускании переменного тока частотой 1-30 МГц. Таким образом, аналитическим сигналом служит изменение полной электропроводности ячейки;
2.6.5.1. Теоретические основы метода. При повышении частоты внешнего электрического поля электропроводность растворов электролитов уменьшается. Кроме того, высокие частоты деформируют молекулу, поляризуя ее и вызывая явление деформационной поляризации. Под действием высокой частоты полярная молекула может также определенным образом перемещаться, в результате чего возникает ориентационная поляризация. В результате этих поляризационных эффектов в растворе возникают кратковременные токи (продолжительностью порядка миллионных долей секунды), изменяющие электропроводность, диэлектрические свойства и магнитную проницаемость.
Измеряемая в этих условиях полная электропроводность электролитической ячейки λячейки состоит из активной составляющей λакт (истинная проводимость раствора) и реактивной составляющей λрвакт (мнимая электропроводность, зависящая от частоты тока и типа ячейки):
![]() (68)
(68)
Сложность зависимости этих величин от состава раствора затрудняет проведение прямого высокочастотного анализа, поэтому высокочастотный метод, как правило, применяют в виде высокочастотного, кондуктометрического титрования.
Широкое распространение получил метод высокочастотного титрования с использованием реакций кислотно-основного взаимодействия, осаждения, комплексообразования, окисления-восстановления.
Форма кривых высокочастотного титрования зависит от многих факторов - частоты переменного тока, концентрации анализируемого раствора и титранта, типа ячейки. Наиболее четко точка эквивалентности определяется при частотах порядка 5 и 20 МГц (рис.58.а), а изменение концентрации исследуемого раствора и титранта может кардинально изменить ход кривой (рис.58,б). Для точного проведения анализа необходимо подбирать частоту, переменного тока, концентрацию анализируемого раствора и тип ячейки.

Рис.58. Зависимость формы кривых высокочастотного титрования растворов НСl растворами NaOH от частоты
(а): 1-5 МГц, 2-10 МГц, 3-20 МГц; и концентрации
(б): 1- СHCl = 0.02М, СNaOH = 0.2 М; 2- CHCl = 0,02 М, СNaOH = 0,2 М
2.6.5.2. Особенности аппаратурного оформления. Схема установки для высокочастотного кондуктометрического титрования изображена на рис.59. Источником тока служат высокочастотные ламповые генераторы с частотой тока от 0,1 до 40 МГц. В качестве регистрирующего устройства обычно используются микроамперметры.
Важной частью высокочастотной установки является сама ячейка. Характерная ее особенность заключается в том, что в ней электроды не соприкасаются с исследуемым раствором. Стекло стенок ячейки не оказывает существенного сопротивления прохождению токов высокой, частоты, поэтому электроды располагаются снаружи ячейки и могут быть выполнены из любого металла.

Рис.59. Схема установки для высокочастотного титрования:
1- генератор высокой частоты; 2-ячейка; 3- постоянное сопротивление;
4-измарительный мост; 5-выпрямительный диод; 6-микроамперметр
Ячейки для высокочастотного кондуктометрического титрования делятся на два типа: емкостные и индуктивные (рис.60). В емкостной С - ячейке кольцевые электроды контактируют снаружи со стенками стеклянного стакана, заполненного раствором электролита. Электроды и слой электролита составляют обкладки конденсатора, стенки служат диэлектриком. В индуктивной L - ячейке сосуд из диэлектрика, заполненный электролитом, помещен в магнитное поле катушки. В проводящем анализируемом растворе, не обладающем магнетизмом, будут наводиться токи.
В зависимости от типа ячейки реактивная составляющая электропроводности при работе в высокочастотном режиме может являться функцией либо емкости С:
![]() .
(69)
.
(69)
либо индуктивности L:
![]() .
(70)
.
(70)

Рис.60. Емкостная (а) и индуктивная (б) ячейки для высокочастотного титрования
Емкостные ячейки (С-ячейки) применяют для анализа растворов с низкой электропроводностью. Чувствительность емкостной С-ячейки может быть повышена при уменьшении толщины стенок сосуда и увеличении площади электродов.
Индуктивные L - ячейки применяются для анализа растворов с высокой электропроводностью. Точность этого типа ячеек может быть повышена с увеличением размеров сосуда.
Чувствительность при измерениях активной составляющей. Чувствительность емкостной ячейки по активной составляющей полной проводимости увеличивается с возрастанием частоты и с уменьшением отношения емкости раствора к емкости стеной (С1/С2). Увеличению чувствительности емкостных ячеек также способствует уменьшение толщины стенок сосу да, увеличение площади электродов ячейки и применение материала для сосуда с возможно большей диэлектрической проницаемостью. Кроме того, большей чувствительности можно ожидать при титровании в средах с возможно низкой диэлектрической проницаемостью.
Чувствительность при измерениях реактивной составляющей. Чувствительность емкостной ячейки при диэлькометрическом титровании по реактивной составляющей не зависит от рабочей частоты тока. Повышению чувствительности способствует увеличение С1 и уменьшение С2. Чувствительность емкостной ячейки несколько повышается при титровании веществ с меньшим значением диэлектрической проницаемости.
Отечественной промышленностью выпускаются высокочастотные титраторы типа ТВ-6Л с емкостной ячейкой. Хорошие результаты высокочастотного анализа можно получить на венгерских осциллотитраторах системы «Пунгор» марки OK-301.
Метод высокочастотного кондуктометрического титрования недостаточно избирателен и позволяет проводить определения, нижний предел которых 10-3. Недостатком метода является сложность аппаратуры и трудность ее настройки. Однако он имеет ряд существенных преимуществ по сравнению с другими методами анализа:
- отсутствует соприкосновение металлических электродов с исследуемым раствором, в результате чего исключается явление поляризации электродов и их химическое взаимодействие с раствором;
- возможен анализ сильно агрессивных растворов, паст, эмульсий и в присутствии диэлектриков (масел, смол);
- титрование в неводных средах может быть осуществлено более просто.
В результате высокочастотное кондуктометрическое титрование находит все более широкое применение для анализа, как в лабораторных, так и в промышленных условиях.