

Гетерогенно - каталитические процессы
.pdf11
двум видам взаим одействия. Кривая 2 см иним ум ом А характеризует ван-дер- ваальсову ф изическую адсорб цию, акривая 1 сб олееглуб оким м иним ум ом Б
— хим ическую адсорб цию. Равновесное расстояние, соответствующее м иним ум у потенциальной энергии, вслучаеф изической адсорб ции (r1) всегда
б ольш е, чем |
при хим ической адсорб ции |
(r2). Кривые 1 |
и 2 |
разделены |
||
потенциальным б арьером , |
т.е. переход |
от ф изической |
к |
хим ической |
||
адсорб ции, какправило, треб ует энергии активации. |
|
|
|
|||
Химическ ая адсо рб ц ия (хемо со рб ц ия) |
представляет соб ой хим ическую |
|||||
реакцию м олекулы (атом а) с поверхностью твердого тела, |
в результате чего |
|||||
об разуются |
поверхностные |
хим ические |
соединения. |
Адсорб ированную |
||
м олекулу и твердоетело надо рассм атривать какединую квантово-хим ическую систем у, в которой необ ходим о учесть и изм енения в электронном состоянии м олекулы в процессе хем осорб ции, и соответствующие изм енения в твердом теле. Квантово-хим ические исследования хем осорб ционной связи развивались вдвухосновныхнаправлениях: 1) об щееописаниесистем ы « м олекула-твердое тело» , рассм атривалось изм енение энергетического спектра м олекулы и твердого тела в зонном приб лиж ении; 2) выделение локальных особ енностей хим ической связи воб разовавш ем ся адсорб ционном ком плексе, ограничиваясь рассм отрением сравнительно неб ольш ого количестваатом ов.
Д есо рб ц ия. Д есорб ция есть отрывчастиц от поверхности вгазовую ф азу или враствор. В б ольш инствекаталитических реакций десорб ция приводит к об разованию конечного продукта. Д есорб ция всегдаэндотерм ична, потом у что энергия активации десорб ции равна сум м еэнергии активации адсорб ции Еа и теплоты адсорб ции Qa: Ed = Ea + Qa.
К И Н Е ТИ К А К АТАЛ |
И ТИ ЧЕ С К И Х Р Е АК ЦИ Й |
С тац ио н арн ы й и к вазистац ио |
н арн ы й реж имы к атализа. В об щем виде |
стационарным является такой реж им , когда в заданном интервале врем ени в каж дой точке реакционного пространства свойства систем ы не изм еняются. Т аким и свойствам и м огут б ыть состав реакционной см еси, скорость реакции,
состояние поверхности катализатора. Ч астным и предельным |
случаем |
стационарности является хим ическоеравновесиевзаданныхусловиях. |
|
П онятие « стационарность» относится и к об разованию, и |
к распаду |
пром еж уточных соединений. Д ля концентрации z каж дого из пром еж уточных
соединений встационарном |
реж им есоб людается равенство |
|
|
||
|
|
dzi/dt = 0, |
|
|
(1) |
означающее, что алгеб раическая сум м а скоростей об разования и |
распада |
||||
пром еж уточных соединений равна нулю. |
Е сли составить такие равенства для |
||||
каж дого |
из пром еж уточных соединений, |
для линейных м еханизм ов м ож но |
|||
выразить |
концентрации |
пром еж уточных соединений |
через |
текущие |
|
концентрации реагентов, |
определяем ые эксперим ентально, |
далее получить |
|||
аналитическое выраж ение для скорости |
реакции и в первом приб лиж ении |
||||
подтвердить или опровергнуть предполагаем ый м еханизм реакции. Э то м ет од квазист ацион арн ы х кон цен т раций , илип рин ци М . Боден шт ей н а (1913 г.).
Рассм отрим , наприм ер, реакцию А → В , протекающую нацентрахZ через двеоб ратим ыестадии:
|
|
12 |
1. |
A + Z Ђ |
AZ |
2. |
AZ Ђ В + Z |
|
О б означим |
концентрации реагентов через а и b, концентрации |
|
пром еж уточныхвеществ, своб одныхцентровкатализатораZ через z, азанятых центров AZ — через z1, константы скорости прям ой и об ратной реакций — через k1 и k–1, k2 и k–2 для реакций (1) и (2) соответственно. О б щая концентрация всехцентровz0 встационарном реж им епостоянна:
|
|
|
|
|
|
|
|
|
z + z1 = z0. |
|
|
|
|
|
систем у уравнений: |
||||||
Т огда, используя принципБоденш тейна(1), напиш ем |
|||||||||||||||||||||
|
|
|
|
|
dz / dt = −k1az + k−1z1 |
+ k2z1 − k−2bz = 0, |
|
|
|
|
|
(2) |
|||||||||
|
|
|
|
|
dz1 / dt = k1az + k−2bz − k−1z1 − k2z1 = 0. |
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
П ри |
|
условии |
равенства |
(1) |
реш ение диф ф еренциальных уравнений |
||||||||||||||||
зам еняется нареш ениеалгеб раическихуравнений. И з систем ы (2) получаем |
|||||||||||||||||||||
|
|
z = |
|
k2 + k−1 |
|
z0, |
z1 = |
|
|
k1a + k−2b |
z0. |
|
|
|
|||||||
|
|
k1a |
|
|
|
|
|
k2 + k−2b + k2 + k−1 |
|
|
|
||||||||||
|
|
|
|
+ k−2b + k2 + k−1 |
|
|
|
|
|
|
|
||||||||||
П ром еж уточные соединения, |
удовлетворяющие |
условию |
(1), |
иногда |
|||||||||||||||||
называют б оденш тейновским и |
продуктам и. |
Квазистационарный |
реж им , при |
||||||||||||||||||
котором |
соб людается условие(1), осуществляется, если врем я ж изни каж дого |
||||||||||||||||||||
из пром еж уточных соединений (τ*) |
значительно м еньш е врем ени протекания |
||||||||||||||||||||
реакции, |
|
т.е. |
|
необ ходим о, |
чтоб ы |
пром еж уточные |
соединения |
|
м огли |
||||||||||||
м ногократно |
об разовываться |
и |
разлагаться |
с |
сохранением |
постоянной |
|||||||||||||||
концентрации во врем ени. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
В принципестационарность характерна для открытых систем |
(проточные |
||||||||||||||||||||
реакторы). |
О днако принцип Боденш тейна (1) |
прим еняли и |
для закрытых |
||||||||||||||||||
систем . |
Квазистационарный реж им |
в них |
м ож ет |
осуществляться, |
если |
||||||||||||||||
изм енение концентрации |
пром еж уточных |
соединений |
в каж дый |
м ом ент |
|||||||||||||||||
врем ени б удет отвечать условию (1) |
по отнош ению к такж е изм еняющим ся |
||||||||||||||||||||
концентрациям |
|
реагентов. |
В |
условиях реакции |
м ож ет возникнуть |
такая |
|||||||||||||||
ситуация, |
когда |
лиш ь |
часть |
пром еж уточных |
соединений |
удовлетворяет |
|||||||||||||||
условию |
|
стационарности, |
а |
другая |
часть |
|
— |
это |
б олее |
долгож ивущие |
|||||||||||
соединения. Е стественно, |
при этом |
сниж ается |
число |
диф ф еренциальных |
|||||||||||||||||
уравнений |
типа (2). В |
систем е останутся |
|
только уравнения для |
истинно |
||||||||||||||||
боденш тейновскихпродуктов.
Принцип квазистационарных концентраций Боденш тейна ш ироко
использовался для получения кинетических уравнений и проверки гипотез о
м еханизм е реакции, о возм |
ож ном участии в реакции того или иного |
пром еж уточного соединения. |
О сновные слож ности в прим енении его для |
катализа связаны с отклонениям и концентрации пром еж уточных веществ от
постоянного их значения вследствие изм енения активности |
катализатора, |
||
отравления поверхности, изм енения внеш нихусловий и т. д. |
|
|
|
В последнее врем я принцип Боденш тейна прим еняется |
для |
изучения |
|
м еханизм а редко. Э то |
вызвано тем , что в катализе (как и |
в гом огенной |
|
кинетике) появились |
ф изические м етоды, позволяющие прям о |
изм ерить |
|
13
пром еж уточныесоединения, вм есто того чтоб ы строить различныегипотезы об ихоб разовании наосновепринципастационарности.
Зак о н |
действую щ их по верхн о стей. Ф орм альным аналогом |
закона |
|||||||
действующих м асс в кинетике гетерогенно-каталитических реакций |
или, |
в |
|||||||
об щем |
случае, в кинетикепроцессов на твердых поверхностях является закон |
||||||||
дей ст вующих п оверхн ост ей . Э тот терм ин б ылпредлож ен И . Л енгм юром |
в1918 |
||||||||
г. прим енительно |
к реакциям |
частиц (м олекул или атом ов) на поверхности |
|||||||
твердого |
тела, |
состоящей |
из определенного |
числа |
статистически |
||||
располож енных адсорб ционных центров, каж дый |
из которых удерж ивает в |
||||||||
адсорб ированном |
состоянии |
одну частицу |
(м олекулу |
или |
атом ). |
||||
Каталитическая реакция протекает за счет превращения этой |
м олекулы |
на |
|||||||
данном |
центре или за счет взаим одействия с другой м олекулой на соседнем |
||||||||
центре. |
П ри этом |
частицы, |
адсорб ированные на |
соседних центрах, |
не |
||||
взаим одействуют друг с другом |
(если они не вступили в реакцию). В |
об щем |
|||||||
случаевреакции уч аствуют какадсорб ированныем олекулы А, В и т.д. , таки своб одныецентры Z
ν1A + ν2B + ν3Z + ... → продукты
и для скорости каталитической реакции rK на адсорб ционных центрах Z поверхности по закону действующихповерхностей м ож но написать
r |
= k |
|
ν |
θ |
ν |
ν |
, |
(3) |
s |
zθ 1 |
2 |
θ 3 |
|||||
K |
|
A |
|
B |
Z |
|
|
гдеz — об щеечисло адсорб ционныхцентровнаединицеповерхности; θА, θВ — доля центров, заполненных адсорб ированным и веществам и А и В , θZ — доля своб одныхцентров. Реакция протекает вм оном олекулярном слое, поэтом у θА + θВ + θZ + θ... = 1. В частном случаезакон действующихповерхностей описывает и процессы адсорб ции, если ν1 = ν2 = 0, и десорб ции, если ν2 = ν3 = 0.
О граничений на |
прим енение закона действующих поверхностей к |
|
кинетике каталитических реакций еще б ольш е, чем |
на прим енение закона |
|
действующих м асс |
к кинетике гом огенных |
реакций. В о-первых, |
адсорб ционный слой неявляется идеальным и предполож ениео равноценности всех центров не подтверж дается эксперим ентам и. В о-вторых, как правило, заполнения θi эксперим ентально изм ерить трудно. Л иш ь в последнее врем я с появлением спектральных м етодов исследования in situ (которые м огут прим еняться вусловияхпротекания каталитической реакции) стало возм ож ным определениечислаадсорб ированныхм олекул. В -третьих, вреальныхреакциях на поверхности, кром езаполнений θi и концентрации своб одных центров θ, в уравнение(3) часто входят такж епарциальныедавления ком понентоврi , когда м олекула реагирует прям о из газовой ф азы. П оследние достиж ения в исследовании кинетики гетерогенно-каталитических реакций показывают, что они м огут протекать на границах островков, поверх прочносвязанного адсорб ционного слоя, по типу цепныхреакций синициированием и об рывом .
К И Н Е ТИ К А И М Е ХАН И ЗМ Э Л Е М Е Н ТАР Н Ы |
Х АК ТО В |
|
Н А П О ВЕ Р ХН О С ТИ |
|
|
Тео рия аб со лю тн ы х ск о ро стей |
и ее примен ен ие к |
к атализу. Т еория |
активированного ком плекса, теория (м |
етод) переходного состояния, или теория |
|
|
|
|
|
|
|
|
14 |
|
|
|
|
|
|
|
|
|
аб солютных скоростей |
реакции, б ыла |
|
|
|
|
|
|
|
|
|||||||
созданав1930-хгодахГ . Э йрингом , М . |
|
|
|
|
|
|
|
|
||||||||
Э вансом и М . |
П оляни. О на позволяет |
|
|
|
|
|
|
|
|
|||||||
рассчитывать |
|
скорости |
элем ентарных |
|
|
|
|
|
|
|
|
|||||
хим ических |
реакций |
на основании |
|
|
|
|
|
|
|
|
||||||
знания об |
электронном |
строении |
и |
|
|
|
|
|
|
|
|
|||||
свойствахм олекулреагентов. |
|
|
|
|
|
|
|
|
|
|
||||||
П о верхн о сти |
по тен ц иаль н о й |
|
|
|
|
|
|
|
|
|||||||
эн ергии. В основетеории аб солютных |
|
|
|
|
|
|
|
|
||||||||
скоростей |
|
|
реакций |
|
леж ит |
|
|
|
|
|
|
|
|
|||
представление |
о |
|
м ногом ерной |
|
|
|
|
|
|
|
|
|||||
поверхности |
потенциальной |
энергии |
|
|
|
|
|
|
|
|
||||||
реакционной систем ы. |
В об щем |
случае |
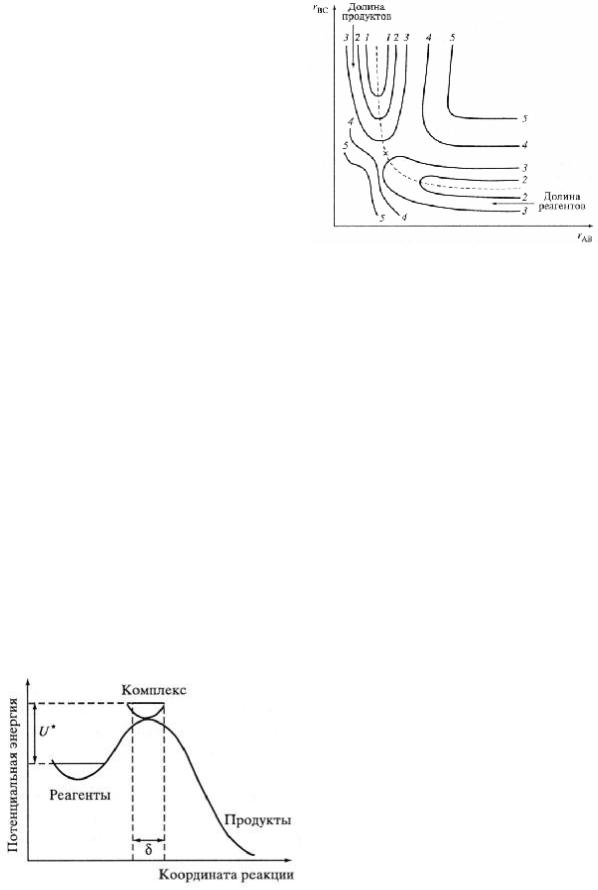
Рис. |
3. |
П ростейш ая |
двухм ерная |
||||||||||
м ногом ерная |
|
|
|
поверхность |
поверхность потенциальной |
энергии для |
||||||||||
потенциальной |
энергии систем ы из N |
реакции |
А + |
В С |
→ AB |
+ |
С |
при |
||||||||
атом ов |
представляет |
ф ункцию |
располож ении всех трех атом ов на одной |
|||||||||||||
потенциальной энергии U от их |
прям ой. |
|
П о |
осям координат |
— |
|||||||||||
внутренних координат или внутренних |
м еж атом ныерасстояния rBC и rAB. Кривые |
|||||||||||||||
степеней |
своб оды (вращательных |
и |
1— 5 — |
уровни |
постоянной |
энергии; |
||||||||||
ш триховая линия — |
координата реакции; |
|||||||||||||||
колеб ательных). В систем еиз N атом ов |
||||||||||||||||
число внутренних степеней своб оды п |
= 3N –6 (или |
= 3N – 5, если ядра |
||||||||||||||
располож ены наодной линии). |
|
|
|
|
|
|
|
|
|
|
||||||
П ростейш ая двухм ерная поверхность потенциальной энергии (п |
= 2) для |
|||||||||||||||
реакции А + В С → АВ + С при располож ении атом овА, В и С наодной прям ой
изоб раж ена на рис. 3. |
П о осям координат — расстояния (r) А— В и В — С . |
Реагентам и продуктам |
реакции наней соответствуют относительно неб ольш ие |
значения потенциальной энергии (долины), разделенныеоб ластью повыш енной потенциальной энергии (потенциальный б арьер). Ш триховую кривую, проходящую по дну долин через потенциальный б арьер, называют координатой
реакции. |
|
|
|
|
|
|
|
|
|
Ч асто |
используют одном ерные схем ы, |
представляющие сечение вдоль |
|||||||
координаты реакции (рис. 4). Н а этих схем ах состояния А + В С |
и АВ |
+ С |
|||||||
являются |
устойчивым и м иним ум ам и, а верш ине потенциального |
б арьера |
|||||||
|
|
|
соответствует |
седловинная точка, |
|||||
|
|
|
или |
точка |
перевала (×). |
В ысота |
|||
|
|
|
потенциального |
|
|
б арьера |
|||
|
|
|
определяется конф игурацией частиц, |
||||||
|
|
|
величиной энергии, необ ходим ой для |
||||||
|
|
|
преодоления |
|
отталкивания, |
и |
|||
|
|
|
некоторым и |
другим и |
ф акторам и. |
||||
|
|
|
Каж дом у |
расстоянию |
|
м еж ду |
|||
|
|
|
реагирующим и |
частицам и |
отвечает |
||||
|
|
|
точканаповерхности потенциальной |
||||||
|
|
|
энергии. |
|
|
|
|
|
|
Рис. 4. П роф иль поверхности потенциальной |
|
|
|
|
|
|
|||
энергии |
вдоль |
координаты |
реакции. |
|
|
|
|
|
|
В ертикальные линии ограничивают об ласть |
|
|
|
|
|
|
|||
активированного ком плекса на |
координате |
|
|
|
|
|
|
||
реакции разм ером |
δ, горизонтальные — |
|
|
|
|
|
|
||
15 |
|
|
Х им ическая реакция рассм атривается как переход |
от конф игурации |
|
реагентов к конф игурации продуктов через точку ABC. Э ту точку (или некий |
||
м алый отрезок траектории реакции длиною |
δ) называют акт ивирован н ы м |
|
комп лексом , или п ереходн ы м сост оян ием . |
Разность E0 |
м еж ду энергиям и |
начального состояния и активированного ком плекса ABC представляет соб ой |
||
энергию активации элем ентарной реакции А + В С . Координата реакции — |
||
наиб олее выгодный путь протекания |
реакции, |
треб ующий наим еньш их |
энергетическихзатрат. |
|
|
Ак тивиро ван н ы й к о мплек с. В |
основе |
т еории акт ивирован н ого |
комп лекса, или т еориип ереходн ого сост оян ия (она ж е— теория аб солютных
скоростей), леж аттри предполож ения. |
|
|
|
||||
1. |
С об людается |
м аксвелл-б ольцм ановское |
равновесие |
м еж ду |
|||
активированным |
ком плексом |
и реагентам и; поэтом у их концентрацию м ож но |
|||||
вычислить спом ощью ф ункции распределения М |
аксвелла-Больцм ана. |
|
|||||
2. |
С корость |
реакции |
отож дествляется |
со |
скоростью |
распада |
|
активированного ком плекса. Реакция протекает спреодолением сам ого низкого потенциального б арьера в точке активированного ком плекса или вб лизи от него.
3. П реодоление |
потенциального б арьера |
вб лизи |
активированного |
||
ком плекса |
описывается как поступательное |
движ ение |
систем ы |
вдоль |
|
координаты |
реакции. |
Д виж ение систем ы (протекание |
реакции) |
вдоль |
|
координаты реакции возм ож но только в направлении об разования продуктов реакции. Э то значит, что активированный ком плекс, если уж он об разовался, не
мож етпревращаться об ратно висходныевещества.
Это свойство коренным об разом отличает активированный ком плекс, описывающий элем ентарный акт реакции, от свойств пром еж уточных
продуктов, описывающих путь хим ического превращения и об наруж иваем ых
ф изическим и |
м етодам и |
исследования. |
У ж е сам ого |
об разования |
активированного ком плексадостаточно для осуществления реакции. |
||||
Активированныеком плексы — это теж ечастицы или ком плексы частиц, |
||||
отличающиеся |
только конф игурацией с повыш енным запасом |
энергии и |
||
неустойчивыевнаправлении координаты реакции. И хсреднееврем я ж изни
τ* = 2πh/kT,
гдеh и k — постоянныеП ланкаи Больцм анасоответственно. П ри об ычныхдля хим ических реакций тем пературах τ* = 10–13 с, т.е. б лизко к врем ени одного колеб ания. Т акие врем ена стали доступны эксперим ентатору относительно
недавно |
с появлением фемт осекун дн ой |
сп ект роскоп ии (ф ем то |
— 10–15), в |
|||||
которой |
для |
идентиф икации частиц |
прим еняли лазеры |
с |
им пульсам и |
|||
продолж ительностью до 10–14 с, т.е. м еньш еврем ени одного колеб ания. |
||||||||
Тео рия |
аб со лю тн ы х |
ск о ро стей |
для |
реак ц ий |
н а |
по верхн о сти. |
||
Активированный ком плекс приним ается адсорб ированным |
на поверхности с |
|||||||
одной существенной особ енностью. О на заключается в том , |
что необ ходим о |
|||||||
учитывать возм ож ность занятия нескольких |
центров поверхности одним |
|||||||
активированный ком плексом . |
Кром е того, в отличие от гом огенных реакций, |
|||||||
16
активированные ком плексы на поверхности, как правило, не об ладают поступательным и вращательным движ ением .
Концентрация активированных ком плексов на поверхности долж на зависеть от числа центров, занятых активированным ком плексом , от числа способ ов осуществления конф игурации активированного ком плекса, от его структуры, структуры поверхности, парам етрареш етки твердого тела, разм еров м олекулы, об щего числацентровнаповерхности.
Рассм отрим об щий случай для элем ентарной реакции наповерхности aA + bB + x(ZC) + y(ZD) + zZ → продукты.
Здесь А и В — вещества, реагирующиеиз газовой ф азы; С и D — вещества, вступающиевреакцию из адсорб ированного состояния; Z — своб одныецентры; а, b, х, уи z — стехиом етрическиекоэф ф ициенты; z — число своб одныхцентров, необ ходим ых для участия в реакции. В действительности, в элем ентарной реакции участвует не б олее трех частиц, считая и своб одные центры. С ледовательно, a + b + x + y + z = 3 и некоторыеиз членовлевой части равны нулю.
С огласно теории аб солютных скоростей |
реакций, |
константа скорости |
|||||
реакции наповерхности |
F y |
|
)e-E0 / kTa, |
b x |
|
||
k |
c |
= χ(kT / h)N(gF ¹ / F F F |
|
(4) |
|||
|
A B ZC |
ZD |
|
|
|
||
гдеχ — трансм иссионный коэф ф ициент, который учитывает возм ож ность того, что не каж дый активированный ком плекс, достигш ий верш ины б арьера и движ ущийся по координатереакции, действительно б удет распадаться и давать
продукты реакции, |
N — число центров на |
единице поверхности; |
g — число |
|
возм ож ных располож ений активированного |
ком плекса на поверхности; |
если |
||
ф иксирован один |
конец активированного ком плекса g = |
1; |
если |
|
активированный ком плексзаним ает двацентра, g = 2, 4 или 6 взависим ости от
сим м етрии реш етки поверхности; F ¹ — статистическая сум м а активированного ком плексаб ез вклада координаты реакции; FA, FB, FZC и FZD
— статистические сум м ы веществ А, В , ZC и ZD, рассчитанные из энергий м икросостояний, отсчитанных от нулевой энергии данной частицы в газовой или адсорб ированной ф азе; E0 — энергетический б арьер реакции (энергия активации при Т = 0).
П ользуясь уравнением (4), м ож но найти разм ерность и порядоквеличины k0 простейш ихгетерогенныхреакций, которыем огут б ыть элем ентарным и. Э ти разм ерности, отнесенные к одном у центру поверхности, приведены в таб л. 1. Адсорб ционный коэф ф ициент выраж ается соответственно всм 3/м олек.
С опоставляя эксперим ентальные значения k0 с расчетным и из таб л. 1, м ож но определить, по каком у м еханизм у протекает реакция.
Т аблиц а 1
Р азмерн о сть и ин тервалы измен ен ия предэк спо н еиц иаль н ы х мн о ж ителей для про стейш их гетеро ген н ы х реак ц ий
Г етерогенная реакция |
Разм ерность |
П орядок |
|
константы скорости |
величины ka |
||
|
|||
1 |
2 |
3 |

|
17 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
1. |
О дноцентровая адсорб ция атом ов |
см 3/(ат• с) |
10–11 — |
10–12 |
||||
2. |
О дноцентровая адсорб ция м олекул |
см 3/(м олек•с) |
10–11 — |
10–15 |
|
|
||
3. |
Д иссоциативная двухцентровая |
см 5/(м олек•с) |
10–28 — |
10–30 |
||||
адсорб ция линейныхм олекул |
|
|
|
|
|
|
|
|
4. |
Д иссоциативная двухцентровая |
см 3/(м олек2 • с) |
10–39 — |
10–42 |
||||
адсорб ция нелинейныхм олекул |
|
|
|
|
|
|
|
|
5. |
М оном олекулярная десорб ция |
–1 |
|
10 |
13 |
|
|
|
с |
|
|
|
|
||||
6. |
Реком б инационная десорб ция |
см 2/(м олек•с) |
10–1 — |
10–8 |
|
|
||
7. |
П оверхностная м играция атом ов |
–1 |
|
10 |
13 |
|
|
|
с |
|
|
|
|
||||
|
|
Т аблиц а 1 |
(п ро д о лж ение) |
|||||
|
1 |
2 |
|
|
3 |
|
|
|
8. |
П оверхностная м играция м олекул |
–1 |
10 |
10 |
— |
13 |
|
|
с |
|
10 |
|
|
||||
9. |
М оном олекулярная реакция |
–1 |
10 |
10 |
— |
13 |
|
|
вадсорб ированном слое |
с |
|
10 |
|
|
|||
|
|
|
|
|
|
|
||
10. Бим олекулярная реакция по ударном у |
см 3/(м олек•с) |
10–10 — |
10–19 |
|||||
м еханизм у (м еханизм И ли-Ридила) |
|
|
|
|
|
|
|
|
11. Бим олекулярная реакция в |
см 2/(м олск•с) |
10–1 — |
10–8 |
|||||
адсорб ированном слое(м еханизм Л енгм юра- |
||||||||
Х инш елвуда) |
|
|
|
|
|
|
|
|
12. Реком б инация радикаловсучастием |
см 2/(м олек•с) |
10–33 — |
10–47 |
|||||
« стенки» кактретьей частицы |
|
|
|
|
|
|
|
|
|
Число ак тивн ы х ц ен тро в. Ч исло активных центров поверхности м ож но |
|||||||
оценить, разделивэксперим ентальноезначениеk0 нарассчитанноепо ф орм уле
(4) по теории аб солютных скоростей. О б щий результат таких расчетов для разных каталитических реакций сводится ктом у, что число активных центров
получается значительно м еньш им , чем |
число атом ов поверхности (1015 см –2). |
|||||||
П ри этом |
на м еталлах число |
активных центров сравнительно велико и |
||||||
приб лиж ается к числу поверхностных атом ов м еталла. |
В озм ож но такж е, что |
|||||||
эксперим ентальные значения k0 заниж ены не только |
из-за м алого |
числа |
||||||
активных |
центров, |
но |
и вследствие низких значений трансм иссионного |
|||||
коэф ф ициентаχ. |
|
|
|
|
|
|
||
Гран иц ы |
примен ен ия |
тео рии |
аб со лю тн ы х |
ск о ро стей. |
Т еория |
|||
аб солютных скоростей |
прим еним а к адиаб атическим процессам , а степень |
|||||||
отклонения |
от |
неадиаб атичности |
учитывается |
трансм иссионным |
||||
коэф ф ициентом |
χ. |
П оследний |
включает в себ я такж е конечное значение |
|||||
вероятности туннельного перехода, т.е. переходавоб ласть продуктоввслучае систем ы, им еющей энергию ниж енулевой энергии активированного ком плекса. Количественный учет неадиаб атичности и туннельного эф ф екта в настоящее врем я возм ож ен лиш ь для простейш ихм оделей газовыхреакций.
В теории аб солютных скоростей нерассм атривается процессоб разования активированных ком плексов. В м есто этого приним ается, что их концентрация соответствует равновесном у распределению М аксвелла-Больцм ана. Н е рассм атривается в этой теории и дальнейш ая судьб а реагирующей систем ы
18
атом ов после пересечения потенциального б арьера. С читается, что такой переход автом атически приводит к об разованию продуктов элем ентарной реакции. О б а эти допущения им еют свои границы прим еним ости и за их пределам и изм еняется не только выраж ение для константы скорости, но и об щий вид кинетического уравнения.
Координата реакции лиш ь внекоторыхслучаяхявляется плавной линией,
какна рис. 4. О б ычно она — |
кривая вм ногом ерном |
пространствевнутренних |
|||
перем енных, отраж ающая |
слож ную ком б инацию |
элем ентарных движ ений |
|||
(поступательных, вращательных, |
колеб ательных, |
электронных), которая |
|||
неодинакова на различных участках. И з-за криволинейности |
координату |
||||
реакции нельзя рассм атривать как независим ую степень своб оды. |
В о-первых, |
||||
м ож ет наруш иться первоначально |
терм ически |
равновесное распределение |
|||
энергии по поступательным |
степеням своб оды, |
а, во-вторых, систем а м ож ет |
|||
вернуться в об ласть реагентов даж е после того, как она уж е прош ла через конф игурацию активированного ком плексавнаправлении продуктовреакции.
Э Л Е К ТР О Н Н АЯ ТЕ О Р И Я К АТАЛ И ЗА Н А П О Л УП Р О ВО Д Н И К АХ
Н аиб олее детально разраб отанной и популярной теорией элем ентарного акта окислительно-восстановительного катализа на полупроводниках б ыла
элект рон н ая |
т еория кат ализа. |
Благодаря простоте и конкретности в 1950- |
|
1960-е годы |
она приоб рела |
б ольш ую популярность. Е е авторы |
. . |
В олькенш тейн, К Х ауф ф е и другие считали, что теория им еет б олее об щий характери относится нетолько кокислительно-восстановительным реакциям .
П олупроводники являются наиб олее распространенным классом катализаторов окислительно-восстановительных реакций. О тсюда вытекает вывод о связи каталитической активности сполупроводниковым и свойствам и катализатора.
С вязь к аталитическ их сво йств по лупро во дн ик о в с уро вн ем Ферми.
Е ще в 1928 г. С .З. Рогинский и А.Ф . И оф ф е высказали м ысль о связи каталитической активности твердого тела счислом электронов проводим ости. В 1933 г. Рогинский показал распространенность полупроводников среди катализаторовокислительно-восстановительного класса. В 1938 г. К. В агнери К. Х ауф ф еэксперим ентально доказали изм енениеэлектропроводности NiO во врем я протекания наего поверхности каталитическихреакций окисления С О и
разлож ения |
N2O. Н аиб олее |
последовательно связь |
м еж ду |
электронным и |
свойствам и |
твердого тела — |
полупроводника — |
и его |
адсорб ционной |
способ ностью и каталитической активностью б ыларассм отренавраб отах . .
Волькенш тейна(1940-1950-егоды).
Согласно В олькенш тейну, возм ож на связь хем осорб ированной частицы с
поверхностью оксида-полупроводника |
трех видов: 1) « слаб ая» связь; 2) |
|
« прочная» акцепторная связь; |
3) « прочная» донорная связь. В первом случае |
|
электрон хем осорб ированной |
частицы |
затягивается на катион реш етки или |
электрон анионареш етки затягивается нахем осорб ированную частицу, которая
остается |
электрически нейтральной. |
В о |
втором |
случае |
электрон |
адсорб ированной на катионе частицы |
взаим одействует со |
своб одным |
|||
электроном |
полупроводника, осуществляя таким |
об разом |
хим ическую связь с |
||

19
Рис. 5. |
Зонная схем а полупроводника (а) и изм енение концентрации адсорб ированных |
||||
частиц |
при |
изм енении полож ения |
уровня Ф ерм и F— F в запрещенной |
зоне |
|
полупроводника (б ). Адсорб ция: η0 — |
« слаб ая» , η– — |
« прочная» донорная, |
η+ — |
||
« прочная» акцепторная. С— С — серединазапрещенной зоны |
|
|
|||
реш еткой. В |
третьем случае атом |
(или м олекула) |
адсорб ируется на анионе |
||
реш етки и вступает во взаим одействиесо своб одной дыркой полупроводника.
Хем осорб ированная м олекула об разует локальныеэнергетическиеуровни
Аи D в запрещенной зоне (рис. 5, а). О б разованию « прочной» акцепторной связи соответствует переход электронанаакцепторный уровень A об разованию « прочной» донорной связи — удаление электрона с донорного уровня D, т.е. переход нанего дырки. С огласно статистикеФ ерм и, относительноесодерж ание
частиц на поверхности, находящихся в состоянии « слаб ой» (h0), « прочной» акцепторной (h+) и « прочной» донорной связи (h–), описывается соответственно ф орм улам и (5)— (7):
η |
0 = |
N0 |
= |
|
|
|
1 |
|
|
|
, |
|
(5) |
||||||
N |
1+ exp{é(v+ - EF )/ kT ù |
+ exp é(w - EF− )/ kT ù} |
|
||||||||||||||||
|
|
|
|
|
|
|
|
ë |
û |
|
ë |
û |
|
|
|
|
|
||
|
+ |
|
N |
+ |
|
|
|
|
|
|
exp é(v+ - E+ )/ kT ù |
|
|
|
|
|
|
||
η |
= |
|
|
|
= |
|
|
|
ë |
F |
û |
|
|
|
, |
|
(6) |
||
|
N |
|
|
|
|
1+ exp{é(v+ - EF+ )/ kT ù |
+ exp é(w - EF− )/ kT ù} |
|
|||||||||||
|
|
|
|
|
|
|
|
ë |
û |
|
ë |
û |
|
|
|
|
|
||
|
− |
|
N |
− |
|
|
|
|
|
|
exp é(w - E− )/ kT ù |
|
|
|
|
|
|
||
η |
= |
|
|
= |
|
|
|
ë |
F |
û |
|
|
, |
|
(7) |
||||
|
N |
|
|
1+ exp{é(v− - EF+ )/ kT ù |
+ exp é(w - EF– )/ kT ù} |
|
|||||||||||||
|
|
|
|
|
|
|
|
ë |
û |
|
ë |
û |
|
|
|
|
|
||
гдеN0, N+, N– — число частиц на поверхности, находящихся вкаж дом из этих |
|||||||||||||||||||
состояний; |
v– — |
|
|
|
расстояние от донорного |
локального уровня D |
до |
зоны |
|||||||||||
проводим ости, |
v+ |
|
|
— |
|
|
расстояние от акцепторного |
локального уровня А до |
|||||||||||
валентной |
зоны; |
|
|
|
EF+ и EF– |
— расстояние от |
уровня Ф ерм и |
до |
зоны |
||||||||||
20
проводим ости или соответственно до валентной зоны (таким об разом EF+ + EF–
равно Еg — ш иринезапрещенной зоны). О чевидно, что η0 + η+ + η– = 1.
И з ф орм ул(5)— (7) видно, что относительноесодерж аниеразличныхф орм
хем осорб ированных частиц |
на поверхности полупроводника при |
установивш ем ся электронном |
равновесии определяется полож ением уровня |
Ф ерм и. Н а рис. 5,б показано изм енениевеличин η0, η+ и η– при перем ещении уровня Ф ерм и в запрещенной зоне от валентной зоны к зоне проводим ости
согласно |
ф орм улам |
(5)— (7). |
Е сли |
предполож ить, что |
каталитическая |
||
активность определяется содерж анием |
какой-либ о из этиххем осорб ированных |
||||||
частиц, м ож но получить уравнение, |
непосредственно связывающее скорость |
||||||
каталитической |
реакции с полож ением |
уровня Ф ерм и. П ри перем ещении |
|||||
уровня |
Ф ерм и |
от |
валентной |
зоны |
к зоне проводим ости |
каталитическая |
|
активность м ож етвозрастать, ум еньш аться или проходить через м аксим ум . |
|||||||
У величение концентрации электронов в полупроводнике, |
т.е. увеличение |
||||||
электронной проводим ости (наприм ер, |
введение прим еси |
Ga2O3 в ZnO), |
|||||
см ещает уровень Ф ерм и (EF) вверх, б лиж е к зоне проводим ости. У величение концентрации дырок, т.е. дырочной проводим ости (наприм ер, введениеLi2O в NiO) см ещает уровень Ф ерм и вниз, б лиж е к валентной зоне. О тсюда на основании сказанного выш евозникает зависим ость каталитической активности от величины и знака электропроводности, от количества донорной или акцепторной прим еси, введенной воксид.
В раб отах В олькенш тейна и других исследователей приведено б ольш ое число схем каталитических реакций на оксидах-полупроводниках, основанных
на представлениях электронной |
теории. Н априм ер, предлагается м еханизм |
||
окисления С О сучастием своб одныхэлектронови дырок: |
|||
1) |
О (1/2O2) + е = O–, |
2) |
O– + С О = С О 2–, |
3) |
С O2– + р = С O2 адс, |
4) |
С O2, адс= С O2. |
В о всехподоб ныхсхем ахпостулировались переходы электронови дырокв различных стадиях реакции. Т аким об разом , электронная теория описывала в основном окислительно-восстановительныереакции.
Е сли принять вданном прим еревкачествелим итирующей стадию 1 или 3, то м ож но получить какрост, таки ум еньш ениекаталитической активности при изм енении полож ения уровня Ф ерм и в запрещенной зоне. Количественных (а
зачастую и качественных) |
подтверж дений электронной |
теории в б ольш инстве |
случаев эксперим ентально |
не получали. В м есто ож |
идавш ихся изм енений |
каталитической |
активности на несколько порядков, а энергий активации |
||||||
каталитической |
реакции |
на 1— 3 |
эВ , |
что соответствовало |
б ы |
ш ирине |
|
запрещенной зоны, |
в эксперим енте наб людали изм енения всего в несколько |
||||||
раз. И зм енения |
электропроводности, |
действительно, происходили в |
|||||
б ольш инстве изученных |
прим еров |
адсорб ции и катализа |
на |
оксидах- |
|||
полупроводниках, но эти изм енения такж еб ыли ниж еож идаем ых.
Уро вен ь Ферми н а по верхн о сти и раб о та вы хо да элек тро н а. В еличины